


Abstract
An NP(X)nY motif is highly conserved among G protein-coupled receptors and is similar to an NPXY motif involved in receptor-mediated endocytosis for several non-G protein-coupled receptors. We investigated the role of this motif in α1B-adrenergic receptor function and regulation. Y348A α1B-adrenergic receptors in which this sequence was mutated from NPIIY to NPIIA were prepared by site-directed mutagenesis and transfected into Chinese hamster ovary cells. Binding of the antagonist prazosin to Y348A receptors was similar to that of wild-type receptors, but affinity of the Y348A receptors for the agonist epinephrine was increased by ∼10-fold. Despite this increase in agonist binding affinity, the Y348A mutation completely uncoupled the receptors from stimulation of phosphoinositide hydrolysis and mobilization of intracellular Ca2+. Exposure of cells expressing Y348A receptors to the agonist epinephrine resulted in receptor “sequestration,” defined as a loss of cell surface receptors accessible to radioligand in binding assays with intact cells on ice, similar to that for the wild-type receptor. In contrast, Y348A receptors did not undergo “endocytosis” into the light vesicle fraction in sucrose density gradient centrifugation assays, as did the wild-type receptor. These results (i) indicate an important role for Tyr348 in coupling the α1B-adrenergic receptor to G protein and subsequent effector activation, (ii) provide further evidence that α1B-adrenergic receptor internalization can be separated into a sequestration step and an endocytosis step, (iii) indicate that effector activation and second messenger formation are not required for the sequestration of these receptors but may be involved in endocytosis, and (iv) provide a useful new tool for further investigation of the nature of the subcellular compartments and the molecular modifications involved in the multiple steps involved in internalization of G protein-coupled receptors.
Binding of agonist ligands to cell surface G protein-coupled receptors leads not only to activation of the signal transduction pathways activated by these receptors but also to a series of adaptive changes that regulate the subsequent responsiveness of the receptor. The adaptive changes that occur on agonist exposure generally lead to a decrease in subsequent responsiveness and have been best characterized in the case of β2-ARs (1-4). These changes include a rapid desensitization of the ability of the receptor to couple to G proteins and to induce effector enzyme activation, often referred to as “uncoupling”; a rapid redistribution of receptors within the plasma membrane and/or into intracellular vesicles, variously referred to as “internalization,” “sequestration,” or “endocytosis”; and a slower loss of radioligand binding sites, referred to as “down-regulation.” The role of receptor phosphorylation by multiple kinases in the rapid uncoupling step of desensitization has been firmly established (1-4). In contrast, the cellular and molecular changes involved in receptor internalization and down-regulation are less clear.
The amino acid sequence NPXY was shown to be an internalization signal for the low-density lipoprotein receptor (5), and related tyrosine-containing internalization signals are present in the intracellular domain of various other receptors that are internalized by a pathway involving clathrin-coated pits and vesicles (6, 7). In these receptors, this motif is present in the cytoplasmic tail but near the single plasma membrane-spanning domain of the receptor. A similar NP(X)nY motif is highly conserved in many G protein-coupled receptors, but in these receptors this domain lies within the putative seventh transmembrane segment near the cytoplasmic face of the plasma membrane. This sequence has been postulated to play a role in the agonist-induced internalization of these receptors, which may also occur via clathrin-coated pits and vesicles. Initial studies with β2-ARs provided evidence for a role of this sequence in receptor internalization (8, 9), but subsequent studies suggested that the effects on internalization caused by altering this sequence were secondary to effects on receptor phosphorylation and subsequent binding of β-arrestin (10-12). Studies with two other G protein-coupled receptors, the AT1 angiotensin receptor (13-15) and the GRP receptor (16), indicated a role for this sequence in receptor signaling but not in internalization.
Several previous studies have provided evidence for sequestration of G protein-coupled receptors within the plasma membrane as a likely intermediate step toward endocytosis of receptors into intracellular vesicles (17-22). Sequestration of these receptors is typically assessed by their relative inaccessibility to hydrophilic or lipophilic ligands in assays with intact cells at low temperature; redistribution to endocytotic vesicles has been assessed by sucrose density gradient centrifugation (1, 2). The existence of this intermediate sequestered form was particularly apparent in our previous studies with α1B-ARs in DDT1 MF-2 cells, in which agonist alone induced only sequestration, and phorbol ester-mediated protein kinase C activation was required along with agonist to induce endocytosis from the plasma membrane into the light vesicle fraction detected with sucrose density gradient centrifugation (21). Recent immunofluorescence microscopy experiments also presented clear evidence for multiple steps in the internalization pathway for β2-ARs (22). In the current study, we investigated the possible roles of the NPIIY sequence of hamster α1B-ARs in receptor function and in both the sequestration and endocytosis steps of internalization by mutating Tyr348 of this motif to alanine (Y348A). Our results provide evidence for a role of Tyr348 in the endocytosis step as well as in functional coupling of these receptors to PI hydrolysis and Ca2+ mobilization.
Experimental Procedures
Materials.
Cell culture medium, serum, trypsin, G418, and LipofectAMINE reagent were from GIBCO BRL (Grand Island, NY). The Muta-Gene In Vitro Mutagenesis Kit was obtained from BioRad (Richmond, CA); other enzymes were from New England Biolabs (Beverly, MA). [3H]Prazosin was from DuPont-New England Nuclear (Boston, MA), and [3H]inositol was from Amersham (Arlington Heights, IL). Fura-2/AM was from Molecular Probes (Eugene, OR). Epinephrine, prazosin, phentolamine, sucrose, and other biochemicals were from Sigma Chemical (St. Louis, MO).
Construction and transfection of wild-type and mutated α1B-AR plasmids.
The cDNA encoding the hamster α1B-AR (23) in the plasmid pRC/CMV was kindly provided by Dr. R. Dale Brown (University of Illinois at Chicago) (24). The cDNA encoding the α1B-AR was cleaved from the plasmid at theHindIII/XbaI sites and subcloned to phage M13mp18 also digested with HindIII/XbaI. The codon change from tyrosine to alanine at position 348 was made by oligonucleotide-directed mutagenesis using the BioRad Muta-Gene M13 kit based on the method of Kunkel (25). The sequence of the mutagenic primer was AACCCCATCATCGCACCGTGCTCCAGC, in which the underlined nucleotides indicate the tyrosine-to-alanine mutation at residue 348. After confirmation of the mutation by DNA sequencing, the mutated α1B-AR was cut from M13mp18 usingHindIII/XbaI and subcloned into the expression vector pRC/CMV. The wild-type hamster α1B-AR also was subcloned into the same vector for parallel transfection.
The wild-type and Y348A plasmids were stably transfected into CHO-K1 Chinese hamster ovary cells. The day before transfection, CHO-K1 cells were plated at 300,000 cells/35-mm dish in F12 medium supplemented with 10% fetal bovine serum. The cells were then transfected using LipofectAMINE according to the manufacturer’s protocol. Clones resistant to G418 (400 μg/ml) were isolated and screened for α1B-AR expression by [3H]prazosin binding to intact cells at 37° as previously described (26).
Cell culture.
Cells were maintained in monolayer culture in Ham’s F12 medium supplemented with 10% fetal bovine serum and 200 μg/ml G418 at 37° in a humidified incubator with a 5% CO2 atmosphere. Cells from confluent flasks were trypsinized and plated in culture dishes at 3000–5000 cells/cm2. Cells were typically used for experiments on the fourth day of culture.
Membrane binding assays.
For membrane preparation, cells grown on 150-mm dishes were rinsed twice with 10 ml of ice-cold lysis buffer (20 mm HEPES, pH 7.4, 2 mm EDTA) and allowed to swell for 10 min on ice. Cells were then scraped from the dish with a rubber policeman and homogenized by 30 strokes with a Teflon/glass homogenizer. The homogenate was centrifuged for 30 min at 20,000 rpm in an SM24 rotor in a Sorvall RC5B refrigerated centrifuge. The membrane pellet was resuspended in binding buffer (20 mm tris[hydroxymethyl]aminomethane, pH 7.4, 2 mm MgCl2, 140 mm NaCl) with a Tissumizer (Tekmar, Cincinnati, OH), and the fresh membrane suspension was used in radioligand binding assays. Membranes were incubated with [3H]prazosin in binding buffer for 60 min at 37° in a shaking water bath. The reactions were stopped by filtration over Schleicher & Schuell no. 30 (Keene, NH) or Whatman (Clifton, NJ) GF/B glass-fiber filters on a Brandel (Gaithersburg, MD) cell harvester and washing three times with 4 ml of wash buffer (10 mmtris[hydroxymethyl]aminomethane, pH 7.4, 140 mm NaCl). Radioactivity associated with the filters was quantified by liquid scintillation counting. For saturation assays, six or seven different concentrations of [3H]prazosin were used. For competition binding assays, [3H]prazosin was used at 1.7 nm, and the concentrations of competing ligands were varied. In all cases, nonspecific binding was defined as that occurring in the presence of 10 μm phentolamine.
PI hydrolysis assays.
Assays were essentially as described previously (26, 27). Cells grown on 35-mm dishes were labeled for 18–24 hr with 2 μCi of [3H]inositol in 1 ml of inositol-free high-glucose DMEM supplemented with 10% fetal bovine serum. After labeling, cells were rinsed once with DMEM/HEPES (DMEM, 20 mm HEPES, pH 7.4) and then stimulated for 20 min with various concentrations of epinephrine in DMEM/HEPES containing 10 mm LiCl. Labeled compounds were then extracted from the cells with methanol, and chloroform and water were added as previously described (27). Inositol phosphates in the resulting aqueous phase were separated on Dowex 1-X8 (formate form) columns. Total inositol phosphates were eluted with 8 ml of 1 m ammonium formate and 0.1 m formic acid. Radioactivity in a 3-ml portion of the eluate (a) and a 0.375-ml portion of the organic phase containing the inositol phospholipids (b) was determined by liquid scintillation counting. The percentage of conversion of inositol phospholipids to inositol phosphates was then calculated by the formula [a/(a + b)] × 100%.
Ca2+ mobilization assays.
Cells grown on glass coverslips were loaded with 7.1 μm Fura-2/AM for 30 min at 37° in Ringer’s solution containing 148 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1.6 mm Na2HPO4, 0.4 mmNaH2PO4, 1.5 mm CaCl2, and 5 mm d-glucose. After loading, cells were washed twice and then incubated again for 20 min in Ringer’s solution to allow intracellular dye cleavage. The coverslips were inserted into the chamber, and Fura-2 was excited at wavelengths of 350 and 380 nm using a PTI Deltascan System as previously described (28). The values for Ca2+ were calculated as follows: [Ca2+] =Kd [(R − Rmin)/Rmax − R)]×(380min/380max), where Rminand Rmax are the fluorescence ratios in the absence (with 3 mm EGTA) and presence of saturating Ca2+ (3 mm), respectively, and Kd = 224 nm.
Cell surface accessibility by assays of radioligand binding on ice.
In the assays of cell surface accessibility of binding sites, we used [3H]prazosin binding to intact cells on ice, similar to previous studies (21). Cells in growth medium on 35-mm dishes were exposed to 10 μm epinephrine plus 1 mm ascorbate for 30 min at 37° to induce redistribution. Control cells were exposed only to the 1 mm ascorbate vehicle. Cells were rinsed twice with 2 ml of Ham’s/HEPES (Ham’s F12 medium, 20 mm HEPES, pH 7.4, 2 mm EDTA) and then incubated on ice for 4 hr with 1.7 nm[3H]prazosin in Ham’s/HEPES. Cells were then rinsed twice with 2 ml Ham’s/HEPES containing 10 μmphentolamine to remove unbound radioligand and dissolved in 1 ml of 0.2n NaOH. Radioactivity associated with the dissolved cells was assessed by liquid scintillation counting. Nonspecific binding was defined as that occurring in the presence of 10 μmphentolamine.
Receptor redistribution by sucrose density gradient centrifugation assays.
Sucrose density gradient centrifugation assays of receptor endocytosis were similar to those previously described (20, 21, 26). Cells grown on 100-mm dishes were given fresh medium on the day before the experiment. Cells were exposed to 10 μm epinephrine or vehicle for 30 min at 37° to induce internalization. Cells were rinsed twice with 10 ml of ice-cold wash buffer and then twice with ice-cold lysis buffer and allowed to swell for 10 min on ice. Cells were then lysed by scraping from the dishes in 0.8 ml of lysis buffer with a rubber policeman. This lysate was layered on top of a discontinuous sucrose density gradient consisting of 1.7 ml of 15% sucrose, 5.0 ml of 30% sucrose, and 2.5 ml of 60% sucrose. Samples were centrifuged at 28,000 rpm for 60 min at 4° in an SW41 rotor in a Beckman L8–70 refrigerated ultracentrifuge. Fractions of 0.8 ml each were then collected from the top of the tubes. Binding of [3H]prazosin (850 pm) to the membranes in each fraction was then determined essentially as described above.
Data analysis.
Nonlinear regression analysis of saturation and competition binding assay and dose-response curve data was performed with Prism 2.01 (GraphPAD Software, San Diego, CA). Data are presented as the mean ± standard error.
Results
Stable transfection and selection of cells.
CHO-K1 cells were transfected with plasmid pRC/CMV containing either the wild-type or the Y348A-mutated α1B-AR sequence, and stable transfectants were isolated by G418 resistance. Similar numbers of positive clones, assessed by preliminary intact cell [3H]prazosin binding assays, were obtained for both forms of the receptor. The range of stable receptor expression levels was also similar for the wild-type and Y348A clones. All subsequent experiments reported below were performed with multiple clones to ensure that the properties observed were not unique to a specific clone.
Radioligand binding assays.
The affinity for the radioligand [3H]prazosin measured in saturation binding assays with membrane preparations (Table 1) was not significantly different between wild-type and Y348A α1B-ARs. The B max values for the clones characterized in these assays also covered a similar range for wild-type and Y348A receptors (Table 1).
Saturation and competition binding assay data for wild-type and Y348A α1B-ARs
Binding of the antagonist prazosin and the agonist epinephrine to wild-type and Y348A receptors was assessed in membrane competition binding assays (Table 1). As in the saturation assays, binding of the antagonist prazosin was not different for the wild-type and Y348A receptors. In contrast, the Y348A receptors exhibited ∼10-fold higher affinity for the agonist epinephrine than did the wild-type receptors (Table 1).
PI hydrolysis and Ca2+ mobilization assays.
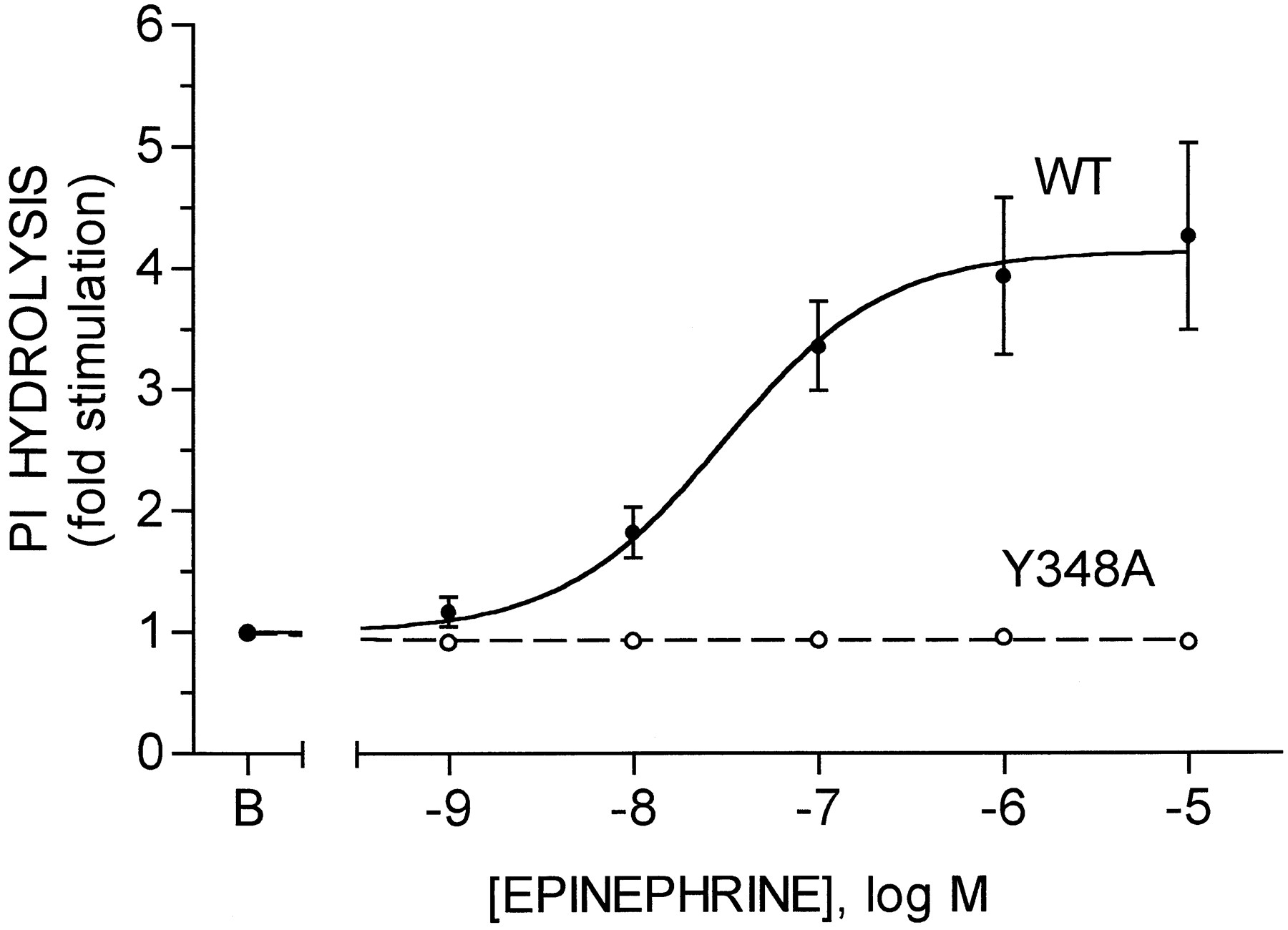
Receptor coupling to PI hydrolysis was assessed as agonist-stimulated formation of [3H]inositol phosphates by cells prelabeled with [3H]inositol (Fig. 1). In cells expressing the wild-type receptor, epinephrine stimulated PI hydrolysis by 4.1 ± 0.1-fold, with half-maximal stimulation observed at 31 ± 2 nm epinephrine (eight experiments, three different clones). In marked contrast, no stimulation of PI hydrolysis was observed with concentrations of epinephrine as high as 10 μm in cells expressing the Y348A receptor (eight experiments, two different clones). However, stimulation of PI hydrolysis by ATP was similar in cells expressing either the wild-type or the Y348A receptor and similar to that induced by epinephrine in cells expressing the wild-type receptor (not shown). Thus, the defect in stimulation of PI hydrolysis in the cells expressing the Y348A receptor is specific for the α1B-AR and not a generalized defect in the PI hydrolysis pathway in these cells.

Stimulation of PI hydrolysis by wild-type and Y348A α1B-ARs. CHO cells expressing the wild-type (WT) or Y348A α1B-ARs and prelabeled with [3H]inositol were incubated for 20 min in the absence [basal (B)] or presence of the indicated concentrations of epinephrine. Formation of [3H]inositol phosphates was then quantified as described in Experimental Procedures. Data are expressed as fold stimulation over basal and are the mean ± standard error from eight experiments with three clones for the wild-type receptor and eight experiments with two clones for the Y348A receptor. Basal activities were 0.31 ± 0.02% and 0.36 ± 0.04% conversion for the wild-type and Y348A receptors, respectively.
Similar results were obtained in studies of agonist-stimulated Ca2+ mobilization assessed by Fura-2 fluorescence (Fig.2). Basal levels of intracellular Ca2+ were ∼85 nm and were not different between cells expressing the wild-type and Y348A receptors. In cells expressing the wild-type receptor, epinephrine increased intracellular Ca2+ by 133 ± 12 nm, with half-maximal increase observed at 9 ± 2 nm epinephrine (six experiments, two different clones). In contrast, essentially no increase in intracellular Ca2+ was observed over the same range of epinephrine concentrations in cells expressing the Y348A receptor (five experiments, three different clones). However, stimulation of Ca2+ mobilization by ATP was similar in cells expressing wild-type or Y348A receptors (not shown), again indicating that the defect in the Y348A cells is specific for the α1B-AR.

Stimulation of Ca2+ mobilization by wild-type and Y348A α1B-ARs. CHO cells expressing the wild-type (WT) or Y348A α1B-ARs and loaded with Fura-2/AM were exposed to the indicated concentrations of epinephrine, and intracellular Ca2+ mobilization was assessed by Fura-2 fluorescence as described in Experimental Procedures. Data are expressed as nm increase over basal and are the mean ± standard error of six experiments with two clones for the wild-type receptor and five experiments with three clones for the Y348A receptor.
Agonist-induced decrease in cell surface receptor binding on ice.
In initial experiments, the ability of epinephrine to induce a decrease in cell surface accessibility of α1B-ARs was assessed in intact cell binding assays on ice using a single concentration of [3H]prazosin (1.7 nm). In these experiments, exposure of cells expressing wild-type receptors to 10 μm epinephrine for 30 min induced a 26 ± 2% (13 experiments, 10 different clones) decrease in [3H]prazosin binding. In cells expressing the Y348A receptor, the agonist-induced decrease in binding was 42 ± 3% (nine experiments, six different clones), which is somewhat larger than that for the wild-type receptor.
In further experiments, saturation binding assays with intact cells on ice were performed to determine the relative contributions of changes in B max and Kd values to the decreased binding. Saturation data from a typical experiment with each clone are presented in Fig.3, and average decreases in Bmax from multiple experiments are presented in Fig.4. In the case of wild-type receptors, the decrease in binding was primarily due to a decrease in Bmax with perhaps a small decrease in Kd. Pooled data from multiple experiments (four experiments, four different clones) revealed a 29 ± 4% decrease in B max values, withKd values for [3H]prazosin of 153 ± 41 pm in control cells and 118 ± 30 pm in epinephrine-treated cells. In the case of Y348A receptors, epinephrine treatment induced a consistent increase in Kd values for [3H]prazosin as well as a decrease inB max values. Pooled data from multiple experiments (four experiments, two different clones) revealed a 32 ± 6% decrease in B max values, withKd values for [3H]prazosin of 125 ± 19 pm in control cells and 269 ± 55 pm in epinephrine-treated cells.

Saturation assays of [3H]prazosin binding to intact control and agonist-treated cells on ice. CHO cells expressing the wild-type (WT; top) or Y348A (bottom) α1B-ARs were incubated for 30 min in the absence [control (CTL)] or presence of 10 μm epinephrine (EPI) to induce internalization. Cells were then washed, and binding of various concentrations of [3H]prazosin to the cells was measured in 4-hr assays on ice as described in Experimental Procedures. Data are presented in the form of Rosenthal (40) transformations and are from representative experiments; average values from multiple experiments with multiple clones are presented in the text. TheK d andB max values for the experiments shown were 101 ± 8 pm and 60.5 ± 1.4 fmol/dish for wild-type control, 93 ± 8 pm and 43.4 ± 1.0 fmol/dish for wild-type epinephrine, 108 ± 16 pm and 16.4 ± 0.7 fmol/dish for Y348A control, and 347 ± 57 pm and 10.7 ± 0.7 fmol/dish for Y348A epinephrine.

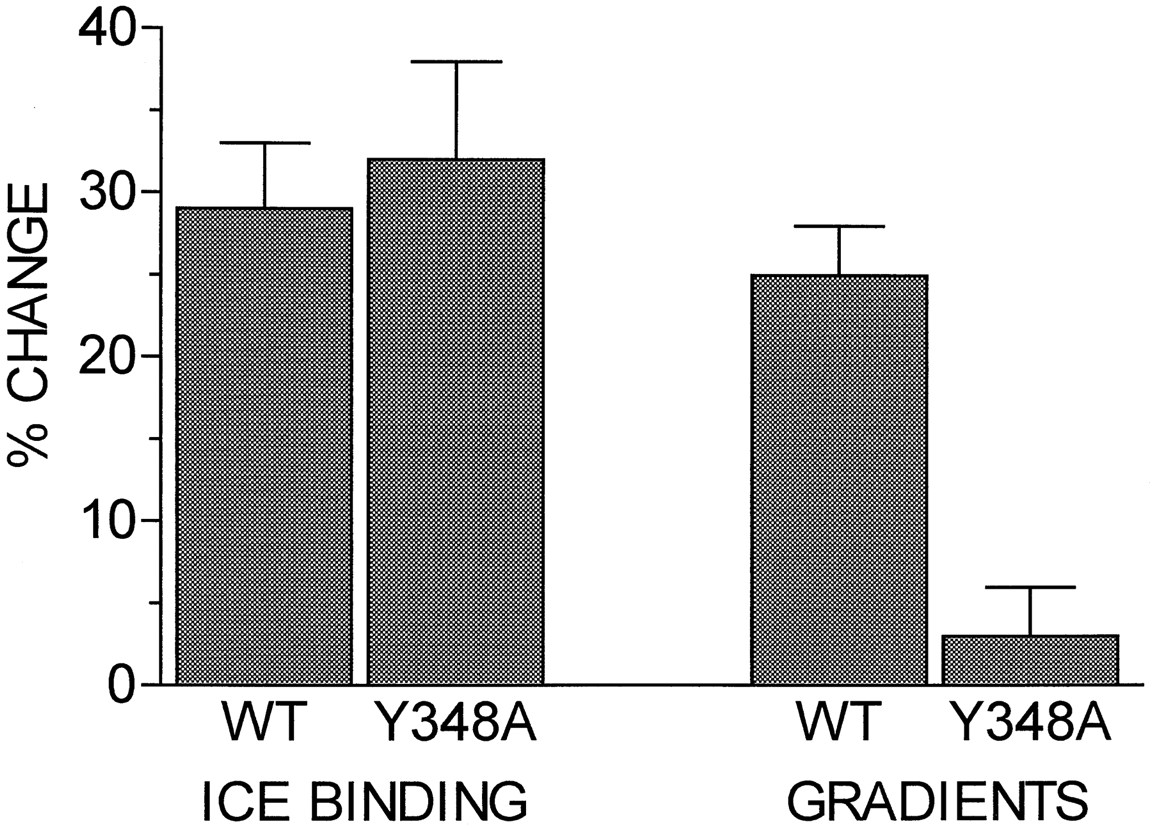
Comparison of sequestration assessed by binding to intact cells on ice with endocytosis assessed by sucrose density gradient centrifugation. Cells expressing the wild-type (WT) or Y348A α1B-AR were incubated for 30 min in the absence or presence of 10 μm epinephrine, and assays of binding to intact cells on ice (ICE BINDING) or sucrose density gradient centrifugation assays (GRADIENTS) were performed. Ice binding data are expressed as the percentage decrease in B maxfrom saturation assays similar to those shown in Fig. 3 and are the mean ± standard error of four experiments with four clones for the wild-type receptor and four experiments with two clones for the Y348A receptor. Data from one clone that showed an atypical increase inB max in some experiments were excluded. Gradient data are expressed as the percentage of the receptors in the plasma membrane fraction (gradient fractions 8–10) that were shifted to the light vesicle fraction (gradient fractions 1–4) after epinephrine pretreatment and are the mean ± standard error of nine experiments with four clones for the wild-type receptor and eight experiments with three clones for the Y348A receptor.
Sucrose density gradient assays of receptor redistribution.
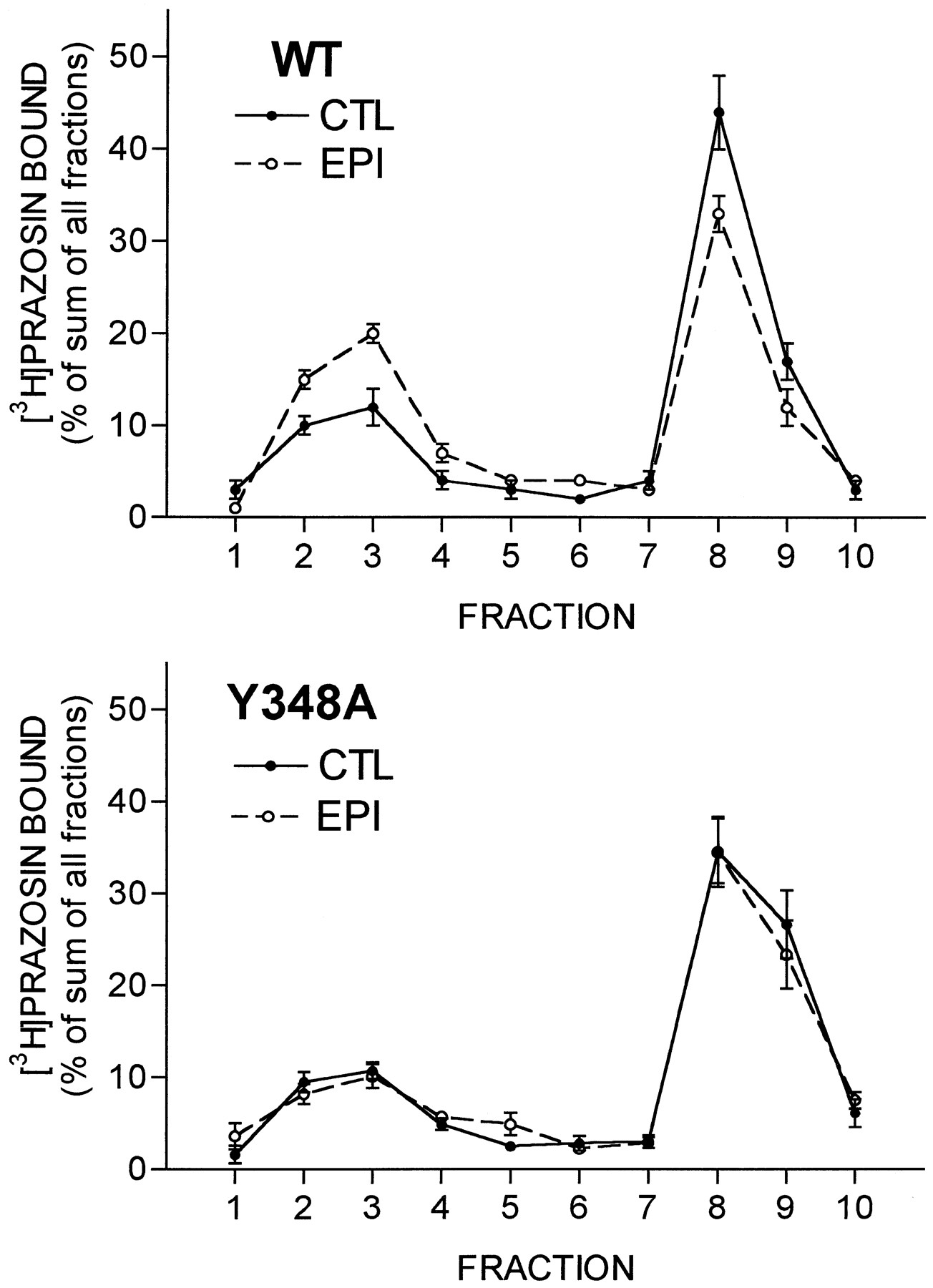
Agonist-induced redistribution of receptors from the plasma membrane fraction to the light vesicle fraction in sucrose density gradient centrifugation assays also was assessed. Gradient profiles are presented in Fig. 5, and the average changes are presented in Fig. 4 for comparison with the changes in intact cell binding assays on ice. Exposure of cells expressing wild-type receptors to 10 μm epinephrine for 30 min induced a shift of 17 ± 2% (nine experiments, four different clones) of the total cellular receptors from the plasma membrane fraction to the light vesicle fraction. In control cells, 69 ± 2% of the receptors were in the plasma membrane fraction, and the remaining 31% were in the light vesicle fraction; with agonist treatment; this ratio shifted to 52 ± 2% in the plasma membrane fraction and the remaining 48% in the light vesicle fraction. Thus, the agonist-induced shift to the light vesicle fraction represents a 25% decrease in the number of receptors in the plasma membrane fraction, similar to the 29% decrease in theB max value for [3H]prazosin binding observed in the ice assays with these cells, suggesting that translocation of receptors to the light vesicle fraction can account for essentially all of the decrease in ice binding for the wild-type receptor.

Sucrose density gradient assays of receptor endocytosis for wild-type (WT) and Y348A α1B -ARs. CHO cells expressing the wild-type (top) or Y348A (bottom) α1B-ARs were incubated for 30 min in the absence [control (CTL)] or presence of 10 μmepinephrine (EPI) to induce internalization. Cells were then lysed, and the lysates were subjected to sucrose density gradient centrifugation. [3H]prazosin binding to each fraction was then determined as described in Experimental Procedures. Binding in each fraction is expressed as the percentage of the sum of binding to all fractions. Data are the mean ± standard error from nine experiments with four clones for the wild-type receptor and eight experiments with three clones for the Y348A receptor.
In marked contrast, there was only a 2% (eight experiments, three different clones) shift of receptors from the plasma membrane fraction to the light vesicle fraction for the Y348A receptor. In control cells, 71 ± 2% of the receptors were in the plasma membrane fraction and the remaining 29% were in the light vesicle fraction; with agonist treatment, this ratio shifted to 69 ± 2% in the plasma membrane fraction and the remaining 31% in the light vesicle fraction. Thus, in Y348A cells, the 32% decrease in the B max value for [3H]prazosin binding observed in the ice assays with these cells occurs with almost no shift of receptors to the light vesicle fraction. In both cases, the total amount of [3H]prazosin binding in all fractions was not decreased, indicating that there was no down-regulation of total receptor number by this short term agonist exposure.
Discussion
These studies document an important role for Tyr348 of the α1B-AR in agonist binding, in functional coupling to PI hydrolysis and Ca2+ mobilization, and in agonist-induced internalization of the receptor. Mutation of Tyr348 to alanine increased agonist binding affinity by ∼10-fold, without altering antagonist binding. This mutation also essentially completely eliminated agonist-induced stimulation of PI hydrolysis and elevation of intracellular Ca2+. Agonist-induced conversion of receptors to a form that is inaccessible to the lipophilic radioligand [3H]prazosin in intact cell binding assays on ice occurred to a similar or greater extent for the Y348A receptor as for the wild-type receptor. In contrast, agonist-induced redistribution of receptors from the plasma membrane fraction to the light vesicle fraction in sucrose density gradient centrifugation assays occurred for the wild-type receptor but not for the Y348A receptor.
Two important conclusions regarding α1B-AR internalization follow from these results. First, these studies provide further evidence indicating that at least two distinct steps in the overall internalization pathway for these receptors can be distinguished experimentally: (i) a step in which the receptors become relatively inaccessible to ligands at the cell surface but remain plasma membrane associated, which we refer to as sequestration; and (ii) a step in which the receptors are translocated from the plasma membrane to another membrane-bound compartment that is physically separated from the plasma membrane and sediments with the light vesicle fraction on sucrose density gradient centrifugation, which we refer to as endocytosis. In previous studies of adrenergic receptor internalization in DDT1 MF-2 hamster vas deferens smooth muscle cells, which express both β2- and α1B-ARs endogenously, we found that agonists alone induced only the sequestration of α1B-ARs, with no endocytosis of receptors to the light vesicle fraction on sucrose density gradients (20, 21). However, when these cells were exposed to agonist plus strong protein kinase C activators, such as phorbol-12-myristate-13-acetate, these α1B-ARs were sequestered away from the cell surface as well as endocytosed to the light vesicle fraction (21). In contrast, agonist alone was sufficient to induce endocytosis of β2-ARs to the light vesicle fraction in these cells, with no further effect with the addition of protein kinase C activators (20, 29). These studies suggested the possibility of somewhat different internalization pathways or mechanisms for these two types of adrenergic receptors. However, when the hamster α1B-AR was transfected and expressed in CHO cells, agonist alone was sufficient to cause both a loss of cell surface binding and a shift of receptors to the light vesicle fraction (Ref. 26 and this study). Interestingly, the behavior of the Y348A-mutated receptor in the transfected CHO cells exposed to agonist alone is similar to that of the endogenous α1B-AR in DDT1 MF-2 cells exposed to agonist alone. The basis for the different behavior of the wild-type α1B-AR endogenously expressed in the DDT1 MF-2 cells versus its behavior when transfected into CHO cells remains to be determined.
Our recent studies using inhibitors of clathrin-mediated receptor internalization to investigate the role of internalization in agonist-induced changes in the binding properties of α1B- and β2-ARs on intact DDT1 MF-2 cells also provided evidence consistent with two separable steps in α1B-AR internalization (30, 31). Hypertonic sucrose and intracellular K+ depletion were able to inhibit the agonist-induced changes in binding properties for both α1B- and β2-ARs (31); however, ATP depletion inhibited these changes for β2-ARs but not for α1B-ARs (30). Hypertonic sucrose and K+depletion have been shown to inhibit clathrin-coated pit formation, an early step in the clathrin-mediated internalization pathway for non-G protein-coupled receptors (32, 33), whereas ATP seems to be required for later steps of internalization (34). Thus, we proposed that the steps we define as sequestration and endocytosis for α1B-ARs may be analogous to the early and later steps previously defined for clathrin-mediated internalization of non-G protein-coupled receptors (31).
A few previous studies have presented evidence for multiple steps or compartments in the internalization pathway for β-ARs. Sequestered β-ARs that were associated with the plasma membrane after freeze/thaw lysis but were dissociated from the plasma membrane following osmotic lysis were observed by Strader et al. (17) in an early study with frog erythrocytes. In our previous studies with 1321N1 human astrocytoma cells, we showed that the bulk of the β-ARs that were inaccessible to hydrophilic ligands in 37° assays (18) and to lipophilic radioligands in assays on ice (19) migrated in the light vesicle fraction on sucrose density gradients; however, in both of these studies, a subpopulation of cell surface-inaccessible (sequestered) receptors was found to migrate with the plasma membrane fraction. More recently, von Zastrow and Kobilka (22) used immunofluorescence and electron microscopy with epitope-tagged receptors to identify two separable steps in the β2-AR internalization pathway in transfected 293 cells: (i) a clustering of receptors within the plasma membrane that was agonist dependent, occurred at 16°, and did not require ATP; and (ii), an endocytosis of receptors into intracellular vesicles that was agonist-independent, did not occur at 16°, and required ATP. It is tempting to speculate that the two steps identified by our assays and differentiated by the Y348A mutation of the α1B-AR are the same as these two steps identified morphologically for the β2-AR. The correlation of our two steps with those of von Zastrow and Kobilka (22), and the possible role of Tyr348 of the α1B-AR in regulating the second, agonist-independent step identified in their studies, will be a goal of future studies.
The exact physical nature of the sequestered and endocytosed or light vesicle receptor compartments remains to be established. In the case of β2-ARs, immunofluorescence microscopy localization studies (35) showed that transfected epitope-tagged β2-ARs redistribute into the same endocytotic vesicles as transferrin, which is internalized by the well-characterized receptor-mediated endocytosis pathway involving clathrin-coated pits and vesicles (6, 7). A similar immunofluorescence microscopy study revealed internalization of α1B-ARs into the same intracellular vesicle population as that containing internalized transferrin (24). Based on these studies and on studies of the effects of clathrin-mediated endocytosis inhibitors on adrenergic receptor internalization (1, 24, 36), it seems likely that the light vesicle compartment isolated in our sucrose density gradient assays may be the same endocytotic vesicles that mediate internalization of transferrin and various other peptide ligands via clathrin-coated pits and vesicles. However, the nature of the sequestered receptor compartment within the plasma membrane remains to be determined. Interestingly, a previous study using site-directed mutagenesis of insulin receptors found that deletion of the NPXY sequence allowed the early steps of insulin receptor redistribution within the plasma membrane to occur but prevented endocytosis into intracellular vesicles (37). These results thus appear to be analogous to ours with the Y348A α1B-AR.
A second major conclusion from our study is that the sequestration step of α1B-AR internalization apparently does not require phospholipase C activation or second messenger generation because the Y348A receptor became sequestered in response to agonist but did not stimulate PI hydrolysis or Ca2+ mobilization. This is similar to results with the β2-AR, in which sequestration of receptors has been shown to occur in the absence of functional G proteins, adenylyl cyclase activation, and cAMP formation (1-4). It is possible that phospholipase C activation and/or second messenger formation is required for the endocytosis step for α1B-ARs because both endocytosis and functional coupling are defective in the Y348A receptor. However, these could also be two unrelated consequences of this mutation. Our previous studies with DDT1 MF-2 cells indicate an involvement of second messenger generation and protein kinase C activation in the endocytosis step because inclusion of phorbol esters to activate protein kinase C specifically promoted endocytosis, which was not induced by agonist alone in these cells (21). Recent preliminary experiments indicate that the inclusion of phorbol-12-myristate-13-acetate along with agonist can induce endocytosis for the Y348A receptor,1 which is similar to results with the wild-type receptor expressed in DDT1MF-2 cells. Enhancing phosphorylation may thus rescue the endocytosis defect of the Y348A α1B-AR, similar to the rescue of the defect of the Y326A β2-AR by overexpression of β-AR kinase (11). Further studies of the role of protein kinase C in sequestration and endocytosis of α1B-ARs are in progress. Studies with other mutated α1B-ARs defective in either coupling and/or endocytosis will also shed additional light on the roles of G protein coupling and second messenger generation in the multiple steps of α1B-AR internalization.
The Y348A mutation also altered the binding properties of the α1B-AR. Agonist binding affinity was increased ∼10-fold, but antagonist binding affinity was unaltered. Despite the higher binding affinity for the agonist epinephrine of the Y348A receptor, agonist binding did not lead to functional coupling of the receptor to the PI hydrolysis signal transduction pathway. Although binding of the radioligand [3H]prazosin to the Y348A receptor in control cells was not different from that for wild-type receptors, agonist exposure led to an apparent decrease in affinity of the Y348A receptor for the radioligand [3H]prazosin in intact cell binding assays on ice, a change that was not observed for the wild-type receptor. The basis for this decrease in affinity is not clear. One possibility is that it results from residual epinephrine from the agonist pretreatment competing with the [3H]prazosin during the binding assay, thus causing an apparent decrease in radioligand affinity. The 10-fold higher affinity of the Y348A receptor for epinephrine would make residual epinephrine more likely to interfere in assays with the Y348A receptor than with the wild-type receptor. The Y348A mutation clearly causes multiple defects in the receptor; it is therefore also possible that the multiple changes in ice binding observed for the Y348A receptor result from an agonist-induced conformational change other than or in addition to sequestration that occurs for the Y348A receptor but not for the wild-type receptor.
Mutations analogous to the Y348A mutation in the α1B-AR have been generated and characterized for several other G protein-coupled receptors. Original studies of this mutation in the β2-AR revealed an almost complete lack of receptor sequestration, suggesting that the NP(X)nY motif might in fact be an internalization or sequestration signal for β2-ARs (8, 9). However, subsequent studies have suggested that the sequestration defect in the Y326A β2-AR is secondary to a more general conformational disruption that leads to a defect in receptor phosphorylation (10, 11). More recent studies suggest a role for β-AR kinase-mediated phosphorylation of β2-ARs in facilitating receptor interactions with β-arrestin, which seems to be required for sequestration of β2-ARs (12, 38, 39). The Y326A β2-AR also showed decreased coupling to stimulation of adenylyl cyclase (8-10), although it was not as extensive as the coupling defect observed for the Y348A α1B-AR. Studies of the NP(X)nY motif have also been conducted for the AT1 angiotensin receptor (13-15) and the GRP receptor (16), two receptors that couple to PI hydrolysis. The studies with angiotensin receptor mutants indicated that the NPFLY sequence of these receptors was important for agonist binding and functional coupling to PI hydrolysis but was not an important determinant of receptor sequestration (defined by resistance to acid-salt washing). In the case of the GRP receptor, agonist binding, functional coupling, and receptor sequestration were unaltered in the Y324A mutant. Together, these results indicate that the NP(X)nY motif in multiple G protein-coupled receptors is in a domain that may be important for specifying appropriate conformations for ligand binding, receptor function, and some aspects of receptor internalization, but this sequence is not a specific signal for receptor sequestration.
In summary, Tyr348 in the α1B-AR seems to be an important residue for agonist binding, functional coupling, and receptor endocytosis. The Y348A α1B-AR provides a unique new line of evidence for multiple steps in the G protein-coupled receptor internalization pathway. This mutated receptor should also be a powerful tool for use in further dissection of the multiple cellular compartments and molecular modifications involved in agonist-induced redistribution of this important class of receptors.
Acknowledgments
We are grateful to Drs. R. D. Brown for providing the plasmid containing the α1B-AR cDNA, D. R. Cerutis for assistance with site-directed mutagenesis, and P. Carmines and V. Andalaro for assistance with the Ca2+ mobilization assays all from the University of Nebraska Medical Center, Omaha, NE.
Footnotes
- Received January 21, 1997.
- Accepted April 21, 1997.
-
Send reprint requests to: Myron L. Toews, Ph.D., Department of Pharmacology, University of Nebraska Medical Center, 600 South 42nd Street, Omaha, NE 68198-6260. E-mail:mtoews{at}mail.unmc.edu
-
↵1 J. Wang, unpublished observations.
-
This work was supported in part by National Institutes of Health Research Grant GM34500.
Abbreviations
- AR
- adrenergic receptor
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- GRP
- gastrin-releasing peptide
- CHO
- Chinese hamster ovary
- PI
- phosphoinositide
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- DMEM
- Dulbecco’s modified Eagle’s medium
- AM
- acetoxymethyl ester
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}