


Abstract
The β2-adrenergic receptor (β2AR) is prototypic of the large family of G protein-coupled receptors (GPCRs) whose desensitization and resensitization are regulated by intracellular kinases, arrestin proteins, phosphatases, and ill-defined components of the cellular endocytic machinery. The study of β2AR signal transduction and behavior in living cells is technically difficult because of the relatively low cellular expression of the receptor and a lack of useful biological reagents. Availability of a functional β2AR tagged with the highly sensitive Green Fluorescent Protein (GFP) could allow measurements of the various properties of the β2AR. We demonstrate that a fully functional β2AR/GFP can be engineered. In mammalian cells, β2AR/S65T/GFP demonstrates strong, diffuse plasma membrane fluorescence when observed with 480 nm excitation. The fluorescent receptor binds agonist and antagonist, stimulates adenylyl cyclase, undergoes phosphorylation, and is internalized in a manner indistinguishable from wild-type receptor. We then show that its internal trafficking and surface mobility can be determined by measuring only the endogenous fluorescence of the conjugate. β2AR/S65T/GFP was found to be localized on endosomal membranes in living cells within minutes of agonist treatment, and within 15 min it is observed in more complicated structures formed from fusion of multiple endosomes. Finally, its free diffusion (diffusion coefficient, 4.0–12 × 10−9 cm2/sec) was assessed on living cells using photobleaching recovery measurements. This approach and the fidelity of the biochemical properties of the β2AR/S65T/GFP demonstrate that real-time optical measurements of β2AR (as well as other GPCR) interactions and dynamics on living cells are feasible.
Structural features that are common to members of the GPCR superfamily include an extracellular amino terminus, seven transmembrane helical domains connected by alternating intracellular and extracellular loops, and an intracellular carboxyl terminus (tail) of variable length (1-3). The β2AR, which was the first to have its primary structure elucidated by cloning techniques, often has served as a paradigm for other GPCR family members in the elucidation of the structural determinants necessary for normal signal transduction (4). Sections of the β2AR that are crucial for ligand binding are found in the transmembrane domains, and elements that regulate G protein binding and signaling include the intracellular loops and cytoplasmic portions of the transmembrane domains (1, 5). Also, multiple serine and threonine residues in the β2AR carboxyl tail are phosphorylated by GRKs in response to agonist (1, 6). Presumed conformational changes caused by phosphorylation increase receptor affinity for arrestins, proteins that not only desensitize receptor response but also initiate both internalization and resensitization processes (7-9).
Optical methods such as video microscopy, fluorescence recovery after photobleaching, and resonance energy transfer, are available for studying protein dynamics and interactions in intact cells (10-12) but are generally not very useful for GPCRs. In comparison to abundant intracellular proteins such as actin or tubulin, GPCRs are expressed at relatively low levels and so produce marginal signals when tagged with fluorophores or labeled with fluorescent agonists or antagonists, a procedure that often modifies the behavior of these compounds (13). The introduction of foreign epitopes onto receptor cDNA is now a standard technique used to enhance their detection, permitting monoclonal antibody recognition of β2ARs and other GPCRs in flow cytometry or fluorescence microscopy (14). However, even this technique has major limitations, including applicability to living cells, nonstoichiometric labeling of receptors, the eventual dissociation of the antibody from the receptor, and an inability to label intracellular receptors in nonpermeabilized cells.
GFP from the jellyfish Aequorea victoria has been demonstrated to function as a reporter molecule in the fluorescent localization of cytoplasmic proteins, secretory proteins, and locomotor proteins and the monitoring of cell transfection (15-18). It has an inherent green bioluminescence that can be excited optically by blue light or by nonradiative energy transfer (19, 20), and it stoichiometrically labels when integrated into cDNA as either an amino- or a carboxyl-terminal fusion protein. However, its size (238 amino acids) (21) in comparison with the overall size of the β2AR (413 amino acids) and other GPCRs (3, 4) makes it an unlikely candidate for creating a normally behaved GPCR/GFP fusion protein. Interestingly, we report the characterization of what seems to be a fully functional carboxyl-terminal β2AR/GFP fusion protein, β2AR/S65T/GFP (Fig. 1). β2AR/S65T/GFP transiently expressed in HEK 293 cells has normal antagonist and agonist binding, undergoes phosphorylation, activates adenylyl cyclase, and is sequestered (internalized) in response to agonists like wild-type β2AR. Endosomes containing sequestered β2AR/S65T/GFP rapidly fuse into larger vesicular organelles during the early stages of endocytosis, in a manner that has been described for clathrin-mediated non-GPCR internalization. Furthermore, β2AR/S65T/GFP diffusion can be readily measured on the cell surface without the use of exogenous fluorescent tags. The observed behavior of β2AR/S65T/GFP indicates it interacts appropriately with G proteins, GRKs, and arrestins, suggesting that β2AR/S65T/GFP and other similarly conjugated GPCRs should be important tools in vitro or in vivo for optical measurement of biochemical and biophysical processes that are relevant to GPCR signal transduction.

Linear representation of the β2AR/S65T/GFP construct in the plasmid pGFP-N3. As described in Experimental Procedures, the β2AR with its terminal stop codon replaced by a SalI restriction site was inserted in-frame into GFP in the pGFP-N3 polylinker between theSacI and SalI sites. Indicated are positions of the 12CA5 epitope after the amino-terminal methionine and glycine residues of the receptor and the S65T mutation in GFP.
Experimental Procedures
Materials.
125I-Pindolol and [3H]adenine were purchased from DuPont-New England Nuclear (Boston, MA). Propranolol, isoproterenol, and anti-mouse antibody were obtained from Sigma Chemical (St. Louis, MO). Antibody against the 12CA5 epitope was obtained from Boehringer-Mannheim (Indianapolis, IN), and anti-Flag antibody was from Kodak IBI (Rochester, NY). Cell culture media and physiological buffers were obtained from Gibco Life Technologies (Baltimore, MD). Restriction enzymes were obtained from Promega (Madison, WI) or New England Biolabs (Beverly, MA). T4 ligase and plasmid Megaprep kits were from Promega, and Hot Tub DNA polymerase was from Amersham (Arlington Heights, IL). Plasmid-containing variants of GFP were from Clontech (Palo Alto, CA) Methylase negative (Dam−) Escherichia coli, Epicurian SCS 110, were obtained from Stratagene (La Jolla, CA).
Plasmid construction.
A mutant GFP (pS65T-GFP) with a red shifted excitation spectrum and enhanced fluorescence compared with wild-type GFP was attached to the carboxyl terminus of the β2AR (Fig. 1). The starting cassette of the 12CA5 epitope (YPYDVPDYA) tagged β2AR was from the plasmid pBC (22). The (TAA) stop codon following the carboxyl-terminal leucine was replaced using site-directed mutagenesis (23) with an in-frameSalI (GTCGAC) restriction site, and the entire receptor containing the cassette was cut out with SacI andSalI. A variant pS65T-GFP-N3 was constructed from the Clontech vectors pGFP-N3 (amino-terminal protein fusion vector) and pS65T-C1 (carboxyl-terminal protein fusion vector) as follows. The Clontech vectors pS65T-N3 and pGFP-N3 were grown in Dam− E. coli and then digested with the restriction enzymes Msc1 and BspE1. The cassette containing the S65T mutation from pS65T-C1 (base-pairs 784-1328) was ligated at 12° overnight using T4 ligase with the pGFP-N3 backbone along with the second BspE1/Msc1 cassette (1328–2835) isolated from the original digest of pGFP-N3. The resultant vector (pS65T-GFP) was digested withSacI/SalI to remove theSalI/SacI amino-terminal polylinker cassette. The purified vector backbone was ligated with theSalI/SacI cassette containing the 12CA5, epitope-tagged β2AR (plasmid, pβ2AR/S65T/GFP).
Cell culture and transfection.
HEK 293 cells maintained in minimum essential medium or Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 1:100 (v/v) penicillin/streptomycin in a 5% CO2 incubator at 37° were transfected with either 2.5 μg of plasmid containing β2AR/S65T/GFP cDNA or 2.5–5 μg of pBC containing the β2AR using coprecipitation with calcium phosphate (7). All experiments were performed with receptor expressed in transiently transfected cells.
Binding studies.
Binding with radioligand was performed on cell membranes purified from HEK 293 cells transiently expressing epitope-tagged wild-type β2AR or the pS65T-GFP receptor construct as follows.
Membranes were prepared from 100-mm plates of confluent cells that were first washed with PBS and then incubated with 4 ml of ice-cold lysis buffer (10 mm Tris, 2 mm EDTA, pH 7.4) for 10 min. The cells were gently scraped into lysis buffer and further homogenized by treatment with a Brinkman Instruments (Westbury, NY) Polytron PT-3000. The resulting suspension was spun at 40,000 ×g for 20 min at 4° and washed once in lysis buffer, and the membrane pellet was resuspended in 1 ml of lysis buffer.
For antagonist binding, membranes typically expressing 1–3 pmol of receptor/mg of protein in lysis buffer were added to an ice-cold 0.1-ml solution of binding buffer (75 mm Tris·HCl, 12 mm MgCl2, 2 mm EDTA, pH 8). Fifty microliters of binding buffer containing various concentrations of the antagonist 125I-pindolol (0–530 pm) was then added, and the mixture was incubated at room temperature for 90 min. Nonspecific binding was determined in the presence of 20 μm propranolol. Binding was terminated by washing five times with vacuum filtration over Whatman GF/C glass-fiber filters using ice-cold wash buffer consisting of 50 mm Tris and 120 mm NaCl, pH 7.2. Triplicate samples were counted on a Wallace gamma counter.
For agonist competitive binding, fresh membranes with receptor, prepared as above, were incubated in 0.25 ml of binding buffer for 90 min at room temperature in the presence of 185 ± 10 pm 125I-pindolol and of increasing concentrations (0–100 μm) of the agonist isoproterenol. Binding was terminated as described above. Samples were assayed in duplicate or triplicate.
For basal sequestration (22), transiently transfected, washed cells containing receptor were detached using ice-cold PBS and aliquoted into tubes. Total receptor was measured by incubation with the hydrophobic ligand 125I-pindolol alone for 3 hr at 13°, whereas internal receptor was determined by 125I-pindolol binding in the presence of a 100 nm concentration of the hydophilic ligand CGP-12177 and nonspecific binding in the presence of 20 μm propranolol. Basal receptor sequestration was defined as the fraction of specific radioligand binding not competed for by CGP-12177. Data were analyzed with Prism (GraphPAD Software, San Diego, CA).
Whole-cell phosphorylation.
HEK 293 whole-cell phosphorylation was performed as described previously (7) using cells transfected with plasmid-containing wild-type β2AR cDNA or S65T/GFP-conjugated β2AR cDNA. In brief, cells previously labeled for 45 min at 37° with 100 μCi/ml [32P]orthophosphate were treated or not treated for 15 min at 37° with 10 μm isoproterenol in serum-free medium containing 100 μm ascorbate. The cells were washed three times in ice-cold PBS, scraped into a solution (containing 150 mm NaCl, 50 mm Tris, 5 mm EDTA, 10 mm NaF, 10 mm disodium pyrophosphate, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, 0.1 mmphenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ml pepstatin A, pH 7.4), and solubilized for 1 hr at 4° on an inversion wheel. Epitope-tagged receptor was immunoprecipitated on protein A/Sepharose beads using 12CA5 monoclonal antibody. Bound receptor was eluted in 50 μl of SDS sample buffer, and equivalent amounts of receptor protein were loaded onto each lane of an SDS-polyacrylamide gel. The extent of phosphorylation was quantified with a Molecular Dynamics PhosphorImager (Sunnyvale, CA).
Immunofluorescence, interference contrast, and video microscopy.
Cells plated onto ethanol-sterilized glass coverslips were fixed with 3% formaldehyde for 10 min at room temperature. Cells labeled with R-phycoerythrin-conjugated secondary antibody were then washed twice in PBS after formaldehyde fixation, labeled for 30–40 min at room temperature with a 1:300 dilution of anti-12CA5 antibody or a 1:500 dilution of anti-Flag antibody, washed three times in PBS, and then incubated for 30–40 min with secondary antibody. Coverslips were photographed through a Leica epifluorescence microscope connected to an Optronics VI-470 CCD video camera system. Video images were captured or printed with a Sony model UP-5600 MD color video printer with a UPK-5502SC digital interface board, and analyzed with IP Labs software (Signal Analytics, Vienna, VA).
Adenylyl cyclase.
Transfected HEK 293 cells were plated onto 12-well Falcon (Falcon Plastics, Oxnard, CA) dishes 24 hr after transfection at a density of 125,000–150,000 cells/well and treated 4–6 hr before the experiment with 1–2 μCi of [3H]adenine/ml/well. The cells were washed once with fresh medium and treated with varying concentrations of isoproterenol in 0.5 ml of medium containing 10 mm HEPES, 1 mm 1-methyl-3-isobutylxanthine, and 100 μmascorbate, pH 7.4, for the indicated times. The reaction was stopped with a 0.5-ml solution of ice-cold 5% v/v perchloric acid, 0.2 mm cAMP, and 4 μCi of [14C]cAMP/500 ml. The wells were incubated for 20–30 min on ice, and the cell lysate was added to tubes containing 100 μl of 4.2 m KOH and analyzed as described previously (22).
Sequestration.
Flow cytometry analysis was performed as described previously (22) using 250,000–400,000 cells/well and goat anti-mouse R-phycoerythrin-conjugated antibody; 50,000 cells were analyzed for each condition using 520 nm (green) excitation.
Diffusion measurements.
Receptor diffusion was measured by fluorescence recovery after photobleaching (10, 24) using a confocal microscope on the apparatus described previously (25). Diffusion data were analyzed under the following assumptions: (a) diffusion occurred in the x,y plane, (b) the bleach beam was gaussian with diameter ω, (c) photobleaching occurred in a time that was much less than the characteristic diffusion time, (d) the sampling beam had average intensity Iavg and diameter ω̄≪ ω, and (e) the measured signal was the integral of the intensity sampled from the strip defined by x = 0 and −l < y < l. The experiment thus measures how diffusion affects the amplitude of a gaussian photobleached area. A theoretical expression for the intensity at a given point (x,y) can be written as:
Results
Comparison of wild-type β2AR and β2AR/S65T/GFP ligand binding.
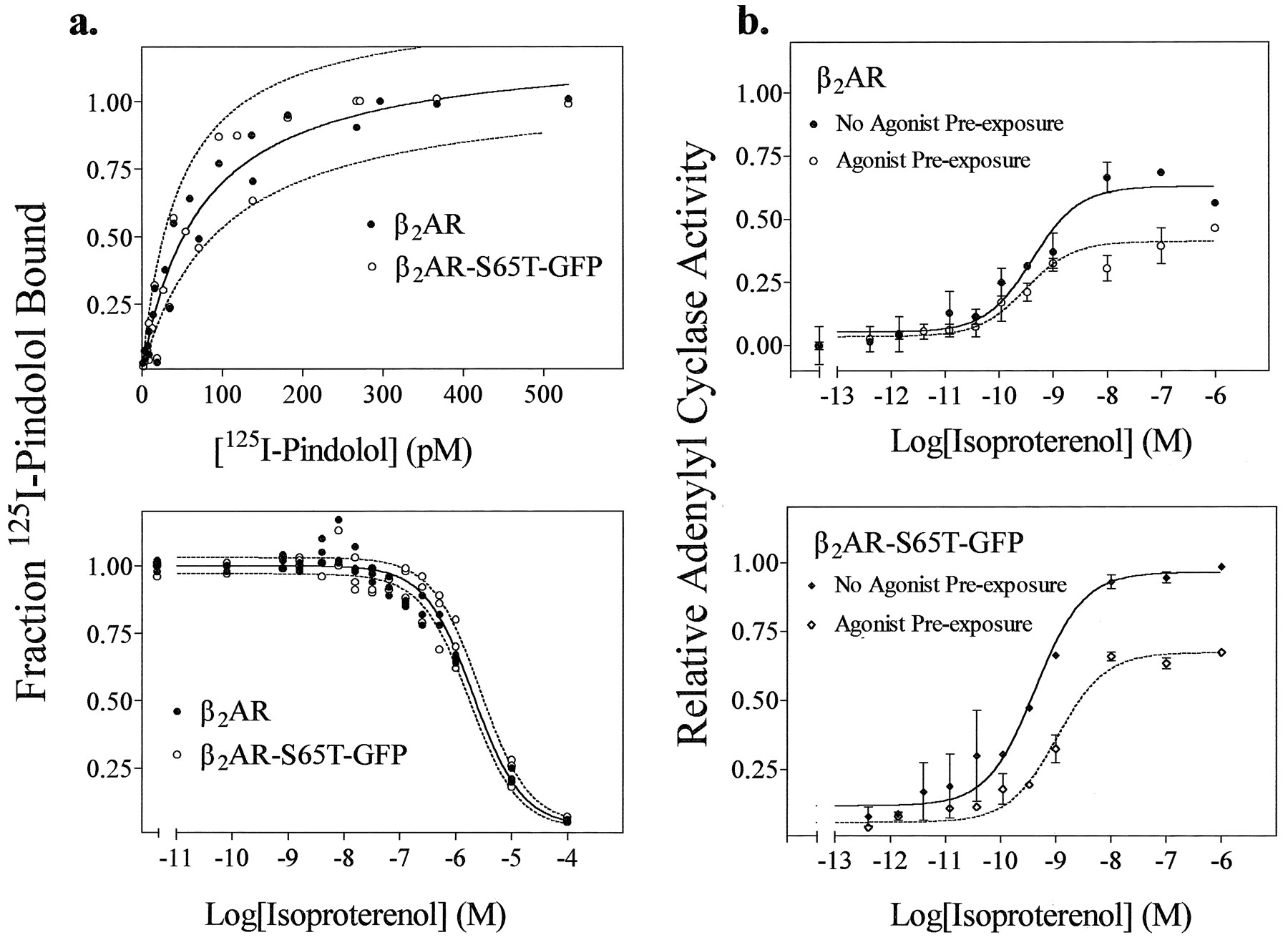
Receptors were transiently expressed in HEK 293 cell membranes to characterize their binding properties to the antagonist 125I-pindolol and the agonist isoproterenol. The antagonist binding isotherms for both receptors are nearly identical, as shown in Fig. 2a (top), with dissociation constants of 73 ± 13 pm for wild-type β2AR and 78 ± 16 pm for the β2AR/S65T/GFP conjugate. The agonist binding characteristics of each receptor were likewise similar (Fig. 2a, bottom). Results were analyzed by a one-site rather than a two-site model. The apparent lack of agonist/high affinity receptor/G protein interactions is commonly observed in transient expression systems with wild-type β2AR or other GPCRs and is presumably the result of the high level ofexpression of receptors (27). In the presence of increasing amounts of the agonist isoproterenol, IC50values for wild-type and conjugated receptor were 2.1 ± 0.3 × 10−6 m and 2.3 ± 0.4 × 10−6 m, respectively corresponding to aKd value for isoproterenol of ∼6.8–7.6 × 10−7 m.

Comparison of the agonist and antagonist binding and adenylyl cyclase activation properties of β2AR- and β2AR/S65T/GFP-expressing cells. a, Membranes prepared from HEK 293 cells containing either wild-type β2AR (2.25 ± 0.95 pmol/mg) or β2AR/S65T/GFP (1.59 ± 1.10 pmol/mg) were exposed to (top) increasing concentrations of the radiolabeled antagonist 125I-pindolol (range, 0–530 pm) to evaluate antagonist binding or a single concentration of 125I-pindolol (185 ± 10 pm) and (bottom) increasing concentrations of the agonist isoproterenol (0–100 μm) to evaluate agonist binding. Paired dashed lines, 95% confidence intervals. Data are from three separate experiments. b, Levels of cAMP were measured in whole HEK 293 cells expressing (top) β2AR or (bottom) β2AR/S65T/GFP that were exposed to increasing concentrations of the agonist isoproterenol for 15 min. Desensitization of the cAMP responses (dashed lines) are shown in cells preexposed to 100 nm isoproterenol for 5 min at room temperature, washed three or four times, and then reexposed to increasing concentrations of isoproterenol for 15 min. The net amount of cAMP accumulation for the desensitization experiments was calculated by subtracting the cAMP accumulation (after preexposure) measured at zero time. Results for each receptor are normalized to the maximal cAMP accumulation of β2AR/S65T/GFP with no isoproterenol before exposure. This maximal cAMP accumulation for the β2AR/S65T/GFP conjugate (b, bottom) is assigned a value of 1, and all other cAMP results are plotted relative to it. The non-normalized levels of cAMP accumulation expressed as 1000 × (total counts of [3H]cAMP accumulation/well of cells)/(total uptake of [3H]adenine/well of cells) were for wild-type β2AR (basal, 4.1 ± 1.4; maximal, 60 ± 9.4; prestimulated basal, 27 ± 14; prestimulated maximal, 54 ± 14) and for β2AR/S65T/GFP (basal, 3.4 ± 2.9; maximal, 80 ± 12; prestimulated basal, 32 ± 12; prestimulated maximal, 81 ± 13). Results are representative of four experiments.
Whole-cell adenylyl cyclase stimulation.
The coupling to G protein of the wild-type β2AR and β2AR/S65T/GFP was investigated by measuring their ability to stimulate whole-cell adenylyl cyclase (Fig. 2b). Basal levels of cAMP production were unaffected by the presence of the GFP, and the conjugated receptor activated adenylyl cyclase as well if not better than the wild-type receptor, for approximately equal receptor expression. The EC50 values for adenylyl cyclase stimulation by isoproterenol were similar: (1.6 ± 2) × 10−10 m for wild-type and (2.0 ± 2) × 10−10 m for the conjugate. In experiments to verify the desensitization of the adenylyl cyclase response (Fig. 2b,dashed lines), the extent of inhibition of the maximal cyclase response was 44 ± 4% for the β2AR and 39 ± 3% for β2AR/S65T/GFP, with no significant shift in EC50 values.
Phosphorylation.
The presence of the large GFP group at the carboxyl terminus of the receptor might be expected to inhibit agonist-mediated receptor phosphorylation (Fig. 3). The agonist-independent and -dependent phosphorylations of wild-type receptor are shown (lanes 1 and 2). The glycosylated form of β2AR migrates on the gel between 56 and 85 kDa and is phosphorylated in response to agonist to a relative level of 129. A nonglycosylated form of the receptor migrates more rapidly below this range (28). β2AR/S65T/GFP phosphorylation is shown (lanes 3 and 4). As expected from the addition of the 28-kDa GFP group, the conjugated receptor migrates more slowly, and this is reflected by the upward displacement of the phosphorylated bands. The faster migrating, nonglycosylated bands are absent in the β2AR/S65T/GFP-expressing cells, suggesting that receptors are either glycosylated more efficiently or not processed to the plasma membrane where they can be phosphorylated. The maximal extent of β2AR/S65T/GFP phosphorylation is 80% that of wild-type, whereas the agonist-mediated increase (maximal - basal) is 60%. Although we cannot formally exclude the possibility that some phosphorylation can occur on the added GFP, it is interesting that the phosphorylation signal remained agonist dependent.

Comparison of the agonist-induced phosphorylation properties of wild-type β2AR and β2AR/S65T/GFP. Receptor expressed in HEK 293 cells transfected with plasmid-containing cDNA for either receptor was assayed for whole-cell phosphorylation as described (see Experimental Procedures). Autoradiograph shows a representative gel of β2AR (lanes 1 and 2) and β2AR/S65T/GFP (lanes 3 and 4) after incubation for 15 min in the absence (−) or presence (+) of 10 μm isoproterenol (Iso). Numbers below the lanes, net phosphorylation (corrected for nonspecific background). The major species of β2AR has a molecular mass ranging from 56 to 85 kDa. β2AR/S65T/GFP has a molecular mass corresponding to 85–110 kDa and is maximally phosphorylated to 80% (101/129) of the wild-type receptor.
Receptor sequestration.
Sequestration of the receptor is an important process in the desensitization/resensitization process, but it can also serve as a measure of how well the phosphorylated receptor can interact with β-arrestin proteins (8, 9). The steady state decrease in surface receptor, as determined by flow cytometry, after 30 min of agonist exposure was 57 ± 5% for wild-type and was 62 ± 11% for GFP-conjugated receptor in HEK 293 cells. Basal sequestration as determined by radioligand binding reflects the agonist-independent intracellular pool of receptor and was estimated at 32 ± 4% for wild-type and 24 ± 1% for β2AR/S65T/GFP, which is in agreement with previous data for transiently transfected wild-type β2AR (7).
Immunofluorescence.
The similar biochemical properties exhibited by wild-type β2AR and β2AR/S65T/GFP suggest that their cellular distribution and trafficking might be similar. The series of micrographs shown in Fig. 4 demonstrate this and also show that β2AR/S65T/GFP can reveal details of β2AR trafficking not readily seen with the use of standard immunofluorescence techniques. In viewing these fluorescence micrographs, it is important to consider that HEK 293 cells normally grow in layers. Therefore, fluorescence from planes above and below the actual focal plane of the lens may contribute to the image. The images in Fig. 4 are representative of what was observed with many cell preparations. Fig. 4, a—d, demonstrates the general appearance and membrane distribution of wild-type β2AR or β2AR/S65T/GFP in different populations of formaldehyde-fixed, nonpermeabilized HEK 293 cells. Wild-type β2ARs (Fig. 4a) were labeled by anti-12CA5 epitope mouse monoclonal IgG, followed by an R-phycoerythrin secondary antibody. These phycoerythrin-labeled β2ARs appear orange when excited by green light. β2AR/S65T/GFP fluorescence (Fig.4b) is excited strongly by blue light and appears green. The fluorescence distributions of both receptors are characteristic of a plasma membrane labeling pattern. Many cells in the plane of focus have enhanced peripheral staining or uniform staining over the nuclear region. The absence of enhanced perinuclear fluorescence, a characteristic of cytoplasmic distribution, is expected for the wild-type receptor because the cells were not permeabilized. The similarity in the pattern for the β2AR/S65T/GFP distribution in Fig. 4b suggests that β2AR/S65T/GFP is also membrane bound. Fig. 4, c and d, shows interference contrast images of fields a and b. Fig. 4, e–h, further demonstrates the plasma membrane distribution of the β2AR/S65T/GFP by exploiting the photoinstability of R-phycoerythrin. Fig. 4e shows an image of a fixed, nonpermeabilized, green-illuminated field of cells transfected with only 12CA5 epitope-tagged β2AR/S65T/GFP. These cells were labeled with mouse monoclonal anti-12CA5 antibody followed by R-phycoerythrin-conjugated goat anti-mouse antibody. The fact that these cells are not permeabilized indicates that only plasma membrane receptors, not internalized receptors, are detected. Before this cell field was viewed with blue light to excite the S65T/GFP fluorescence, the R-phycoerythrin dye was photolyzed with a 30-sec exposure to green light. After this treatment, the R-phycoerythrin-labeled receptors are no longer visible with green illumination (Fig. 4g). (After a similar photolytic treatment with green light, phycoerythrin-labeled wild-type β2ARs were not visible with green or blue illumination; data not shown.) With blue illumination of the same field (Fig. 4f), these S65T/GFP-conjugated β2ARs produced a strong green fluorescence and seemed to be distributed on the cells in a pattern nearly identical to that produced by the antibodies. Fig. 4h is an interference contrast image of the field. This indicates that β2AR/S65T/GFP is indeed predominantly membrane bound because fluorescence detectable from internal β2AR/S65T/GFP should have significantly changed the appearance of the image. Although Fig. 4, e–h, shows that the agonist-independent localization of β2AR/S65T/GFP is similar to that of wild-type receptor, Fig. 4, i–l, further suggests an equivalence between the agonist-mediated translocation of these receptors. Shown are images of a single cell population previously transfected with cDNA for both Flag epitope-tagged wild-type β2AR and 12CA5 epitope-tagged β2AR/S65T/GFP. These cells were exposed to the agonist isoproterenol for 30 min and then fixed and permeabilized for R-phycoerythrin antibody labeling using an anti-Flag primary antibody. The round endocytic vesicles observed by phycoerythrin fluorescence (Fig. 4i) are still visible in the β2AR/S65T/GFP image (Fig. 4k) despite phycoerythrin photolysis (Fig. 4j), indicating that the receptors internalize along the same pathway. However, the S65T/GFP internalized receptor seems to produce a sharper, higher-contrast image than dye-conjugated receptor, which is indicative of a better signal-to-noise ratio and perhaps also of a decreased labeling efficiency of antibody for internalized epitope. The internalization of β2AR/S65T/GFP in response to agonist can be monitored in real time. Fig. 4l shows the interference contrast image of the field in Fig. 4, i–l. The cells in Fig. 4, m–p, are representative of how the cells appear after 5 or 15 min of agonist exposure. Cells containing β2AR/S65T/GFP exhibit numerous well-defined, internalized vesicles that form against the plasma membrane within 5 min of agonist treatment (Fig. 4m). Within 15–30 min, they seem to coalesce into vesicles that are fewer, brighter, and larger (Fig. 4n). Computerized edge enhancement of digitized images (Fig. 4, o and p) demonstrate the differences in the vesicular organelles. The vesicles observed at 5 min are spherical with a clear center (Fig. 4o), which contrasts with the ellipsoidal, multiwalled vesicles that develop later (Fig. 4p) and can be observed to form in real time.

Interference contrast and fluorescence micrographs of wild-type β2AR and β2AR/S65T/GFP distribution in HEK 293 cells. a–d, Images of one cell population containing only antibody-labeled wild-type β2AR and another cell population containing only β2AR/S65T/GFP. Fluorescence (a and b) and corresponding interference contrast (c and d) fields are shown. e–h, Distribution of only β2AR/S65T/GFP in a single cell population. In these unpermeabilized cells, the receptor is visualized using (e) fluorescent antibody (e) or (f) the endogenous receptor fluorescence. g, Same field as in e but after the R-phycoerythrin conjugated secondary antibody signal was bleached. h, Interference contrast field. i–l, Agonist-mediated distribution of internalized wild-type β2AR and β2AR/S65T/GFP in a single population of HEK 293 cells. Cells were transiently transfected with cDNA for (i) wild-type, Flag epitope-tagged β2AR or (j) 12CA5 epitope-tagged β2AR/S65T/GFP. After incubation at 37° for 15 min in medium containing 10 μmisoproterenol, the cells were fixed with 3% formaldehyde in PBS. Both primary antibody labeling by mouse monoclonal anti-Flag epitope antibody and secondary antibody labeling by R-phycoerythrin-conjugated goat anti-mouse antibody were in a permeabilization buffer (0.1% saponin/PBS). j, Intrinsic GFP fluorescence of the field shown in i after photobleaching (k). m–p, Time dependence of agonist-mediated β2AR/S65T/GFP distribution using endogenous receptor fluorescence. Note that vesicle appearance and distribution change between 5 min (m and o) and 15 min (n and p). These 760 × 494-pixel camera images do not fully reflect the quality of the fields viewed through the microscope, although the quality of the fluorescence images using GFP-conjugated receptor is better than that obtained using antibody. Computer enhancement of the vesicles (o and p) was performed to bring out details observed by the eye. Magnification: a–d, 76×; e–l, 380×; and m–p, 500×.
β2AR/S65T/GFP diffusion.
The room temperature translational diffusion of the receptor on live HEK 293 cells was measured using fluorescence recovery after photobleaching. A 10-μm2 area of cell membrane was exposed to a brief high intensity laser pulse to photolyze a fraction of the illuminated GFP conjugate, resulting in a rapid decrease in the monitored fluorescence intensity (Fig. 5, top). The recovery curve reflects the rate of return of nonphotolyzed β2AR/S65T/GFP to the bleached area (bottom,dark-gray spot). The results indicate that free β2AR/S65T/GFP diffuses at ∼4–12 × 10−9 cm2/sec with a mobile fraction of 75 ± 20%.

Diffusion of β2AR/S65T/GFP on the plasma membrane of HEK 293 cells. Top, characteristic appearance after photobleaching of a typical β2AR/S65T/GFP recovery curve. Bottom, gray-scale representation of the confocal, fluorescence image of the cell just after GFP photolysis (occurring in the site outlined by thewhite square) by a gaussian-profile laser beam.
Discussion
Optical studies in cultured cells of β2ARs in particular and of GPCRs in general are difficult due to the small number of expressed membrane receptors. Neither fluorescent ligands nor antibody techniques have adequately overcome this constraint. An ideally labeled receptor should be relatively unperturbed by its fluorescent tag, exhibit little or no change in its biochemical or biophysical behavior, have a large fluorescence signal above background when excited by visible light in addition to being photostable, and be stoichiometrically labeled. We characterized a fluorescent β2AR analogue, β2AR/S65T/GFP, that exhibits all of these properties. This is the first biochemical characterization of a mammalian membrane receptor or GPCR fused to GFP (29). Our data demonstrate that the ligand binding, second messenger stimulation, agonist-mediated phosphorylation, and sequestration properties of β2AR/S65T/GFP closely resemble those of wild-type receptor. Furthermore, the conjugate produces a highly specific fluorescent signal that permits identification of a coalescence phase of endocytic vesicles containing sequestered receptors in either live or minimally fixed cells with standard, visible light epifluorescence microscopy. These data suggest that the size of the GFP adduct does not significantly change the inherent physical or biochemical behavior of the β2AR.
Two characteristics of GFP found in earlier studies that diminished its usefulness in mammalian cell lines were the need to excite it at UV wavelengths to obtain a maximal fluorescent signal (major excitation peak at 395 nm and minor excitation peak at 470 nm) and decreased fluorescence at temperatures of >23° (30-32). Both problems are apparently corrected by the use of the S65T/GFP mutant. The serine-to-threonine mutation improves its visible excitability, and very adequate fluorescent signals can be obtained with non-UV observation (33). Also, colocalization with antibody did not reveal any pockets of nonfluorescent receptor, suggesting that the oxidation and formation of the GFP peptide chromophore (33) may be enhanced by the local environment of the β2AR.
Glycosylation of the β2AR plays a significant role in its appropriate trafficking to the plasma membrane (28). The gel in Fig. 3shows that β2AR/S65T/GFP expresses less nonglycosylated receptor compared with the glycosylated form than wild-type receptor, suggesting that the conjugate is either more efficiently glycosylated or it is not processed to the plasma membrane where it can be phosphorylated. These results suggest that the β2AR carboxyl tail may play a role in the normal processing of the receptor before membrane insertion. It has been reported that some cytosolic GFP fusion proteins form insoluble aggregates (33, 34) after synthesis. However, this does not seem to be a problem with the β2AR conjugate because essentially uniform membrane staining has been observed in the absence of agonist.
Characterization of the agonist-mediated internalization pathways of the β2AR has recently been of particular interest. We were recently able to demonstrate that the proteins that regulate rapid β2AR agonist-mediated desensitization, GRKs and arrestins (8), and that are expressed endogenously in 293 cells1 are also responsible for direction of receptor internalization through probable dynamin- and clathrin-dependent mechanisms (35). Our data showing that β2AR/S65T/GFP is phosphorylated and sequestered normally indicate the transmembrane regions or intracellular loops responsible for G protein, GRK, or arrestin binding are probably still accessible despite the presence of GFP. Also, the observation that β2AR-containing endosomes rapidly fuse with larger structures is similar to what has been observed for non-GPCRs that constitutively internalize in clathrin-coated pits (36). Further studies with β2AR/S65T/GFP in permanently transfected cells should make it possible to microscopically follow nearly the entire receptor sequestration and recycling pathway in a single experiment. It might also be possible to identify the cellular organelles used in receptor down-regulation and to finally determine whether sequestration and down-regulation use the same endocytic pathways. Furthermore, using this approach, it might be possible to identify other trafficking pathways that are used by other members of the GPCR family, such as the angiotensin II type 1A receptor (35).
In an earlier study, the measurement of β2AR diffusion on live cell membranes was reported using a fluorescent analogue of alprenolol (37). However, controversy subsequently developed regarding the specific-to-nonspecific labeling by this probe of the β2AR in intact cells (13, 38). In contrast, the GFP tag is incorporated into each β2AR/S65T/GFP expressed by the cell, and short of significant receptor proteolysis, all labeling is specific. The diffusion rate of the mobile GFP-conjugated receptor fraction is consistent with the free diffusion rates found for other membrane proteins, such as the low density lipoprotein receptor in membrane vesicles (39). Thus, if the unconstrained mobility of β2ARs in the plane of the membrane reflects their diffusibility after agonist-mediated arrestin desensitization, their diffusion to coated pits or other vesicles would not be a rate-limiting step in sequestration (35).
Many of the molecular components mediating agonist-dependent GPCR coupling to heterotrimeric G proteins, activation of effector molecules, desensitization, and resensitization have been elucidated. However, determination of the kinetics as well as the biophysical properties of these interactions, especially in living cells, has not been possible. The demonstration that a GPCR tagged with GFP is essentially unperturbed in its cellular and biochemical properties and the availability of other variants of GFP with distinct spectral properties suggest that it may be possible to probe these questions in whole cells using real-time measurements by resonance energy transfer. Our data with the β2AR/S65T/GFP demonstrating that the biochemistry of a representative GPCR/GFP conjugate can be sufficiently preserved to investigate important biological questions in signal transduction is a necessary first step in this endeavor.
Footnotes
- Received September 10, 1996.
- Accepted October 30, 1996.
-
Send reprint requests to: Dr. Marc G. Caron, Depts. of Cell Biology & Medicine, Duke University Medical Center, Box 3287, Durham, NC 27710. E-mail: caron002{at}mc.duke.edu
-
↵1 L. Menard, personal communication.
-
This work was supported by National Institutes of Health Grant NS19576 and a Bristol Myers Squibb Unrestricted Grant Award (M.G.C) and by National Institutes of Health Grant HL03422 (L.S.B.). S.S.G.F. was the recipient of a Michael Smith Postdoctoral Fellowship from the Medical Research Council of Canada.
Abbreviations
- β2AR
- β2-adrenergic receptor
- GPCR
- G protein-coupled receptor
- GRK
- G protein-coupled receptor kinase
- GFP
- Green Fluorescent Protein
- HEK
- human embryonic kidney
- PBS
- phosphate-buffered saline
- SDS
- sodium dodecyl sulfate
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}