


Abstract
We characterized participation of the stress-activated protein kinase (SAPK) cascade in the lethal actions of the cytotoxic lipid messengers ceramide and sphingosine in U937 human monoblastic leukemia cells. Acute exposure of U937 cells to either lipid resulted in loss of proliferative capacity, degradation of genomic DNA, and manifestation of apoptotic cytoarchitecture. Ceramide robustly stimulated p46-JNK1/p54-JNK2 activity and increased expression of c-jun mRNA and c-Jun protein; in contrast, sphingosine moderately stimulated p46-JNK1/p54-JNK2 and failed to modify c-jun/c-Jun expression. Dominant-negative blockade of normal c-Jun activity by transfection with the TAM-67 c-Jun NH2-terminal deletion mutant abolished the lethal actions of ceramide but was without effect on those of sphingosine, indicating that ceramide-related apoptosis is directly dependent on activation of c-Jun, whereas sphingosine-induced cell death proceeds via an unrelated downstream mechanism. Characterization of the mitogen-activated protein kinase (MAPK) cascade in these responses revealed a further functional disparity between the two lipids: basal p42-ERK1/p44-ERK2 activity was gradually reduced by ceramide but immediately and completely suppressed by sphingosine. Moreover, blockade of the MAPK cascade by the aminomethoxyflavone MEK1 inhibitor PD-98059 unexpectedly activated p46-JNK1/p54-JNK2 and induced apoptosis in a manner qualitatively resembling that of sphingosine. Both lipids sharply increased p38-RK activity; selective pharmacological inhibition of p38-RK by the pyridinyl imidazole SB-203580 failed to mitigate the cytotoxicity associated with either ceramide or sphingosine, suggesting that p38-RK is not essential for lipid-induced apoptosis. These findings demonstrate that reciprocal alterations in the SAPK and MAPK cascades are associated with the apoptotic influence of either lipid inasmuch as (i) ceramide-mediated lethality is primarily associated with strong stimulation of SAPK and weak inhibition of MAPK, whereas (ii) sphingosine-mediated lethality is primarily associated with weak stimulation of SAPK and strong inhibition of MAPK. We therefore propose that leukemic cell survival depends on the maintenance of an imbalance of the outputs from the MAPK and SAPK systems such that the dominant basal influence of the MAPK cascade allows sustained proliferation, whereas acute redirection of this balance toward the SAPK cascade initiates apoptotic cell death.
The physiological regulation of such diverse processes as cell death, proliferation, and differentiation requires the in
tegration of distinct, and potentially conflicting, signals. Elucidation of the intracellular systems involved in cell survival has identified specific roles for the SAPK cascade in the initiation of apoptosis and for the MAPK cascade in the maintenance of proliferation and/or differentiation. Accordingly, the balance between the SAPK and MAPK systems has been proposed as a fundamental determinant of cell survival (1). Numerous lines of evidence indicate that these cellular processes are in many instances governed proximally by multiple lipid effectors. We and others have described the induction of programmed cell death or apoptosis by the cytotoxic lipid messengers ceramide (2-4) and sphingosine (5, 6) in human myeloid leukemia cells; however, the downstream systems underlying the lethal actions of these lipids have not been completely delineated.
Several subcellular targets for ceramide have been described, including a membranal proline-directed serine/threonine protein kinase (CAPK) (7), which was recently reported to be identical to KSR (8), and a cytosolic class 2A protein phosphatase (CAPP) (9, 10). None of these signaling elements has been identified as an essential proximal effector for the induction of apoptosis; however, the SAPK activities p46-JNK1 and p54-JNK2 (also referred to as JNKs) have been implicated as essential downstream effectors for the apoptotic influence of ceramide (11). Phosphorylation of c-Jun within the amino-terminal transactivation domain (on residues 63 and 73) is essential for maximal AP1-dependent transcriptional activation (12). Both endogenous ceramides and synthetic ceramide analogs promote activation of p46-JNK1 and p54-JNK2 (12-15) and expression of c-jun/c-Jun (13,15); similarly, these enzymes are engaged by many lethal ceramide-dependent stimuli, including activation of cytotoxic receptor systems [e.g., CD120a (p55, or “type-I” TNF receptor) (13-16), CD95 (APO1/FasR) (14, 17)] and environmental stresses [e.g., ionizing radiation, oxidative stress, and heat shock (14)]. Interruption of the SAPK cascade abrogates the apoptotic influence of ceramide (14), indicating that this signaling system is essential for ceramide-mediated lethality.
The biological actions of sphingoid bases are most frequently ascribed to inhibition of the phorboid-sensitive PKC subfamilies (18), although additional subcellular targets for these lipids have been identified. Inhibition of PKC results in the initiation of apoptotic cell death in most proliferating cell types; consistent with the established importance of PKC activity in cell survival (reviewed in Ref. 19), dramatic apoptotic responses may be elicited by sphingoid bases (5, 6) as well as by pharmacological PKC inhibitors (20, 21). Initiation of cell death by sphingoid bases thus derives from acute suppression of various cPKC/nPKC isoforms and the consequent loss of a poorly defined cytoprotective influence of these enzymes, presumably reflecting disruption of one or more uncharacterized downstream elements essential for survival (e.g., the MAPK cascade).
To date, involvement of the SAPK cascade in the apoptotic actions of sphingosine and other sphingoid bases has not been addressed. In addition, the newly discovered SAPK, p38-RK (also referred to as reactivating kinase) (1), is a presumptive regulatory element in the induction of apoptosis; however, this enzyme does not recognize c-Jun as a physiological substrate in all cellular settings. Currently, the role of p38-RK in lipid-mediated cytotoxicity is uncertain. The current study was undertaken to compare the relative involvement of SAPK activities in the induction of apoptosis by ceramide and sphingosine in U937 cells. The results demonstrate that the lethal actions of these lipids entail reciprocal, but coordinated, alterations in SAPK and MAPK activities such that both ceramide and sphingosine stimulate p46-JNK1/p54-JNK2 but suppress p42-ERK1/p44-ERK2, albeit to varying degrees. These findings suggest that the apoptotic effects of these cytotoxic lipids are subserved by acute redirection of the balance between the SAPK and MAPK cascades.
Materials and Methods
Drugs and reagents.
Synthetic preparations of ceramide (N-acetylsphingosine), sphingosine, dihydroceramide (N-acetyldihydrosphingosine), and dihydrosphingosine (BIOMOL Research Laboratories, Plymouth Meeting, PA) were initially dissolved in 100% ethanol and stored at −70°. For experimental use, concentrated ethanol stocks of various sphingolipids were complexed at a 1:1 molar ratio with delipidated bovine serum albumin [Fraction V, fatty acid free; 2 mm in PBS (138 mm NaCl, 3mm KCl, 10 mm Na2HPO4, 2 mm NaH2PO4)] by vigorous mixing for 90 min at 37°; stable protein-bound sphingolipid preparations were stored at −20°. Fumonisin B1(Sigma Chemical, St. Louis, MO), oleoylethanolamine (Sigma), and bothd-erythro- andl-erythro-MAPP (BIOMOL) were dissolved in ethanol; these agents were tested after delivery as either organic stocks or complexed to delipidated bovine serum albumin as described above for sphingolipids. Sphingomyelinase (from Staphylococcus aureus; Sigma) was prepared and diluted in sterile 50% glycerol/250 mm phosphate buffer, pH 7.5. Recombinant human tumor necrosis factor-α (R and D Systems, Minneapolis, MN) and monoclonal antibody directed against human Fas (derived from clone CH-11; Kamiya Biomedical, Thousand Oaks, CA) were dissolved in sterile physiological saline. Calphostin C and chelerythrine (Alexis Biochemicals, San Diego, CA) were dissolved in sterile water. SB-203580, SKF-105809 (SmithKline-Beecham, King-of-Prussia, PA), and PD-98059 (Calbiochem, San Diego, CA) were dissolved in sterile dimethylsulfoxide. All vehicles were found to be without discernible biological effect in U937 cells. Test reagents were diluted to final concentrations in medium at 37°.
Cell culture.
The human monoblastic leukemia cell line U937 was derived from a patient with diffuse histiocytic lymphoma (22). U937 cells were previously transfected by electroporation with pMexMth metallothionine-inducible vectors without or with the insertion for the c-Jun deletion mutant TAM-67, giving rise to the stable sublines U937/136–4 and U937/101–2-1 (designated U937/TAM, respectively); U937/136–4 cells were tested in parallel with wild-type parental cells (referred to as U937/WT) and consistently exhibited identical biological responses. All cell lines were grown in complete RPMI-1640 medium (phenol red-free formulation supplemented with 1.0% sodium pyruvate, nonessential amino acids, l-glutamine, penicillin, and streptomycin; Life Technologies, Grand Island, NY) and 10% heat-inactivated fetal bovine serum and maintained under a fully humidified atmosphere of 95% room air/5% CO2 at 37°. Transfected cell lines were grown in the presence of G418 (400 μg/ml; Life Technologies). Cultures were passed twice weekly. Cell densities were determined by Coulter counter and cell viability was assessed by trypan blue exclusion.
Test exposures.
All experimental incubations were performed as described previously (2, 6, 20). Cells in log-phase growth were pelleted, washed twice in complete medium, resuspended at a density of 4 × 105 cells/ml), and maintained as indicated above. Cells were exposed to test agents for appropriate intervals in complete medium; loss of cells under these conditions due to either washing or cell adherence was negligible (≤5%). Test incubations were terminated with gentle pelleting of the cells by centrifugation at 400 × g for 10 min at 4°; in some instances, aliquots of the medium were retained for direct assay of released DNA. After the determination of cell density, the cells were pelleted and prepared as outlined below for agarose gel electrophoresis, spectrofluorophotometric assays of DNA damage, assay of cloning efficiency, examination of cellular morphology, determination of mRNA and protein expression, or assay of SAPK and MAPK activities.
Qualitative analyses of DNA damage.
To assess both early and late aspects of ceramide-related DNA degradation, apoptotic DNA fragments of varied sizes were resolved electrophoretically in parallel studies on both pulsed-field and static-field agarose gels as follows:
Pulsed-field gel electrophoresis.
The formation of rosette (∼300 kbp) and loop (∼50 kbp) DNA fragments was assessed by field-inversion gel electrophoresis as described previously (4, 6). Pelleted cells were resuspended in PBS and lysed by the addition of molten 1.0% low melting-point agarose (In-Cert; FMC Corp. Bioproducts, Rockland, ME) with thorough mixing (yielding a final concentration of 2 × 107 cells/ml); fractions of the lysate mixtures (corresponding to ∼2 × 106cells) were cast into precooled 85-μl block molds and allowed to solidify at 4°. The agarose-imbedded lysates were then treated with 250 mm EGTA, 250 mm EDTA, and 1%N-lauroylsarcosine, pH 8.0, containing proteinase-K (200 μg/ml; Sigma) at 55° for 48 hr. Deproteinated lysate plugs were rinsed in 250 mm EDTA and 250 mm EGTA, pH 8.0, and imbedded in 2.25% agarose gels (Sea-Kem Gold; FMC); high-molecular-weight DNA fragments were resolved by field-inversion electrophoresis at 6 V/cm for 24–28 hr in 0.5× Tris-borate/EDTA buffer at 14°; pulse intervals were ramped from T1 = 0.5 sec to T2 = 50.0 sec, with an F/R ratio of 3.0. Gels were stained in buffer containing 0.5 μg/ml ethidium bromide, and DNA fragments were visualized by UV transillumination. DNA molecular weight reference preparations (48.6-kbp ladder; Life Technologies) routinely were run in parallel for estimation of the size of rosette and loop DNA fragments.
Static-field gel electrophoresis.
The formation of oligonucleosomal DNA fragments (∼0.2–1.2 kbp) was assessed by fixed-field agarose gel electrophoresis as described previously (4, 6). Pelleted cells were resuspended in PBS and lysed by the addition of 10 mm Tris·HCl, 15 mm EGTA, 15 mmEDTA, and 0.1% Nonidet P-40, pH 7.4 (yielding a final concentration of 4 × 107 cells/ml) and mixed thoroughly with gentle mechanical agitation; the lysates were then treated with proteinase-K (200 μg/ml) at 55° for 24 hr. The deproteinated extracts were centrifuged at 30,000 × g for 75 min at 4°, and the pellets were discarded; the supernatants were treated with ribonuclease A (100 μg/ml; Sigma) at 37° for 18 hr. Aliquots of final lysate preparations (corresponding to 2 × 106 cells) were loaded into 2.25% agarose gels (Metaphor; FMC) impregnated with ethidium bromide; low-molecular-weight DNA fragments were resolved by electrophoresis at 6 V/cm for 90–180 min in 1× Tris-acetate/EGTA buffer at 10°. DNA fragments were visualized by UV transillumination. DNA molecular weight reference preparations (100-bp ladder; Life Technologies) were run in parallel for estimation of the size of oligonucleosomal DNA fragments.
Quantitative analyses of DNA damage.
The formation and release of DNA fragments and the corresponding breakage of bulk DNA were assessed by bisbenzimide spectrofluorophotometry as described previously (2, 4, 20). To measure intracellular DNA fragments, pelleted cells (4 × 106 cells/pellet in quadruplicate) were resuspended in PBS and lysed by the addition of 5 mm Tris·HCl, 30 mm EGTA, 30 mmEDTA, and 0.1% Triton X-100, pH 8.0 (yielding a final density of 107 cells/ml), with gentle mechanical agitation. The lysates were centrifuged at 30,000 × g at 4° for 40 min. To measure extracellular DNA fragments, aliquots of incubation medium were adjusted to 5 mm Tris·HCl, 30 mmEGTA, and 30 mm EDTA, pH 8.0, and centrifuged at 20,000 × g at 4° for 40 min. The pellets were discarded, and the presence of nonsedimenting DNA fragments in the supernatant from lysate and medium extracts was quantified after dilution in modified Tris-sodium/EGTA buffer (3 mm NaCl, 10 mm Tris·HCl, 1 mm EGTA, pH 8.0) by spectrofluorophotometry in the presence of Hoechst 33258 (1 μg/ml; λex = 365, λem = 460). Net fluorescence was directly proportional to the presence of DNA fragments; final values were calculated relative to highly purified calf thymus DNA calibration standard and are expressed as ng/μg of DNA recovered or released from 106 cells. To measure the corresponding loss of integrity of bulk DNA, pelleted cells (8.25 × 106 cells/pellet in quadruplicate) were resuspended in cold PBS and subjected to timed alkaline denaturation in 0.1 n NaOH; denaturation was terminated by neutralization in 0.1 n HCl. Cells were then further diluted in PBS and lysed by the addition of 200 mmK2HPO4, 50 mmEDTA, and 0.16% N-lauroylsarcosine with brief sonication. Damage to bulk DNA in cell lysates was quantified by spectrofluorophotometry in the presence of Hoechst 33258 (λex = 350, λem = 450). Net fluorescence was inversely proportional to introduction of strand breaks; final values were standardized against graded DNA strand breakage induced by scaled irradiation from a [137Cs] point source (30–3000 rads) and are expressed as rad-equivalents.
Determination of clonogenicity.
Pelleted cells were washed extensively and prepared for soft-agar cloning as described previously (2, 4, 6). Cells were resuspended in cold PBS and seeded onto 35-mm culture plates at a fixed density (400 cells/ml/well) in complete RPMI-1640 medium containing 20% fetal calf serum, 10% 5637-CM, and 0.3% Bacto agar. Cultures were maintained for 10–12 days before the formation of colonies (defined as groups of ≥50 cells) was scored.
Cytological characterization of apoptosis.
Pelleted cells were resuspended in PBS and fixed in cytocentrifuge preparations according to established procedures (2, 4, 6). For visualization of apoptotic morphological alterations, fixed cells were stained with 20% Wright-Giemsa stain. At least three 100-cell fields were scored for each treatment by conventional light microscopy by assessing the expression of cytoarchitectural characteristics of apoptosis (i.e., condensed nucleoplasm and cytoplasm, formation of membrane blebs, karyolytic degeneration of the nucleus into apoptotic bodies, overall cell shrinkage). For visualization of apoptotic DNA damage, fixed cells were sequentially (a) treated with ethanol/acetic acid (2:1 v/v) at 20° for 5 min, (b) stained for broken DNA by treatment with TdT in the presence of FITC-dUTP (Molecular Probes, Eugene, OR) at 37° for 60 min, and (c) counterstained for intact DNA with 0.01% propidium iodide in sodium citrate at 20° for 10 min. At least three 100-cell fields were scored for each treatment by fluorescent microscopy by assessment of increased direct fluorescence of end-labeled double-stranded DNA.
Determination of c-jun/c-Jun expression.
Steady state levels of c-jun mRNA and c-Jun protein were monitored by conventional Northern and Western analyses as described previously (23).
Northern analysis of c-jun mRNA levels.
Pelleted cells were lysed in 4 m guanethidine containing 0.5% sodium dodecyl sulfate (107 cells/250 μl). Total cellular RNA (∼15 μg) was extracted after ultracentrifugation at 42,000 × g for 1 hr and separated by electrophoresis on 1% agarose-formaldehyde gels. RNA was transferred to nylon membranes by capillary transfer and cross-linked by heating at 80° for 2 hr. Blots were hybridized with a α-32P-dCTP-labeled cDNA probe (3000 Ci/mmol) for human c-jun and then washed in 0.2× standard saline citrate/1% sodium dodecyl sulfate at 65°. Blots were exposed to film with screen intensifiers at −90° for 18 hr; relative intensity in each band determined by quantitative radioautography. Values are expressed as a percentage of untreated controls.
Western analysis of c-Jun protein levels.
Pelleted cells were resuspended in 2× Laemmli’s buffer containing 1 μg/ml aprotinin (107 cells/100 μl), briefly sonicated, and boiled for 20 min; lysate samples were then resolved on 12.5% polyacrylamide gels (5 × 105cell-equivalents/lane). Proteins were transferred onto nitrocellulose membranes and washed in TBST. Membranes were sequentially (a) blocked in TBST containing 5% nonfat dry milk for 1 hr at 22°, (b) exposed to primary antibody (mouse anti-human c-Jun carboxyl-terminal sequence antibody; 1:250) at 22°, (c) washed in TBST, (d) exposed to secondary antibody (goat anti-mouse Ab/HRP conjugate; 1:2000) at 22°, and (e) washed again in TBST. Immunoreactive c-Jun was visualized by enhanced chemiluminescence; relative intensity in each band was quantified by radioautography and digital scanning densitometry. Values are expressed as a percentage of untreated control. To assess in situprotein phosphorylation, c-Jun was immunoprecipitated from cells metabolically prelabeled with [32P]orthophosphate (325 μCi/106 cells); after conventional Western analysis, in situ radiophosphate incorporation into immunoblotted c-Jun was determined by radioautography.
Determination of SAPK and MAPK activities.
Pelleted cells were washed in PBS, repelleted, and flash-frozen. Cell pellets were lysed in 25 mm HEPES, pH 7.4, containing 5 mmEGTA, and 5 mm EDTA and supplemented with protease inhibitors (5 mm benzamidine, 1 mmphenylmethylsulfonyl fluoride, 1 mg/ml soybean trypsin inhibitor, 40 μg/ml pepstatin, 40 μg/ml chymotrypsinogen, 40 μg/ml E64, 40 μg/ml aprotinin, 1 μm microcystin LR), phosphatase inhibitors (0.5 mm trisodium orthovanadate, 0.5 mm tetrasodium pyrophosphate), and 0.05% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, and 0.1% (v/v) 2-mercaptoethanol. Lysates were clarified by centrifugation at 5000 × g at 4° for 5 min. SAPK/MAPK was immunoprecipitated from clarified lysates with protein A/agarose-conjugated antibody/antisera. SAPK activities were then assayed after immunoprecipitation of (a) p54-JNK1/p46-JNK2 using GST/c-Jun 1–169 as substrate or (b) immunoprecipitation of p38-RK using myelin basic protein as substrate; alternatively, MAPK activity was assayed after immunoprecipitation of p42-ERK1/p44-ERK2 using myelin basic protein as substrate. Preimmune controls were also run to ensure selectivity of substrate phosphorylation. Reaction mixtures consisted of immunoprecipitated enzyme, substrate, and [γ-32P]ATP (5000 Ci/pmol) in 25 mm HEPES, pH 7.4, containing 15 mmMgCl2, 100 mm trisodium orthovanadate, 0.01% (v/v) 2-mercaptoethanol, and 1 μmmicrocystin LR. Reactions were initiated by the addition of substrate. SAPK assays were terminated by transfer to 10% polyacrylamide gels; phosphorylated products were resolved by electrophoresis, and appropriate substrate bands were excised. MAPK reactions were terminated by transfer to p81 filter paper; filters were rinsed repeatedly in 185 mm orthophosphoric acid and then dehydrated in acetone. Total radioactivity in gels and filters was determined by liquid scintillometry.
Results
Relative effects of ceramide and sphingosine on SAPK activation.
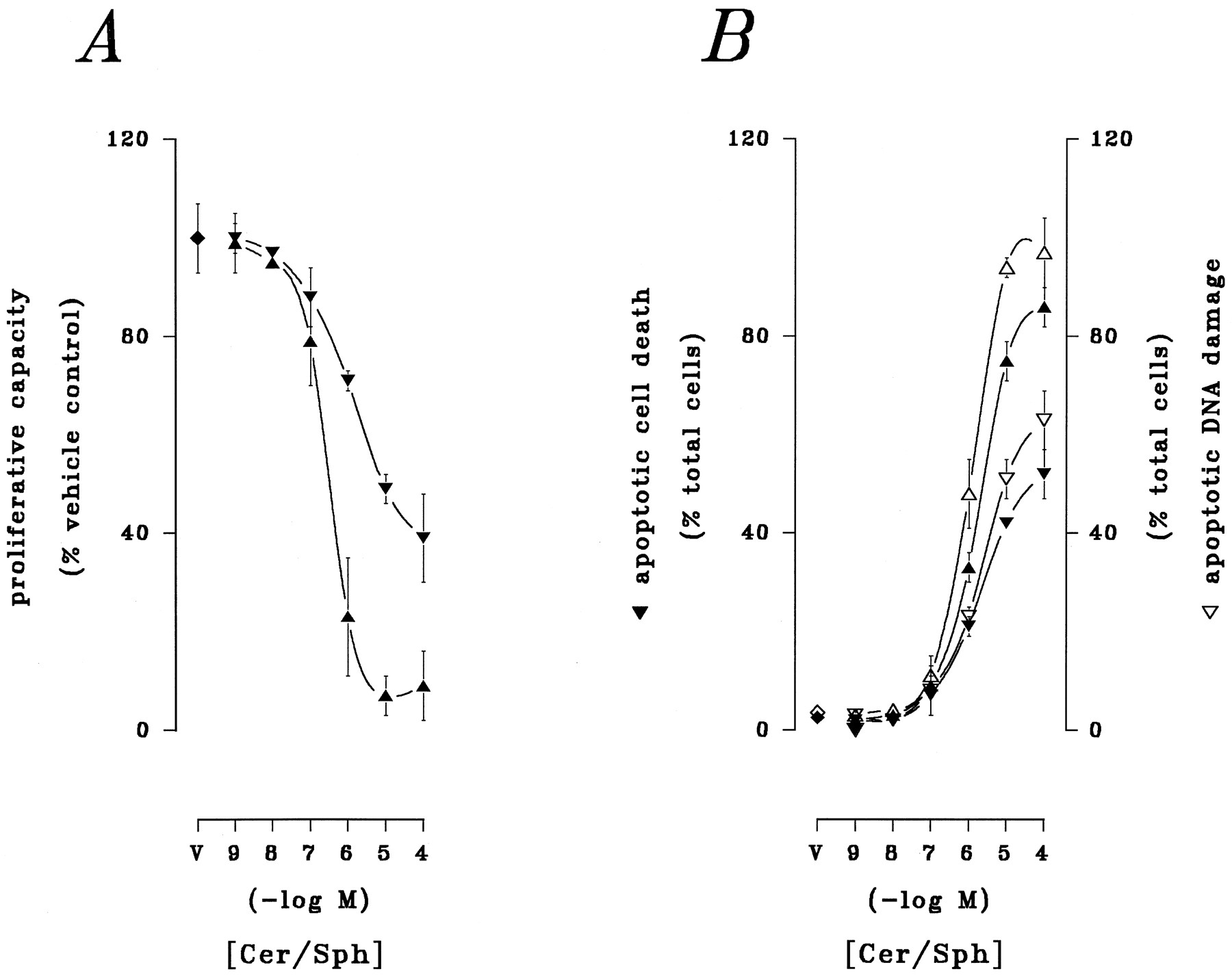
Apoptotic cell death is initiated by ceramide (2, 3) and sphingosine (5, 6) in myeloid leukemia cells. The cytotoxic actions of these lipids were compared directly in U937 human monoblastic leukemia cells in preliminary trials (Fig.1). Proliferative capacity, which provides the most sensitive index of lipid-mediated cytotoxicity in these cells (2), was determined by assay of colony formation by lipid-treated cells chronically sustained in culture. Apoptosis was assessed in fixed preparations through parallel visualization of (a) apoptotic cytoarchitecture by light microscopy and (b) apoptotic DNA damage by fluorescent microscopy. Acute (6-hr) exposure of U937 cells to synthetic preparations of ceramide or sphingosine over a broad range of concentrations (0.001–100 μm) resulted in pronounced loss of proliferative capacity and induction of apoptotic DNA damage and cell death. Both ceramide and sphingosine reduced proliferation in a distinctly concentration-related manner (EC50 = 2.12 μm and 266 nm, respectively; Fig. 1A). Similarly, although spontaneous apoptotic processes were evident in ≤3% of the population, the total fraction of apoptotic cells was sharply increased in a concentration-related manner by exposure to ceramide or sphingosine (apparent EC50 = ∼2.85 and ∼1 μm, respectively; Fig. 1B). Both lipids elicited pronounced apoptotic responses, although the onset of apoptosis was detected with greater sensitivity by DNA damage than by morphological alteration. Given the similar thresholds for maximal cytotoxicity in these responses, subsequent test exposures were confined to a concentration of 10 μm for direct comparisons of ceramide- and sphingosine-related bioactivity at equimolar levels.

Relative cytotoxic potential of ceramide and sphingosine. U937 cells were exposed to synthetic preparations of ceramide (Cer; 10 μm; ▾, ▿) or sphingosine (Sph; 10 μm; ▴, ▵) or to lipid-free vehicle (V) for 6. A, Cells were withdrawn from test agents and transferred to soft-agar assay for colony formation as described in Materials and Methods. B, Cells in fixed preparations were either (i) stained with modified Wright-Giemsa and examined by conventional light microscopy to visualize apoptotic DNA damage (▾, ▴) or (ii) stained with FITC-dUTP in the presence of TdT and examined by fluorescent microscopy to visualize apoptotic DNA damage (▿, ▵), as described in Materials and Methods. Results are from a representative study performed four times with comparable results.
Previously, we have shown that activation of the SAPK enzymes p46-JNK1 and p54-JNK2 is absolutely necessary for ceramide-mediated lethality in U937 cells (14), although corresponding involvement of these signaling elements in sphingosine-mediated lethality is uncertain. Moreover, contribution of the novel SAPK enzyme p38-RK to the lethal influence of either lipid in myeloid cells has not been reported. We therefore compared in the current study the involvement of SAPK activities in the responses to various sphingolipid analogs. The steric aspects of these responses were entirely compatible with the established cytotoxic actions of nonreduced and reduced forms of ceramide and sphingosine. First, SAPK activity was stimulated by ceramide but not by dihydroceramide, which is potentially consistent with stimulation of KSR/CAPK (7, 8) and/or CAPP (9, 10). Ceramide increased activities of both p46-JNK1/p54-JNK2 (by 595%) and p38-RK (by 356%) within 10 min (Table 1), whereas dihydroceramide was ineffective; this pattern was conserved with respect to cytotoxicity in 6-hr exposures (Table 2). Conversely, SAPK activation was comparably increased by sphingosine and dihydrosphingosine, which is consistent with an involvement of cPKC/nPKC inhibition (18). Both sphingosine and dihydrosphingosine elicited similar increases in p46-JNK1/p54-JNK2 (by ∼335%) and p38-RK (by ∼350%) within 10 min (Table 1); both lipids promoted essentially identical increases in apoptotic DNA damage and cell death in 6-hr exposures (Table 2). None of these lipids stimulated MAPK activity in these trials (not shown); in fact, although ceramide and dihydroceramide were completely ineffective, in comparison, sphingosine and dihydrosphingosine decreased p42-ERK1/p44-ERK2 activity (by ∼80%).
SAPK responses to nonreduced and reduced sphingolipid messengers
Cytotoxicity of nonreduced and reduced sphingolipid messengers
Metabolic interconversion of these lipids has been invoked to account for the observed biological actions of ceramide and sphingosine in some settings (24, 25). The extent to which reciprocal interconversion by acylation/deacylation contributed to the actions of a given lipid species was examined in related trials (the metabolizableN-octanoyl-substituted analog of ceramide was used in these studies). Ceramide-mediated apoptosis was unaffected by the ceramide acylhydrolase inhibitor oleoylethanolamine (Table3), indicating that deacylation to sphingosine did not underlie the bioactivity of ceramide. This was confirmed in related studies using more a selective inhibitor (not shown); neitherd-erythro-N-myristoylamino-1-phenyl-1-propanol nor the inactive l-erythro-isomer modified ceramide-related apoptosis. Similarly, sphingosine-mediated apoptosis was unaffected by the ceramide synthase inhibitor fumonisin B1 (Table 3), indicating that acylation to ceramide did not contribute to the bioactivity of sphingosine. These observations confirmed the selectivity of lethal responses to each sphingolipid species tested.
Selectivity of lipid-mediated cytotoxicity in U937 cells
To characterize more precisely involvement of these signaling systems in lipid-mediated cell death, in additional studies, activation of the SAPK cascade and induction of apoptotic DNA damage were examined over extended time courses. U937 cells were exposed to ceramide or sphingosine at equimolar concentrations (10 μm) for 0–9 hr. SAPK activity was determined by an in vitro immune complex assay. Substantive disparities were noted in the kinetic profiles for SAPK activation by the two lipids (Fig.2A). Ceramide produced a large, biphasic stimulation of p46-JNK1/p54-JNK2. Activity increased to a maximal level (890% of vehicle control) within 45 min then decreased to near-basal levels within 90 min. Sphingosine elicited a smaller but more complex stimulation of p46-JNK1/p54-JNK2 activity. Activity rose to an initial maximal level (to 325% of vehicle control) within 30 min and then fell to near-basal levels within 90 min; a slow, progressive rise ensued over the subsequent 3 hr. In contrast, ceramide and sphingosine both produced virtually identical multiphasic increases in p38-RK activity (Fig. 2B), consisting of a prominent transient spike increased by ∼185% in each instance), followed by a smaller, but sustained, plateau (increased by ∼125% and ∼85%, respectively, in response to ceramide and sphingosine).

Relative time courses of SAPK activation. U937 cells were exposed to either ceramide (10 μm; ▾) or sphingosine (10 μm; ▴) for 0–9 hr. SAPK activity was monitored by in vitro immune complex assay as described in Materials and Methods. A, activity of p46-JNK1/p54-JNK2. B, Activity of p38-RK. Values are expressed as a percentage of vehicle-treated control; data shown are from a representative study performed three times with comparable results.
Other disparities in the actions of ceramide and sphingosine were noted. Expression of c-Jun, which represents both an essential component of the AP1 transcription factor complex and a primary substrate for p46-JNK1/p54-JNK2, is increased in response to lethal cellular stresses. Steady state levels of c-jun mRNA and c-Jun protein were determined by quantitative Northern and Western analyses (Fig. 3). Ceramide progressively increased steady state levels of c-jun mRNA and c-Jun protein, whereas sphingosine failed to modify c-jun/c-Jun. Moreover, ceramide-related expression of c-jun/c-Jun was temporally associated with induction of AP1-specific DNA binding by gel mobility shift analysis (not shown), confirming the biological significance of newly synthesized c-Jun in these studies. In parallel trials, the effects were also assessed of ceramide and sphingosine on the expression of another SAPK substrate and potential c-Jun dimerization partner, the transcription factor ATF2 (not shown). ATF2 was not detected in U937 cells under basal conditions by conventional Western analysis, nor was ATF2 expression evident after exposure to ceramide or sphingosine, indicating that induction of this SAPK target did not contribute to lipid-mediated toxicity in these cells.

Time course of c-jun/c-Jun expression. U937 cells were exposed to ceramide (10 μm; ▾, ▿) or sphingosine (10 μm; ▴, ▵) for 0 to 9 hr. Steady state levels of c-jun mRNA (dashed lines) and c-Jun protein (solid lines) as determined respectively by quantitative Northern and Western analyses as described in Materials and Methods. Values are expressed as a percentage of vehicle-treated control and reflect the mean ± standard error of triplicate determinations; data shown are from a representative study performed three times with comparable results.
Ceramide and sphingosine promoted extensive apoptotic DNA damage with similar, but nonidentical, temporal profiles. Both lipids triggered the formation and release of double-stranded DNA fragments (Fig.4A) and corresponding double-stranded breakage of bulk DNA (Fig. 4B). The onset of the response to sphingosine was measurably more rapid than the response to ceramide, as we have noted previously (6). Specifically, ceramide-mediated DNA degradation was evident after only 3 hr, whereas significant sphingosine-related DNA degradation was detectable within ∼1 hr. At subsequent intervals, however, the two responses converged, with both lipids producing maximal genomic damage at 6–7 hr. In parallel trials, the effects of sphingosine and dihydrosphingosine were compared (not shown). Dihydrosphingosine activated both p46-JNK1/p54-JNK2 and p38-RK and induced DNA degradation with kinetics superimposable with those of sphingosine, which is consistent with previous findings from our laboratory that the two sphingoid bases possess comparable apoptotic capacities (6).

Time course of lipid-induced DNA damage. U937 cells were exposed to ceramide (10 μm; ▾) or sphingosine (10 μm; ▴) for 0–9 hr. Apoptotic DNA damage was assessed by quantitative spectrofluorophotometry in the presence of bisbenzimide as described in Materials and Methods. A, Accumulation of double-stranded DNA fragments. Values are expressed as μg of DNA/106 cells. B, Bulk DNA breakage (values expressed as krad equivalents). All values reflect the mean ± standard error of quadruplicate determinations. Data shown are from a representative study performed three times with comparable results.
Interference with SAPK signaling and the apoptotic responses to ceramide and sphingosine.
The lethal actions of ceramide exhibit an absolute requirement for normal p46-JNK1/p54-JNK2 activation (14) and c-Jun function (15). In the current study, failure of sphingosine to induce c-jun/c-Jun expression, and its comparatively weak activation of p46-JNK1/p54-JNK2, suggested that these signaling elements may not directly subserve sphingoid base-mediated cell death. To confirm this possibility directly, lethal actions of ceramide and sphingosine were compared in U937 cells with altered c-Jun function. These studies made use of a stable transfectant subline expressing the transactivation-deficient mutant c-Jun protein TAM-67 (23, 26), which possesses both the leucine-zipper site and carboxyl-terminal DNA-binding domain but lacks residues 3–122 of the amino-terminal transcriptional-activation domain. Because this deletion mutant possesses normal dimerization and AP1 consensus site-recognition functions but is devoid of transactivating activity, expression of TAM-67 results in a dominant-negative system in which normal transactivation properties of native c-Jun are disabled, or “quenched” (26).
The normal apoptotic responses to endogenous ceramide were tested in both wild-type and TAM-67-transfected U937 cell sublines designated U937/WT and U937/TAM, respectively (Table4). Experimental manipulations known to increase endogenous ceramide levels, such as (a) treatment with bacterial SMase (50 munits/ml) for 6 hr or (b) activation of ceramide-driven receptor systems (e.g., on exposure to either recombinant human tumor necrosis factor-α or specific anti-Fas monoclonal antibody) potently induced apoptotic DNA damage in U937/WT cells but were ineffective in U937/TAM cells. These changes were not the result of perturbations in sphingolipid metabolism inasmuch as the generation or subsequent clearance of ceramide was not modified by TAM-67, as we have noted elsewhere (14). Downstream signaling elements involved in ceramide-mediated cell death, such as activation of cPLA2, may be essential for apoptotic cell death in some settings (27, 28), but parallel treatment of U937/WT and U937/TAM cells with bacterial SMase elicited identical increases in both cPLA2 activity and lysophosphatidylcholine accumulation (data not shown), indicating that normal function of cPLA2 was unaffected by expression of TAM-67.
Impaired apoptotic capacity of endogenous ceramide and ceramide-dependent receptor systems in TAM-67 transfectants
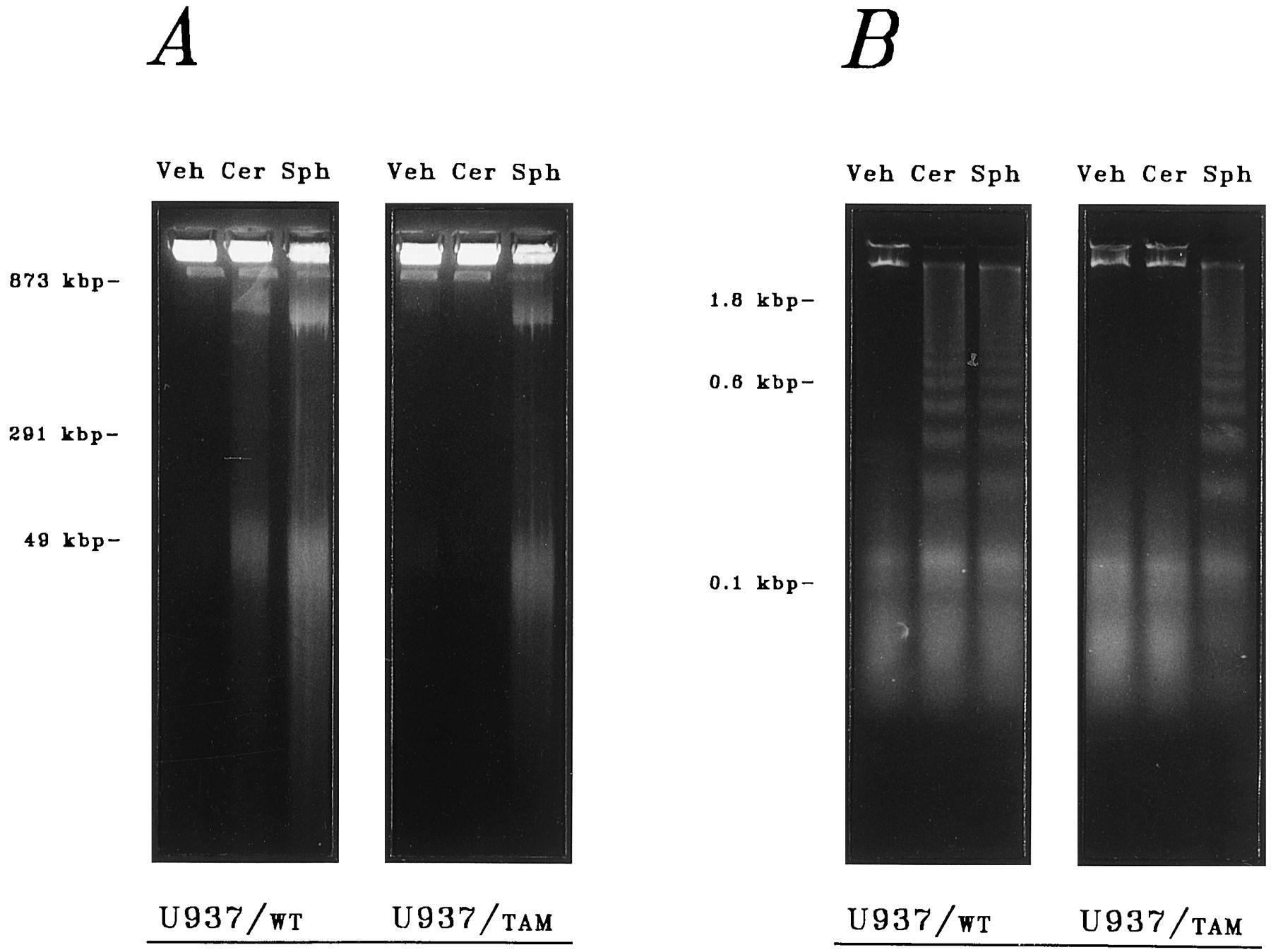
Direct comparison of the relative effects of ceramide and sphingosine in TAM-67-expressing cells revealed a distinctly altered susceptibility to the lethal actions of ceramide but not to those of sphingosine. Extensive degradation of genomic DNA was observed after acute (6-hr) exposure of U937/WT cells to synthetic preparations of ceramide or sphingosine at equimolar concentrations (10 μm), as demonstrated (a) qualitatively by electrophoretic resolution of ∼50-kbp DNA fragments on pulsed-field gels (Fig.5A) or ∼0.2–1.2-kbp DNA fragment ladders on static-field gels (Fig. 5B) and quantitatively by (b) spectrofluorophotometric measurement of the accumulation of double-stranded DNA fragments (Fig. 6A) and corresponding double-stranded breakage of bulk DNA (Fig. 6B). Markedly altered susceptibility to ceramide was observed after parallel exposures in U937/TAM cells. Ceramide-mediated degradation of genomic DNA was abolished by expression of the TAM-67 protein, indicating that normal c-Jun-mediated transactivation (which is initiated through phosphorylation by p46-JNK1/p54-JNK2) is required for ceramide-mediated cell death. In marked contrast, sphingosine-related DNA damage was completely unimpaired in the TAM-67 transfectants, which is consistent with a c-Jun-independent mechanism of sphingosine action as suggested above. In addition, apoptotic DNA damage was closely associated with reduced clonogenic potential. Exposure to ceramide for 6 hr reduced clonogenicity in U937/WT cells by 49%, whereas colony formation in U937/TAM cells was not significantly altered; conversely, parallel exposure to sphingosine reduced clonogenicity in U937/WT cells and U937/TAM cells by 74% and 77%, respectively. Furthermore, the established capacity of highly selective pharmacological PKC inhibitors to promote apoptosis in leukemia cells (20) is also unaffected by the expression of TAM-67, as we reported previously (23); comparable apoptotic responses were noted in both U937/WT and U937/TAM cells after acute (6-hr) exposure to calphostin C (10 nm) or chelerythrine (5 μm) (not shown). On the bases of each of the indices of genomic damage used in these studies, both (a) abrogation of ceramide action in U937/TAM cells and (b) persistence of sphingosine action were unequivocal.

Effect of TAM-67 expression on the apoptotic responses to ceramide and sphingosine. Parental (U937/WT) and transfected (U937/TAM) cell lines were treated with synthetic preparations of ceramide (Cer; 10 μm), sphingosine (Sph; 10 μm), or lipid-free vehicle (Veh) for 6 hr. Apoptotic DNA fragments were analyzed on agarose gels as described in Materials and Methods. DNA loop fragments (50 kbp) were resolved by pulsed-field electrophoresis (A), and oligonucleosomal DNA fragment ladders (200–1200 bp) were resolved by static-field electrophoresis (B). Data shown are from a representative study performed four times with comparable results.

Effect of TAM-67 expression on the induction of DNA damage by ceramide and sphingosine. Parental (U937/WT) and transfected (U937/TAM) cell lines were treated with synthetic preparations of ceramide (Cer; 10 μm), sphingosine (Sph; 10 μm), or lipid-free vehicle (Veh) for 6 hr. Apoptotic DNA damage was quantified by spectrofluorophotometry as described in Materials and Methods. A, Formation (striped bars) and release (cross-hatched bars) of double-stranded DNA fragments. B, Double-stranded breakage of bulk DNA (filled bars). Data shown are from a representative study performed four times with comparable results.
The disparity between the apoptotic responses of TAM-67-negative and -positive U937 cell sublines was further characterized at the cytological level. Cells exhibiting apoptotic DNA breakage were visualized by fluorescence microscopy (Fig.7) after FITC-dUTP staining in the presence of TdT. Under basal conditions, ≤5% of U937/WT cells or U937/TAM cells exhibited spontaneous DNA breakage. DNA breakage was substantially increased after 6-hr exposure to ceramide (10 μm) in U937/WT cells (52%), whereas there was no evidence of increased genomic damage on parallel ceramide exposure in U937/TAM cells. A 6-hr exposure to sphingosine elicited extensive DNA damage in both U937/WT (80%) and U937/TAM (78%) cells. Cells exhibiting apoptotic cytoarchitectural features consistent with apoptosis were visualized by light microscopy after Wright-Giemsa staining in parallel studies (not shown). Under basal conditions, ≤3% of U937/WT or U937/TAM cells spontaneously exhibited apoptotic traits. Pronounced expression of apoptotic cytoarchitecture was noted after 6-hr exposure to ceramide (10 μm) in U937/WT cells (49%), whereas there was no evidence of morphological alterations after parallel ceramide exposure in U937/TAM cells. Conversely, 6-hr exposure to sphingosine elicited profound expression of apoptotic morphology in both U937/WT (71%) and U937/TAM (68%) cells. In parallel trials, the capacity of dihydrosphingosine to limit clonogenicity, induce DNA damage, and elicit apoptotic morphology was also found to be unaffected by expression of TAM-67 (not shown). Direct evidence that p38-RK activation is required for lipid-mediated apoptosis is limited. In the current study, the close association of p38-RK activation with the actions of both ceramide and sphingosine raised the possibility that this signaling element could represent a common or convergent element in the ultimate apoptotic processes initiated by the two lipids; this was of particular interest inasmuch as dominant-negative suppression of c-Jun by TAM-67 was not sufficient to rule out an essential involvement of p38-RK. To examine directly a potential requirement for p38-RK activation in lipid-dependent cell death, the lethal actions of ceramide and sphingosine were tested after pharmacological inhibition of p38-RK using the novel pyridinyl imidazole SB-203580, which acts through competitive inhibition of ATP binding within the catalytic site of the enzyme. Consistent with the reported inhibitory properties of this compound, SB-203580 (5 μm) potently reduced basal p38-RK activity in vitro (by >97%), whereas the inactive analog SKF-105809 had no effect (not shown). Surprisingly, the apoptotic influences of ceramide and sphingosine were unaffected by coexposure to SB-203580. Acute (6-hr) exposure to ceramide and sphingosine increased the fraction of apoptotic cells to 47% and 69%, respectively, as assessed by light microscopy (not shown) and to 51% and 73%, respectively, as assessed by fluorescence microscopy (Table 5). By either index of cell death, the responses to ceramide and sphingosine were completely insensitive to SB-203580, indicating that the lethal influences of these lipids are independent of p38-RK. The cytotoxicity of dihydrosphingosine was equally insensitive to SB-203580 (not shown). The apoptotic capacity of highly selective pharmacological PKC inhibitors was similarly unaffected by inhibition of p38-RK; comparable apoptotic responses were elicited by brief (6-hr) exposure to calphostin C (10 nm) or chelerythrine (5 μm) in the absence or presence of SB-203580 (not shown).

Cytological responses to ceramide and sphingosine: DNA damage. Parental (U937/WT) and transfected (U937/TAM) cell lines were treated with synthetic preparations of ceramide (Cer; 10 μm), sphingosine (Sph; 10 μm), or lipid-free vehicle (Veh) for 9 hr as indicated. After fixation, cells were stained with FITC-dUTP in the presence of TdT and examined by fluorescent microscopy as described in Materials and Methods. Results shown are from a representative study performed four times with comparable results.
Persistence of lipid-induced apoptosis in the presence of SB-203580
In additional experiments, apoptotic responses to ceramide and sphingosine were compared in the presence of both TAM-67 and SB-203580 (not shown). Ceramide-induced apoptosis was abolished in U937/TAM cells as noted above; the addition of SB-203580 failed to alter this response. Conversely, the apoptotic actions of sphingosine, dihydrosphingosine, and pharmacological inhibitors of PKC were unaffected by the combined effects of TAM-67 and SB-203580. The observation that simultaneous interference with both forms of SAPK activity could not block sphingoid base-mediated cell death indicated that p46-JNK1/p54-JNK2 and p38-RK do not mediate redundant cytotoxic signals in the initiation of apoptosis.
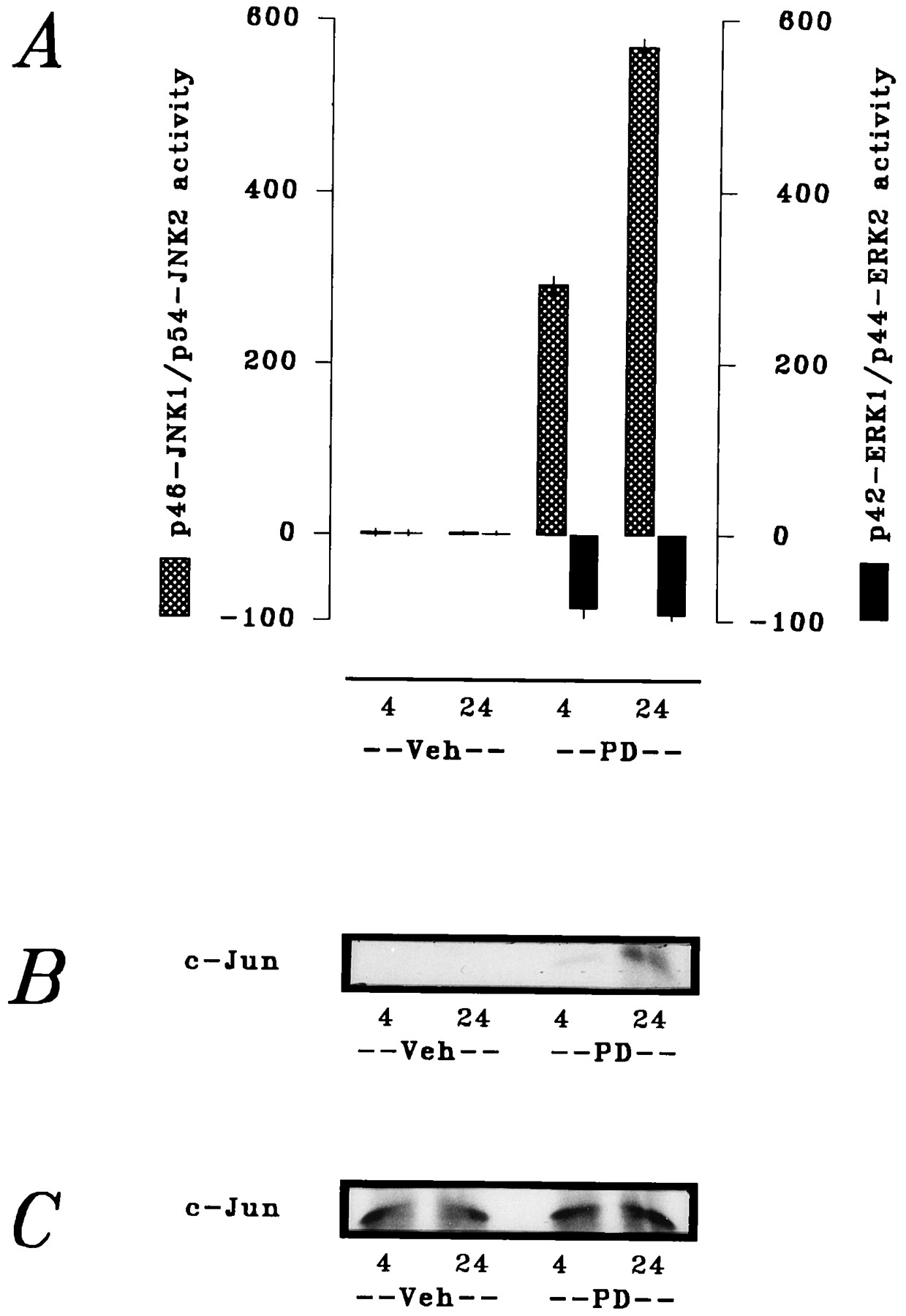
The apparent lack of requirement for p46-JNK1/p54-JNK2 (at least with respect to SAPK-related activation of c-Jun/AP1) in sphingoid base-mediated cell death suggested the participation of a separate downstream signaling process distinct from either of the SAPK activities. Therefore, in final experiments, we addressed the potential involvement of MAPK suppression in the cytotoxic actions of ceramide and sphingosine. In parallel time course studies, U937 cells were exposed to ceramide and sphingosine at equimolar concentrations (10 μm) for 0–60 min. A striking difference was noted between kinetic profiles of the effects of the two lipids on MAPK (Fig.8). Ceramide produced a gradual decline in p42-ERK1/p44-ERK2 activity over the exposure interval, but this suppression was incomplete (i.e., reduced by only 67%). In marked contrast, sphingosine produced an abrupt, virtually complete inhibition (i.e., reduced by 94%) of p42-ERK1/p44-ERK2 activity within 5 min; this inhibition was sustained throughout the exposure interval, with a slight recovery evident after 40 min. Essentially identical responses were obtained with dihydrosphingosine (not shown). In a similar fashion, acute (4-hr) exposure to the highly selective PKC inhibitors calphostin C (1 μm) and chelerythrine (10 μm) also sharply reduced p42-ERK1/p44-ERK2 activity (not shown). These responses collectively suggested that the lethal consequences of acute PKC inhibition, by either physiological or pharmacological agents, are related to a attendant decrease in MAPK cascade function. Consistent with this supposition, inhibition of MEK1 by the aminomethoxyflavone PD-98059 produced reciprocal alterations in MAPK and SAPK function that were qualitatively similar to those noted in response to sphingoid bases. Acute (4-hr) exposure of U937 cells to PD-98059 (10 μm) decreased p42-ERK1/p44-ERK2 activity by 84% and increased p46-JNK1/p54-JNK2 activity by 191% (Fig.9A); this response was accompanied by increased in situ phosphorylation of c-Jun (Fig. 9B). Consistent with the responses to sphingoid bases that have been described, however, PD-98059 failed to modify steady state levels of c-Jun (Fig. 9C) in these exposures. In addition, PD-98059 increased the fraction of cells manifesting evidence of apoptotic DNA damage from 2% to ≥29%. Chronic (24-hr) exposure to PD-98059 elicited more pronounced MAPK and SAPK responses (decreasing activity of p42-ERK1/p44-ERK2 by 94% and increasing activity of p46-JNK1/p54-JNK2 by 467%; Figs. 9, A–C) and further augmented the fraction of cells exhibiting apoptotic DNA damage to ≥58%. These observations demonstrated that interruption of basal MAPK cascade function at the level of MEK1 was sufficient to initiate apoptosis, possibly a consequence of acute derepression of SAPK cascade output.

Time course of MAPK inhibition by ceramide and sphingosine. U937 cells were exposed to ceramide (10 μm; ▾) or sphingosine (10 μm; ▴) for 60 min. Activity of p42-ERK1/p44-ERK2 was determined by in vitro immune complex assay as described in Materials and Methods. Data shown are from a representative study performed three times with comparable results.

Induction of apoptosis by pharmacological interruption of MAPK cascade signaling. U937 cells were exposed to the MEK1 inhibitor PD98059 (PD) (10 μm) for 4 or 24 hr as indicated; cellular responses were then assessed as described in Materials and Methods. A, Activities of p46-JNK1/p54-JNK2 (hatched bars) and p42-ERK1/p44-ERK2 (filled bars); values expressed as percentage of vehicle (Veh)-treated controls. B, In situphosphorylation of c-Jun. C, c-Jun expression.
Discussion
Extensive evidence supports the current recognition of the SAPK and MAPK cascades as opposing effectors in the regulation of cell survival. Multiple sphingophospholipid- and glycerophospholipid-derived messengers mediate cytotoxic and cytoprotective signals that converge at the level of SAPK and MAPK. Previous reports from this and other laboratories have described the lethal effects of ceramide and sphingosine (2-5) in human myeloid leukemia cells. Although the apoptotic responses to these lipids reportedly entail common distal processes [e.g., rapid dephosphorylation of pRb (29-32), suppression of c-myc mRNA expression (5, 29, 31)], substantive differences clearly exist in their respective actions. The action of two parallel, if incompletely delineated, cell death pathways has therefore been inferred in the effector functions of ceramide and sphingosine (reviewed in Ref. 33). The current findings demonstrate that the apoptotic responses to both ceramide and sphingosine entail complementary redirection of SAPK and MAPK activities. Induction of cell death by either lipid effector was associated with coordinately increased p46-JNK1/p54-JNK2 and decreased p42-ERK1/p44-ERK2. Despite qualitative similarities between the two responses, however, it is possible that the reciprocal modulations of SAPK and MAPK actually represent homologous, but distinct, signaling processes inasmuch as (a) the response to ceramide was weighted toward SAPK stimulation, whereas (b) the response to sphingosine was weighted toward MAPK inhibition. Furthermore, the apoptotic signals elicited by these lipids clearly diverged distally. Increased expression and activation of the SAPK substrate c-Jun were absolutely necessary for the induction of cell death by ceramide but not by sphingosine. Although this suggested the participation of one or more additional transregulatory elements downstream of SAPK, we were unable to demonstrate participation of another SAPK substrate, ATF2, in the lethal response to either lipid. Identification of other selectively activated downstream elements will be addressed in future studies.
Ceramide subserves a lethal effector function in the induction of apoptosis by diverse stimuli ranging from activation of cytotoxic receptor systems to various environmental stresses (14). Ceramide induces apoptotic cell death in myeloid leukemia cells (2-4), possibly via direct activation of KSR/CAPK and/or CAPP, although a proximal effector for ceramide in this response remains to be established. Further downstream, however, ceramide-dependent cell death requires activation of the SAPK sequence MEKK1, SEK1, JNK1/JNK2 (13, 14) and, ultimately, proapoptotic transregulatory elements such as c-Jun (14,15). The SAPK cascade is engaged by ceramide and ceramide-dependent lethal stimuli, and experimental interventions that disrupt the primary SAPK signaling sequence [e.g., dominant-negative ablation of SEK1 or quenching of c-Jun (14), antisense blockade of c-jun/c-Jun expression (15)] attenuate or abolish ceramide-mediated lethality. The current findings confirm the importance of SAPK activation in the lethal actions of ceramide. Ceramide rapidly promoted activation of p46-JNK1/p54-JNK2 and increased expression and activity of a primary substrate, c-Jun; these responses correlated temporally with apoptotic DNA damage and cell death. Dominant-negative suppression of normal c-Jun function abolished the apoptotic influence of ceramide and maintained normal proliferative capacity. Ceramide also produced a gradual and moderate reduction in p42-ERK1/p44-ERK2 activity. This contrasts sharply with other reports that MAPK activity is either stimulated (34) or unaffected (13) by ceramide in myeloid leukemia cells, but it is likely that such responses vary with cell lineage and across cell line variants. Ceramide does not seem to modify cPKC/nPKC activity in vitro (18, 35, 36). Nevertheless, ceramide reportedly suppresses diglyceride-dependent translocation of cPKCαin situ (35) and inactivates autophosphorylated cPKCαin situ, responses presumed to derive from CAPP-mediated dephosphorylation (36). These events are effectively reversed by okadaic acid, which may be significant, given that ceramide-induced cell death is partially antagonized in an okadaic acid-sensitive (butnor-okadaone-insensitive) fashion (3). CAPP-related reductions in cPKCα (and possibly of other PKC isoforms as well) may thus underlie ceramide-related reductions in MAPK and protective signaling elements downstream of PKC; an alternative or indirect (i.e., PKC-independent) mechanism negatively coupling ceramide to MAPK has not been described. In any event, ceramide-driven activation of the SAPK cascade would be enhanced by coordinate reduction of the cytoprotective influence of PKC, and such a provision may therefore represent an upstream avenue of repressive cross-talk between the SAPK and MAPK cascades.
Corresponding involvement of the SAPK cascade in sphingoid base action has not been addressed, although these lipids represent lethal effectors in most mammalian cells (18). Sphingosine, dihydrosphingosine, and other lysosphingolipids potently induce apoptotic cell death in myeloid leukemia cells via direct inhibition of cPKC/nPKC (5, 6); these responses are mimicked by selective pharmacological PKC inhibitors (20, 21). Acute reduction of PKC, whether by physiological or pharmacological means, thus constitutes a significant apoptotic stimulus in proliferating cells. Although signaling elements downstream of cPKC/nPKC in this lethal process have yet to be established conclusively, acute inhibition of these enzymes presumably results in attenuation of the MAPK cascade sequence Raf1, MEK1, ERK1/ERK2, and subsequent inactivation of antiapoptotic transregulatory elements such as nuclear factor-κB. In the current study, sphingosine and dihydrosphingosine elicited comparatively modest activation of p46-JNK1 and p54-JNK2 but failed to promote expression of c-Jun. The mechanism through which sphingoid bases engage the SAPK cascade is uncertain, but it may derive from interruption of a tonic suppressive influence exerted over this system by one or more elements comprising the MAPK cascade. The existence of repressive cross-talk pathways between the primary MAPK and SAPK modules has been proposed by others (1). Nevertheless, both ceramide and sphingosine promoted extensive apoptotic DNA degradation and cell death. As noted, the cytotoxic influences of sphingosine and dihydrosphingosine were not associated with induction of c-jun/c-Jun expression, nor were they modified by dominant-negative interference with c-Jun function, indicating that the transregulatory potential of the AP1 transcription factor complex does not contribute to sphingoid base-mediated lethality. It is noteworthy that the apoptotic actions of highly selective pharmacological PKC inhibitors such as calphostin C and chelerythrine were similarly unaffected by quenching of c-Jun activity, a phenomenon that we characterized recently in detail (23). Taken together, these observations demonstrate that the apoptotic responses attendant on acute inhibition of PKC engage a cell death pathway independent of c-Jun-mediated transregulation. The mechanistic importance of this observation is considerable given that c-Jun represents a primary SAPK substrate in most systems; however, there was no apparent involvement of ATF2, another well characterized SAPK substrate. Therefore, these findings point toward the participation of other, as yet unidentified, targets downstream of SAPK that confer selective apoptotic potential in the responses to ceramide and sphingosine. On the other hand, sphingosine and dihydrosphingosine produced immediate and virtually complete suppression of p42-ERK1/p44-ERK2 activity, implicating a direct contribution of MAPK inhibition to the lethal actions of sphingoid bases. This concept is potentially compatible with the established role of the MAPK cascade in protective and/or proliferative actions of other lipid messengers [e.g., sphingosine-1-phosphate (37)].
The novel SAPK isoform p38-RK has been presumed to mediate some aspects of apoptosis (1). Although activation of p38-RK may be associated with the acute actions of ceramide, direct involvement of this enzyme in lipid-mediated apoptosis remains to be established. To that end, we also characterized possible contributions of p38-RK to the cytotoxic actions of ceramide and sphingosine. We found that ceramide and sphingoid bases activated p38-RK with comparable response maxima and superimposable time courses. Pharmacological inhibition of p38-RK by the pyridinyl imidazole SB-203580 has been characterized in vitro, however, and effectively mitigates some aspects of p38-RK activity in situ. The apoptotic capacity of ceramide was not modified by SB-203580, demonstrating that p38-RK does not underlie ceramide-related cytotoxicity, which contrasts markedly with the unambiguous requirement for p46-JNK1 and p54-JNK2 in the actions of this lipid. The apoptotic effects of sphingoid bases and pharmacological PKC inhibitors were similarly unaffected by SB-203580, indicating that none of the SAPK enzymes participate in lethal responses initiated by acute reductions in PKC activity. In fact, other observations from our laboratory indirectly suggest that p38-RK may instead exert a cytoprotective influence in U937 cells. SB-203580 alone is not toxic in acute (i.e., 6-hr) exposures but instead promotes apoptotic cell death extensively in chronic (i.e., 24- or 48-hr) exposures. The complex activation of p38-RK elicited by ceramide and sphingosine may therefore represent a consequence, rather than a cause, of lipid-induced apoptosis and possibly constitutes a cytoprotective response to lethal insult.
Elsewhere, we advanced the concept that the PKC isoenzyme family represents a general and conserved cytoprotective element in the regulation of cell survival (19). Ceramide-driven apoptosis is subject to transmodulation by lipid effectors converging on cPKC/nPKC (e.g., diradylglycerol, sphingosine). More specifically, the apoptotic potential of ceramide is reciprocally (a) attenuated through cPKC/nPKC activation by diglycerides (4, 33) but (b) amplified through cPKC/nPKC inhibition by sphingoid bases (6, 33); furthermore, ceramide action is similarly modulated by pharmacological activators and inhibitors of PKC (6). Thus, although explicitly cytotoxic levels of sphingosine and/or dihydrosphingosine may not often be realized under normal physiological conditions, it is evident that these lipid effectors serve (at sublethal levels) as modulators of ceramide action. Ceramide-mediated lethality is also mitigated by sphingosine-1-phosphate through a PKC-independent mechanism (37). It is significant that all such interactions seem to be manifested through alterations in multiple downstream systems, including the SAPK and MAPK cascades. In the primary induction of apoptosis, we found that (a) ceramide strongly stimulates SAPK and weakly inhibits MAPK, whereas (b) sphingosine weakly stimulates SAPK but strongly inhibits MAPK. Reciprocal SAPK stimulation and MAPK inhibition have been proposed by other investigators (1, 38) and are associated with lipid signaling in various settings (39, 40), including U937 cells (37). Our findings are consistent with this view and demonstrate the participation of a potentially analogous process in the lethal effects of sphingoid bases. These findings also suggest the apparent reciprocity between the MAPK and SAPK cascades is mediated, at least in part, via the actions of mutually inhibitory lateral pathways. As such, the outwardly contrasting effects of ceramide and sphingosine on SAPK/MAPK signaling may represent complementary mechanisms through which diverse signals are integrated to elicit cell death. It is therefore plausible that each of these lipid messengers kills by producing a “weighted average” of opposing cytotoxic and cytoprotective signals with fundamentally similar results: redirection of the SAPK/MAPK output ratio away from p42-ERK1/p44-ERK2 and toward p46-JNK1/p54-JNK2. A dynamic balance between the SAPK and MAPK cascades may thus represent a critical determinant in the lipid-dependent regulation of leukemic cell survival. Further study will be required to confirm this position definitively.
Acknowledgments
The authors thank Dr. Robert Tombes for recommending procedural modifications to improve photomicrography in the cytological studies and providing a careful review of the manuscript.
Footnotes
- Received May 7, 1997.
- Accepted August 14, 1997.
-
Send reprint requests to: Dr. W. David Jarvis, Medical College of Virginia, MED-HEM/ONC, Box 980230, MCV Station, Richmond, VA 23298. E-mail: wjarvis{at}gems.vcu.edu
-
Portions of this work were presented in preliminary form at the Keystone Symposium on Cell Biology entitled Apoptosis (Programmed Cell Death), Tamarron, CO, March 5–11, 1995, and the 87th Annual Meeting of the American Association for Cancer Research, Washington, D.C., April 20–24, 1996.
-
This work was supported primarily by National Cancer Institute Research Grant CA63753 and Leukemia Society of America Award 6405-97 (S.G.). Other support includes National Cancer Institute National Research Service Award CA09380 (W.D.J.), National Heart, Lung, and Blood Institute National Research Service Award HL09241 (F.A.F.), United States Public Health Service Training Grants CA09564 and DK07150 (A.J.F., K.L.A.), and National Cancer Institute Research Grant IN-105V (P.D.). Additional funding was provided by the A. D. Williams Foundation of the Medical College of Virginia; the Robert B. Dalton Endowment Fund and the Thomas F. and Kate Miller Jeffres Memorial Trusts; and by National Cancer Institute Cancer Center Support Core Grant CA-16059 to the Massey Cancer Center.
Abbreviations
- SAPK
- stress-activated protein kinase
- KSR
- kinase suppressor of ras
- CAPK
- ceramide-activated protein kinase
- CAPP
- ceramide-activated protein phosphatase
- ERK
- extracellular signal receptor-activated kinase
- JNK
- c-Jun NH2-terminal kinase
- MAPK
- mitogen-activated protein kinase
- PKC
- protein kinase C
- SMase
- sphingomyelinase
- TAM-67
- c-jun/c-Jun transactivation-deficient mutant
- AP
- activator protein
- cPKC
- group A (conventional) isoform of protein kinase C
- nPKC
- group B (novel) isoform of protein kinase C
- PBS
- phosphate-buffered saline
- TdT
- terminal deoxynucleotidyl transferase
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MAPP
- N-myristoylamino-1-phenyl-1-propanol
- bp
- base pair(s)
- PLA
- phospholipase A
- FITC
- fluorescein isothiocyanate
- TBST
- Tris-buffered saline containing 1% Tween 20
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}