


Abstract
Dopamine D2 receptors contain a cluster of serine residues in the fifth transmembrane domain that contribute to activation of the receptor as well as to the binding of agonists. We used rat D2S dopamine receptor mutants, each containing a serine-to-alanine substitution (S193A, S194A, S197A), to investigate the mechanism through which these residues affect activation of the receptor. Activation of the mutant receptor S194A was abolished in an agonist-dependent manner, such that dopamine no longer inhibited cAMP accumulation in C6 glioma cells or activated G protein-regulated K+ channels in Xenopus laevis oocytes, whereas the efficacy of several other agonists was unaffected. Dihydrexidine did not inhibit cAMP accumulation at either S193A or S194A. The decreased efficacy of dihydrexidine at S193A and S194A and dopamine at S194A was associated with a decreased ability to detect a GTP-sensitive high affinity binding state for these agonists. The ability of dopamine to stimulate [35S]guanosine-5′-O-(3-thio)triphosphate binding via S194A also was decreased by ∼50%. Finally, constitutive stimulation of [35S]guanosine-5′-O-(3-thio)triphosphate binding and inhibition of adenylate cyclase by the D2Sreceptor was reduced by mutation of either S193 or S194. These data support the existence of multiple active receptor conformations that are differentially sensitive to mutation of serine residues in the fifth-transmembrane domain.
D2dopamine receptors, like all catecholamine receptors, contain a cluster of serine residues in the TM5 that contribute to the binding of agonists. Data obtained using mutants of the β2-adrenergic receptor lacking individual TM5 serines support a model in which two of the receptor serine residues form specific hydrogen bonds with each of the catechol ring hydoxyl groups of catecholamine ligands (Strader et al., 1989). The critical role of TM5 serines in agonist binding has been demonstrated for several other catecholamine receptors as well, but it has been difficult to identify invariant interactions between catechol ring hydroxyl groups and particular serine residues as demonstrated for the β2 receptor (Wang et al., 1991; Coxet al., 1992; Pollock et al., 1992; Hwa and Perez, 1996). For example, replacing Ser193 significantly decreases the affinity of the rat D2S and D2L receptors for many, but not all, catecholamine agonists (Cox et al., 1992; Woodward et al., 1996), whereas replacing Ser194 or Ser197 of the rat or human D2 receptor has lesser effects on the binding of some agonists (Cox et al., 1992; Mansour et al., 1992; Woodward et al., 1996).
The conserved serine residues in TM5 also are important for activation of catecholamine receptors. Substitution of one or more of the serine residues decreases or abolishes the ability of several catecholamine receptors to couple to G proteins and to activate intracellular signaling pathways (Strader et al., 1989; Wang et al., 1991; Cox et al., 1992; Mansour et al., 1992; Hwa and Perez, 1996; Woodward et al., 1996). Replacing Ser194 of the rat D2S receptor with an alanine residue prevents activation of the mutant receptor in an agonist-specific manner, such that meta-tyramine, but not dopamine or para-tyramine, inhibits cAMP accumulation (Coxet al., 1992). Although the mechanism through which mutations in TM5 affect receptor function in an agonist-dependent manner is unknown, the observation that mutating Ser188 in a constitutively active mutant of the α1A-adrenergic receptor (corresponding to Ser193 in the D2S receptor) decreases some manifestations of constitutive activity suggests that this serine residue may enable the wild-type receptor to adopt an active conformation (Hwa and Perez, 1996).
In this study, we investigated the mechanism of agonist-specific activation of the rat D2S and mutant receptors in which TM5 serines were replaced by alanine (S193A, S194A, S197A). We characterized agonists from several chemical classes in terms of their ability to modulate intracellular signaling pathways via the receptors by measuring inhibition of cAMP accumulation, activation of G protein-activated K+ channels, receptor coupling to G proteins, and agonist-stimulated binding of [35S]GTPγS. We now report that mutation of Ser193 or Ser194 decreased the constitutive activity of the D2S receptor and prevented modulation of intracellular signaling pathways by some, but not all, D2 receptor agonists. These data suggest that Ser193 and Ser194 in TM5 are important structural determinants of receptor activation and support the hypothesis that G protein-coupled receptors have multiple, distinct active conformations.
Experimental Procedures
Materials.
[3H]Spiperone was purchased from Amersham Life Sciences (Arlington Heights, IL). [3H]cAMP, [3H]adenine, and [35S]GTPγS were from New England Nuclear Research Products (Boston, MA). (+)-Butaclamol, clozapine, haloperidol, spiperone, NPA, quinpirole, and 7-OH-DPAT were purchased from Research Biochemicals International (Natick, MA). DHX and epidepride were generous gifts from Dr. R. Mailman (University of North Carolina, Chapel Hill, NC) and Dr. T. de Paulis (Vanderbilt University, Nashville, TN), respectively. Serum was purchased from HyClone (Logan, UT). Most other reagents, including culture media, dopamine, isoproterenol, and PTX, were purchased from Sigma Chemical (St. Louis, MO).
Cell lines.
The construction and stable expression in C6 glioma cells of rat mutant D2 receptor cDNAs in which Ser193, Ser194, or Ser197 was replaced with an alanine residue, creating S193A, S194A, and S197A, were described previously (Coxet al., 1992). Wild-type and mutant cell lines were maintained in Dulbecco’s modified Eagle’s medium supplemented with 5% iron-supplemented calf bovine serum, 5% fetal bovine serum, 2 μg/ml puromycin, 0.05 unit/ml penicillin, and 50 μg/ml streptomycin at 37° and 10% CO2.
[3H]Spiperone binding assays.
Cells were lysed in ice-cold hypotonic buffer (1 mmNa+-HEPES, pH 7.4, 2 mm EDTA) for 10 min, scraped from the plate, and centrifuged at 18,000 ×g for 20 min. The resulting crude membrane fraction was resuspended with a Brinkmann Polytron homogenizer (Westbury, NY) at setting 6 for 6–10 sec in Tris assay buffer (50 mmTris·HCl, pH 7.4, 0.9% NaCl) for saturation assays or HEPES assay buffer (20 mm K+-HEPES, pH 7.4, 6 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 1 mm dithiothreitol, 0.01% bovine serum albumin, 0.025% ascorbic acid) for competition assays, incubated for 10 min at 37°, centrifuged (18,000 × g for 20 min), and resuspended in the appropriate assay buffer. Membrane proteins (2–20 μg) were incubated in duplicate in a total reaction volume of 1 ml with [3H]spiperone at concentrations ranging from 0.006 to 0.2 nm for saturation binding or 0.05 nm with the appropriate concentration of the competing drug for competition binding. (+)-Butaclamol (5 μm) was used to define nonspecific binding. Reactions were incubated at 37° for 45 min and terminated by filtration (Whatman GF/B filters) using a 96-well Tomtec cell harvester (Orange, CT) and ice-cold saline wash buffer (10 mm Tris·HCl, pH 7.4, 0.15 m NaCl). Filters were allowed to dry, and 50 μl of BetaPlate scintillation fluid was added to each sample. Radioactivity on the filters was measured using a Wallac BetaPlate scintillation counter (Gaithersburg, MD). Data for saturation and displacement binding were analyzed by nonlinear regression using the computer program Prism (GraphPAD Software, San Diego, CA). The free concentration of radioligand was calculated as the concentration added minus the concentration specifically bound. Mean values for units expressed as drug concentration (EC50,KD ,KI ) are expressed as the geometric mean followed by the limits described by the asymmetric standard error.
cAMP assays.
The ability of D2receptor agonists or antagonists to inhibit or potentiate, respectively, isoproterenol-stimulated adenylate cyclase activity or cAMP accumulation via the endogenous β2-adrenergic receptors was measured in intact cells. Cells were plated at a density of 18,000 cells/cm2 onto 6- or 48-well tissue culture plates and used in experiments 2–3 days later. For experiments assessing the effects of PTX, cells were treated for 18–24 hr before the cAMP accumulation assay with 25 ng/ml PTX in Dulbecco’s modified Eagle’s medium, supplemented as described above. Before the assay, cells were preincubated with assay buffer, which was 15 mmNa+-HEPES-buffered L-15, pH 7.4, 0.2% ascorbic acid, with 1 μCi/ml [3H]adenine added for adenylate cyclase activity assays according to the method ofShimizu et al. (1969), or Earle’s balanced salt solution with 0.2% ascorbic acid and 2% calf bovine serum, pH 7.4, for cAMP accumulation assays, for 10 min at 37°. For agonist assays, the plates were incubated in assay buffer with the appropriate agonist concentration and 1 μm isoproterenol for 15 min at 37°. For antagonists, an additional incubation for 10 min was conducted with the appropriate antagonist before the 15-min incubation with isoproterenol. The assay was terminated by decanting the medium, and the cells were placed on ice and lysed with 3% trichloroacetic acid. Lysates were incubated on ice at least 30 min before adenylate cyclase activity was measured as the conversion of [3H]ATP to [3H]cAMP (Shimizu et al., 1969; Cox et al., 1992) or cAMP accumulation was measured using a competitive protein binding assay (Watts and Neve, 1996). Dose-response data were analyzed as described above for radioligand binding.
Oocyte isolation and maintenance.
Adult female Xenopus laevis from Xenopus I (Ann Arbor, MI) were anesthetized by immersion in tap water with 3-aminobenzoic acid ethyl ester (1 g/liter). The ovarian sacs were removed, washed, torn open with forceps, and rinsed several times with OR-2 medium (5 mmNa+-HEPES, pH 7.5, 82.5 mm NaCl, 2 mm KCl, 1 mm MgCl2). Oocytes were enzymatically defolliculated in OR-2 solution containing collagenase A (1–3 mg/ml; Boehringer-Mannheim, Indianapolis, IN) with shaking at room temperature for 3 hr. Oocytes were rinsed four times with fresh ND-96 (5 mm Na+-HEPES, pH 7.5, 96 mm NaCl, 2 mm KCl, 1.8 mmCaCl2, 1 mmMgCl2, 2.5 mm Na-pyruvate, 0.5 mm theophylline, 50 μg/ml gentamycin), selected by visual inspection (stages V and VI), and stored at 16–18° in ND-96.
cRNA preparation and injection.
The Kir plasmid/cDNA constructs used in these studies were kindly provided by Dr. Henry Lester (California Institute of Technology, Pasadena, CA; rat Kir3.1, GenBank Accession No. L25264), Dr. Florian Lesag (Centre Nationale de Recherche Scientifique, Velburne, France; mouse Kir3.2, GenBank Accession No. U11859), and Dr. Berndt Fakler (University of Tübingen, Tübingen, Germany; the X. laevis expression vector pBF). Rat D2S (GenBank Accession No. M36831) and S194A cDNAs (Cox et al., 1992) were subcloned from pRSV into pcDNA-1. Plasmid DNAs were linearized with the restriction endonucleases MluI for Kir3.1/pBF and Kir3.2/pBF andXbaI for D2/pcDNA-1. cRNAs were transcribed in vitro using Sp6 (Kir3 cDNAs) or T7 (D2 cDNAs) RNA polymerases (GIBCO BRL, Grand Island, NY) under standard reaction conditions with the addition of 2.5 mm m7G(5′)ppp(5′)G (Pharmacia, Piscataway, NJ). cRNAs were quantified by absorbance at 260 nm, diluted to concentrations giving empirically determined expression levels, aliquoted, and stored at −80° in 10 mm Tris·HCl, pH 8.0. Oocytes were injected with cRNAs coding for Kir3.1 and Kir3.2 channel subunits together with those coding for D2S or S194A receptors in a total volume of 46 nl with a Nanoject Automatic Oocyte Injector (Drummond Scientific, Broomall, PA) and used for experiments 2–5 days later.
Oocyte voltage-clamp recording and analysis.
Activation of ion channels was measured by standard two-electrode voltage-clamp (−80 mV) using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA). Microelectrode holders and microelectrodes (1–3 MΩ) were filled with 3 m KCl. Oocytes were perfused at 2 ml/min with Ringer’s solutions (low potassium: 10 mmNa+-HEPES, pH 7.5, 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2; high potassium: 10 mm Na+-HEPES, pH 7.5, 2 mm NaCl, 96 mm KCl, 1.8 mmCaCl2). Test compounds were applied via a 1.5-mm tube aimed at the oocyte. Data were low-pass-filtered at 10 Hz (−3 dB; Frequency Devices, Haverville, MA) and collected at 50 Hz using a Macintosh IIvx, MacAdios II/16 analog/digital interface, a 128-kilobyte buffer (first-in, first-out) and Superscope II software (GW Instruments, Somerville, MA).
Concentration curves for stimulation of Kir3 channels were a composite of three to nine experiments for each agonist, with each individual value normalized between experiments using the mean current size for a single agonist concentration representing 80–100% of maximal current (100 nm or 1 μm). Data analysis and curve fits were performed using KaleidaGraph 3.0 (Adelbeck Software, Reading, PA). EC50 values were estimated by fitting to the formula In = I/(1 + (EC50/[A])n), where In is the normalized peak current amplitude at the concentration of agonist [A], I is the maximal peak current amplitude, EC50 is the agonist concentration giving a half-maximal response, and n is the Hill coefficient for the agonist.
[35S]GTPγS Binding.
Methods to measure [35S]GTPγS binding were modified fromLazareno and Birdsall (1993). Membranes were prepared as described for [3H]spiperone binding using hypotonic buffer containing dithiothreitol (1 mm) and phenylmethylsulfonyl flouride (300 μm) and resuspended in 20 mmK+-HEPES, pH 7.4, containing dithiothreitol and phenylmethylsulfonyl flouride. The membranes were recentrifuged at 18,000 × g for 15 min, resuspended at a final concentration of 5 μg/μl, and stored at −80°. Membrane proteins were thawed rapidly, diluted, and added to triplicate reaction mixtures containing the agonist or antagonist of interest in an assay buffer of 25 mm K+-HEPES, pH 7.4 (5 mm MgCl2, 1 mmdithiothreitol, 1% bovine serum albumin, and 0.03% ascorbic acid, with 150 mm NaCl and 10 μm GDP added for agonist assays). After a 10-min incubation at 30°, 1 nm[35S]GTPγS in assay buffer was added to a final volume of 100 μl and incubated at 30° for an additional 60 min. The reactions were terminated by filtration as described above, using a wash buffer of 50 mm Tris·HCl, pH 7.4, 100 mm NaCl, and 5 mm MgCl2. Nonspecific binding was determined in the presence of 10 μm GTPγS. For experiments assessing the effects of PTX on binding, cells were treated 18–24 hr before lysis with the appropriate concentration of PTX.
Results
Receptor expression.
Saturation analysis of [3H]spiperone binding to stably expressed wild-type D2S and mutant receptors used in this study showed that the radioligand bound to one site with similar affinity (KD ) in each cell line. The average KD values (followed by the limits defined by the asymmetric standard error) from at least five independent experiments were 55 (48–63) pm for D2S, 48 (43–54) pm for S193A, 37 (32–43) pm for S194A, and 37 (28–47) pm for S197A. The average density of the receptors was similar (B max = 1119 ± 195 fmol/mg for D2S, 1654 ± 233 fmol/mg for S193A, 1073 ± 223 fmol/mg for S194A, and 1221 ± 223 fmol/mg for S197A; five or more experiments for each cell line).
Agonist-specific inhibition of cAMP accumulation in C6 cells.
In agreement with our previous work (Cox et al., 1992), dopamine produced no inhibition of adenylate cyclase activity in cells expressing S194A (C6-S194A), although maximal inhibition in C6-S193A or C6-S197A cells was similar to that for C6-D2S(Table 1). The D1receptor catecholamine agonist DHX also was an agonist at D2S and S197A receptors but lacked efficacy for inhibition of adenylate cyclase via S193A or S194A. The efficacy of NPA and the noncatecholamine agonists quinpirole and 7-OH-DPAT for inhibition of adenylate cyclase activity was not affected at any of these mutant receptors (Table 1). Inhibition of adenylate cyclase activity by 1 μm 7-OH-DPAT via each of the receptors, including S194A, was prevented by prior inactivation of PTX-sensitive G proteins (25 ng/ml PTX for 18–24 hr, data not shown). Furthermore, inhibition of adenylate cyclase activity by 7-OH-DPAT via S194A was inhibited by dopamine in an apparently competitive manner, as indicated by parallel rightward shifts in the dose-response curve for 7-OH-DPAT in the presence of increasing concentrations of dopamine (Fig.1). The average EC50 values from three independent experiments were 0.7 (0.4–14) nm for 7-OH-DPAT alone, 14 (5–37) nm for 7-OH-DPAT in the presence of 10 μmdopamine, and 71 (40–126) nm for 7-OH-DPAT in the presence of 100 μm dopamine. The pA 2value for dopamine at S194A, calculated by the Schild analysis (Fig. 1,inset; Arunlakshana and Schild, 1959), was similar to the affinity (KH ) for dopamine at this mutant (2.5 μm compared with 4 μm).
Maximal inhibition of isoproterenol-stimulated adenylate cyclase activity by D2S and serine mutant receptors
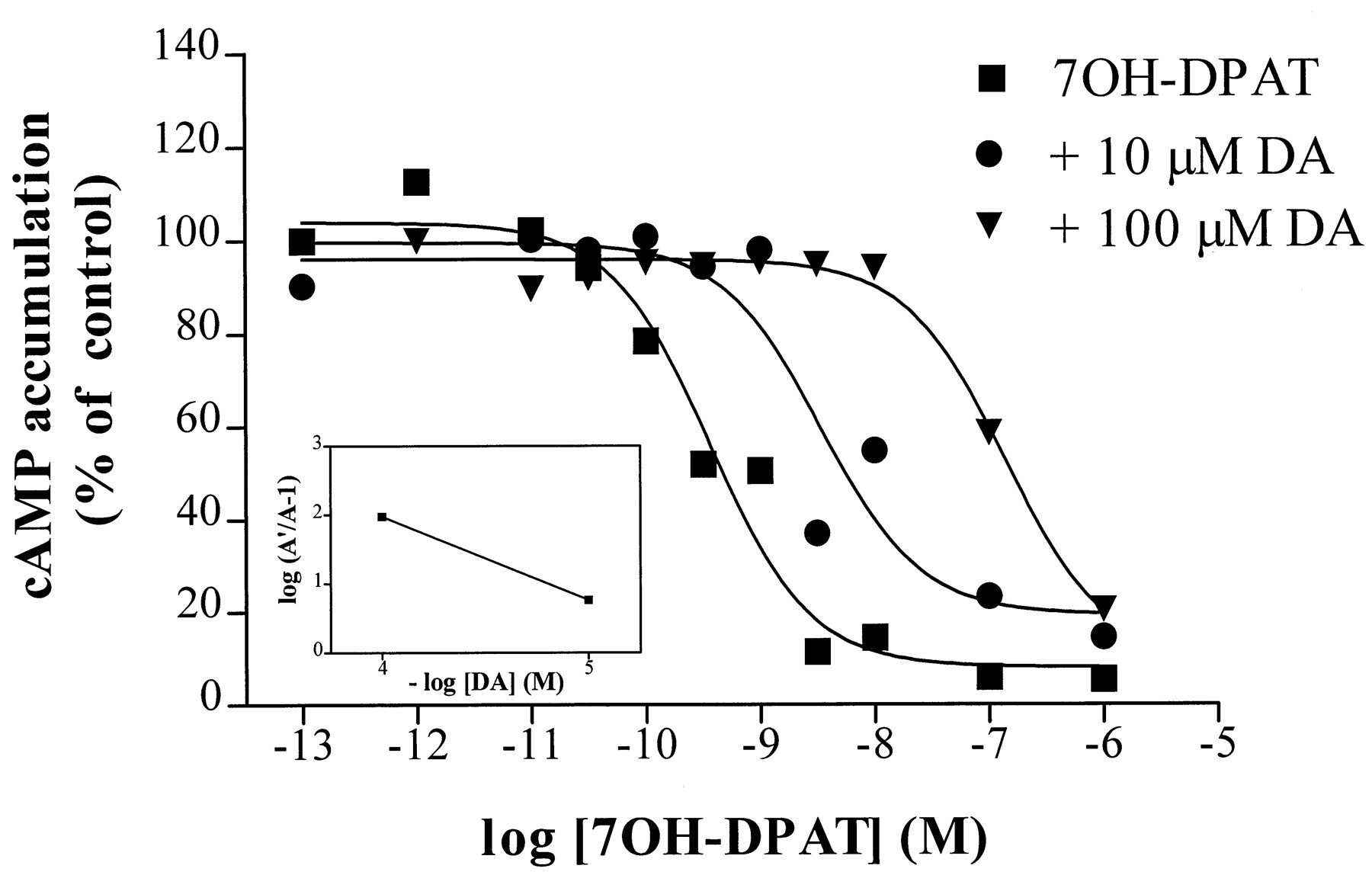
Inhibition of adenylate cyclase activity by 7-OH-DPAT (1 μm) via the S194A mutant receptor is antagonized by dopamine. Data shown are from one experiment that is representative of three, each carried out in duplicate, and are expressed as the percentage of isoproterenol-stimulated cAMP accumulation in the absence of agonist. The EC50 values for the curves shown were 0.4 nm for 7-OH-DPAT alone, 4 nm for 7-OH-DPAT in the presence of 10 μmdopamine, and 139 nm for 7-OH-DPAT in the presence of 100 μm dopamine. Inset, Schild plot of the mean values from three experiments (slope, −1.2 ± 0.006).A′, EC50 value of 7-OH-DPAT inhibition in the presence of dopamine. A, EC50 value in the absence of dopamine.
The effects of the mutations on agonist potency for inhibition of cAMP accumulation generally corresponded to observed changes in affinity determined in radioligand binding assays (Tables2 and 3;Cox et al., 1992). Thus, the potency of dopamine was significantly decreased at S193A compared with that of D2S, and the potency of quinpirole was significantly decreased at S194A compared with that of D2S. NPA had decreased potency for inhibition of adenylate cyclase activity via S193A and S194A, although the decreases were not statistically significant due to higher variability in the responses.
Agonist potency for inhibition of isoproterenol-stimulated adenylate cyclase activity by D2S and serine mutant receptors
Agonist affinity for D2S and serine mutant receptors
Agonist-specific activation of Kir3 potassium channels expressed in oocytes.
The Kir3 potassium channels are directly stimulated by G protein βγ subunits to give fast, inwardly rectifying currents (Kofuji et al., 1995; Krapivinsky et al., 1995). To determine whether activation of signaling pathways in addition to inhibition of adenylate cyclase was altered by mutation of S194, we assessed the ability of D2S and S194A to activate Kir3 potassium channels in X. laevis oocytes expressing cRNAs coding for the receptors and Kir3.1 and Kir3.2 channel subunits. The application of high external K+ (96 mm) caused a rapid inward current (Dascal, 1987), which was potentiated by the addition of agonist (Fig.2A). Ba2+ applied with agonist blocked both the high K+- and agonist-activated currents, except for a minor component due to the Ba2+-insensitive endogenous K+ channel (Fig. 2A; Sharon et al., 1997). Control oocytes expressing Kir3.1/3.2 channels alone displayed large K+-activated currents but not dopamine-activated currents, and oocytes injected with receptors alone showed only small, agonist-insensitive endogenous K+ currents (data not shown).

Agonist-specific activation of Kir3 potassium channels by the D2S and S194A receptors. In vitro transcribed cRNAs coding for Kir3.1 and Kir3.2 channel subunits and D2S or S194A receptors were injected intoX. laevis oocytes, and peak current amplitudes were measured by two-electrode voltage-clamp at −80 mV. A, Representative trace showing Ba2+-sensitive (2 mm) currents activated by high K+ (96 mm) and quinpirole (300 nm) in an oocyte expressing S194A, Kir3.1, and Kir3.2. B–D, Dose-dependent current activation by dopamine, quinpirole, and 7-OH-DPAT, respectively. Values, mean ± standard error of three to nine experiments.
Dopamine produced a concentration-dependent activation of K+ currents with a calculated EC50 value of 39 nm in oocytes expressing the wild-type D2S receptor and Kir3.1/3.2 (Fig. 2B). In oocytes expressing S194A and Kir3.1/3.2, in contrast, small responses were observed only at 1 and 3 μm dopamine. Analysis of the dose -response curve for dopamine at S194A resulted in an EC50 value of 0.2 μm, although this value should be interpreted with caution because of the lack of any data points on the rising portion of the curve. Oocytes expressing D2S or S194A did not significantly differ in their responses to quinpirole (Fig. 2C; EC50 = 36 and 49 nm, respectively) or 7-OH-DPAT (Fig. 2D; EC50 = 87 and 178 nm, respectively).
Agonist binding to D2S and mutant receptors.
We measured agonist inhibition of the binding of [3H]spiperone in the absence of exogenous GTP to assess effects of the mutations on receptor/G protein coupling. Curves for inhibition of radioligand binding to the D2S receptor were fit best by assuming the presence of two classes of binding sites for all five agonists tested (Table 3). The high affinity state, indicative of the G protein-coupled receptor, comprised ∼27–37% of the binding sites for these agonists (Table 4). The addition of GTP eliminated the high affinity component (data not shown). Mutation of Ser197 did not have a significant effect on high or low affinity binding of the five agonists tested. Mutation of Ser193 or Ser194, however, significantly decreased the affinity of the receptor for several agonists (Table 3). In particular, both mutant receptors had decreased affinity for the catecholamine agonists dopamine, NPA, and DHX. The decrease in affinity was greatest for the high affinity (KH ) component of binding, which was completely abolished for DHX at S193A or S194A. Effects of each mutation on the affinity of noncatecholamine agonists were lesser and did not reach statistical significance, with the exception of a significant decrease in the low affinity (KL ) site for quinpirole at S194A. The proportion of sites in the high affinity state for most agonists was not altered (Table 4), except that the high affinity state was not detectable for DHX binding to S193A and S194A in any experiments or for dopamine binding to S194A in three of seven experiments.
Proportion of binding sites in the high affinity state (K H) for wild-type and mutant D2S receptors
Agonist-stimulated binding of [35S]GTPγS.
To confirm the apparent deficit in G protein coupling of S193A and S194A receptors suggested by the decreased affinity of the G protein state for some agonists, we assessed the ability of agonists to modulate G protein activation by quantifying receptor-stimulated binding of [35S]GTPγS. In membranes prepared from C6-D2S cells, dopamine dose-dependently increased [35S]GTPγS binding from mean basal levels of 39 fmol/mg of membrane protein to maximal levels of 76 fmol/mg with an average EC50 value of 355 nm(250–500 nm, three experiments). Dopamine-stimulated [35S]GTPγS binding via S194A was significantly decreased compared with wild-type, S193A, or S197A receptors (Fig. 3). In contrast, 7-OH-DPAT and quinpirole stimulated [35S]GTPγS binding to a similar extent via each of the mutant receptors.

Agonist-stimulated binding of [35S]GTPγS to membranes prepared from C6 glioma cells expressing D2S or mutant receptors. The data for each mutant receptor were expressed as the percentage of basal [35S]GTPγS binding for that cell line and then normalized to the binding stimulated by a given drug at the wild-type D2 receptor, which was 76 fmol/mg of protein for 1 mm dopamine (DA), 57 fmol/mg for 1 mm quinpirole, and 60 fmol/mg for 100 μm7-OH-DPAT. The mean ± standard error is shown for three or four independent experiments. ∗, p < 0.05 compared with D2S (Dunnett’s post hocrepeated-measures analysis of variance).
[35S]GTPγS binding stimulated by dopamine (1 mm) was abolished in C6-D2S cells previously treated with PTX (200 ng/ml overnight), indicating that the response was due to activation of PTX-sensitive G protein α subunits (Fig. 4). PTX treatment also eliminated dopamine-stimulated [35S]GTPγS binding to membranes prepared from C6-S197A cells and significantly decreased dopamine-stimulated binding to C6-S193A membranes. In contrast, the small amount of [35S]GTPγS binding stimulated by dopamine in C6-S194A membranes was completely insensitive to PTX.
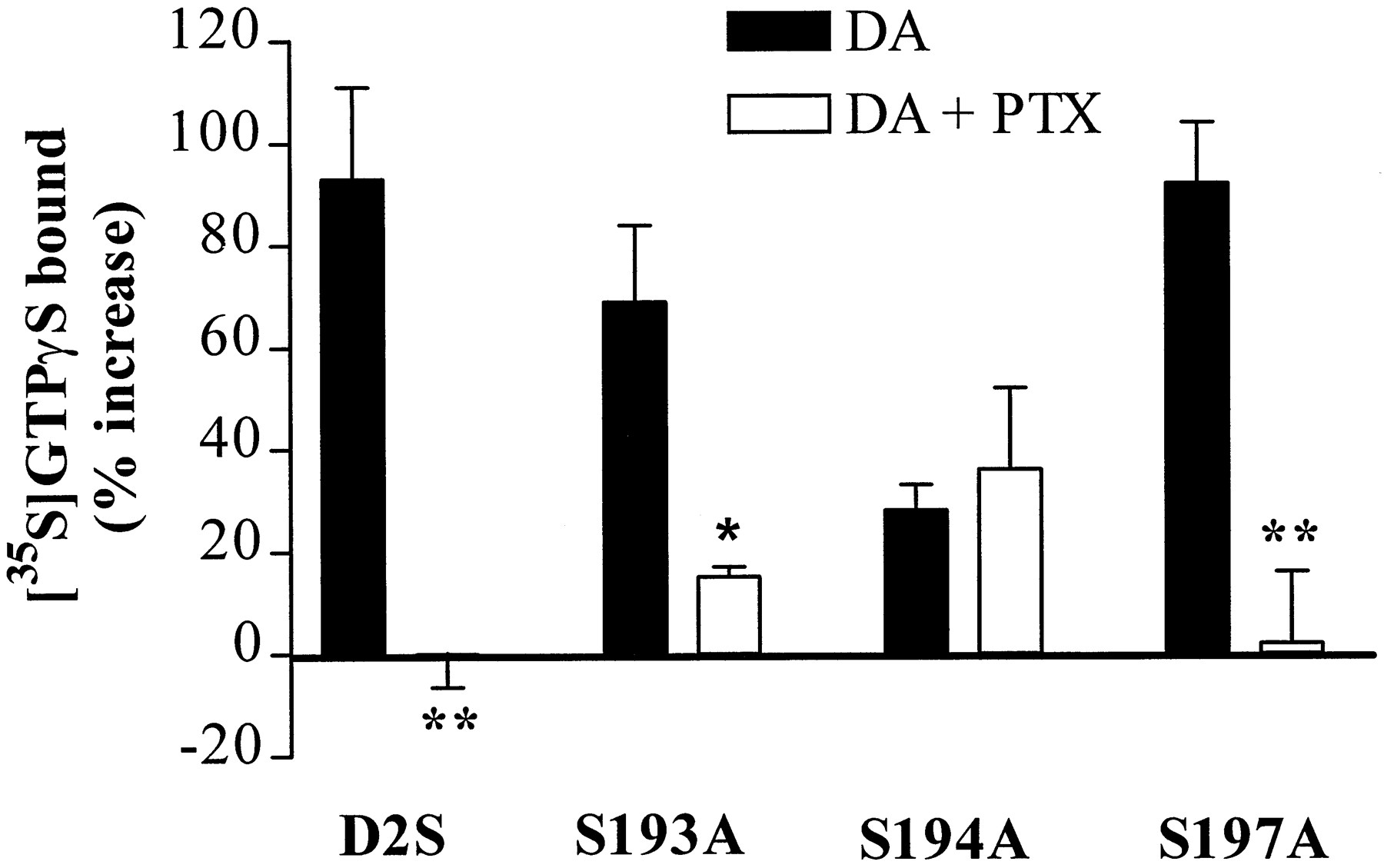
PTX (200 ng/ml) inhibition of dopamine (1 mm)-stimulated [35S]GTPγS binding to membranes expressing D2S or mutant receptors. The data are expressed as the percent increase over binding in the absence of dopamine (DA) for each receptor (D2S, 34 ± 3 fmol/mg; S193A, 40 ± 5 fmol/mg; S194A, 41 ± 2 fmol/mg; S197A, 32 ± 2 fmol/mg). Bars, mean ± standard error of three to six independent experiments, carried out in triplicate. ∗, p < 0.05, ∗∗,p < 0.01 compared with binding stimulated by dopamine in cells not treated with PTX (Student’s ttest).
Inverse agonist and PTX effects on isoproterenol-stimulated cAMP accumulation.
The antagonists butaclamol, clozapine, epidepride, haloperidol, and spiperone acted as inverse agonists at the D2S receptor in that they all significantly potentiated isoproterenol-stimulated cAMP accumulation, presumably due to inhibition of the spontaneous activity of the D2 receptor (Fig.5A). To determine whether the reduced ability of some agonists to activate S194A was accompanied by a decrease in the constitutive activity of the mutant receptor, we compared the responses of the wild-type D2S and serine mutant receptors to inverse agonists. Inverse agonists potentiated isoproterenol-stimulated cAMP accumulation in C6-S197A cells to a similar level (50–100% above control) as in C6-D2S cells (Fig. 5D), but they did not potentiate cAMP accumulation in untransfected C6 cells (data not shown) or in C6-S193A or C6-S194A cells (Fig. 5, B and C).
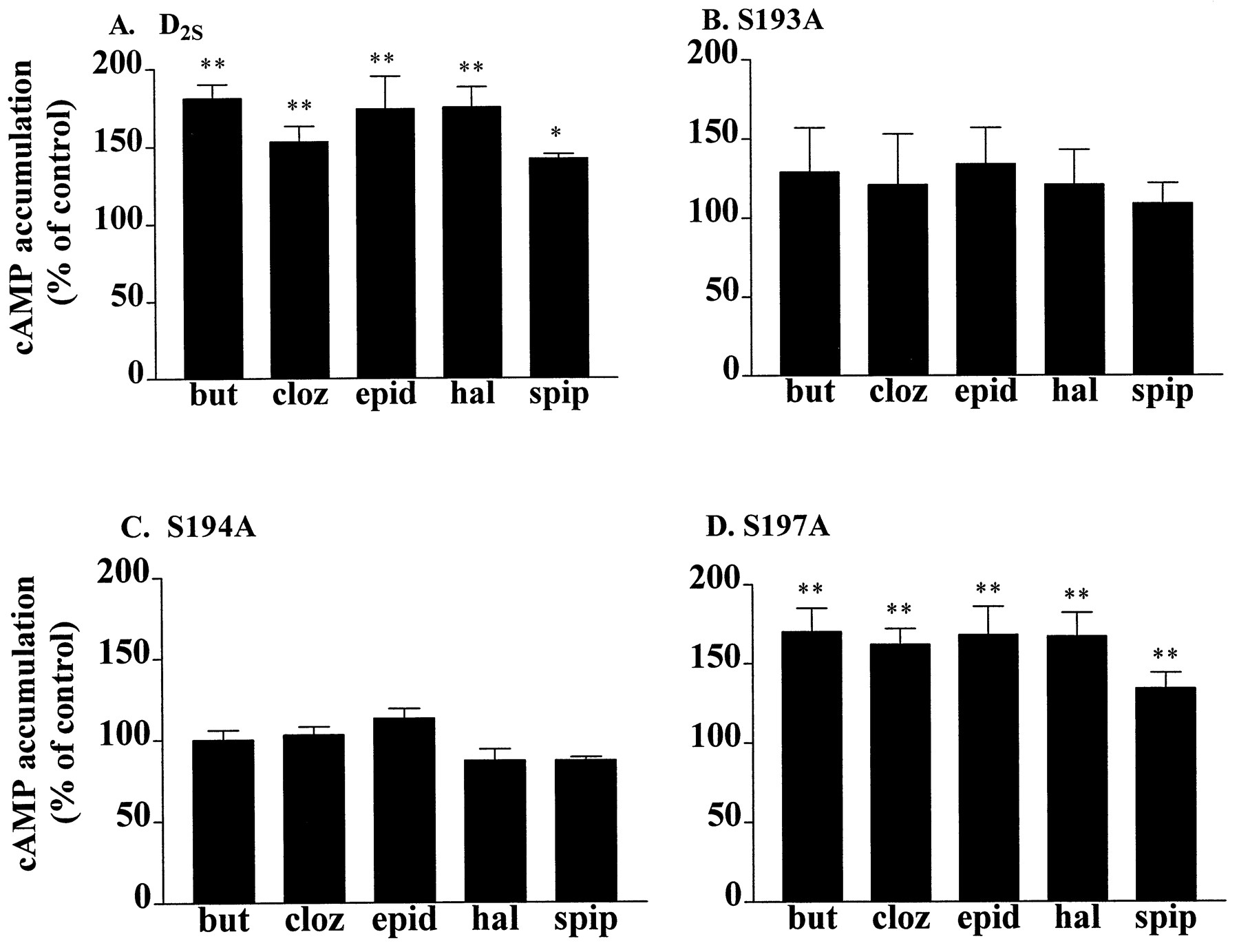
Inverse agonist potentiation of isoproterenol-stimulated cAMP accumulation in C6 cells expressing (A) D2S, (B) S193A, (C) S194A, or (D) S197A receptors. Inverse agonists (10 μm) were incubated with the cells for 10 min at 37° before a 15-min incubation with isoproterenol (1 μm). Data are expressed as the percentage of isoproterenol-stimulated cAMP accumulation in each cell line in the absence of antagonist (D2S, 134 ± 17 pmol/well; S193A, 35 ± 7 pmol/well; S194A, 89 ± 10 pmol/well; S197A, 86 ± 12 pmol/well). Bars, mean ± standard error of three to six independent experiments, each carried out in triplicate. but, butaclamol; cloz, clozapine; epid, epidepride; hal, haloperidol; spip, spiperone. ∗, p< 0.05, ∗∗, p < 0.01 compared with cAMP accumulation in the presence of isoproterenol alone (Dunnett’spost hoc repeated-measures analysis of variance).
The butaclamol-induced potentiation of isoproterenol-stimulated cAMP production via the D2S receptor was concentration dependent (Fig. 6), with an EC50 value of 65 nm (standard error, 26–159 nm, four experiments), producing an average increase of 97 ± 19%. Butaclamol caused a similar response in C6-S197A cells, increasing cAMP accumulation by 67 ± 20% with an average EC50 value of 41 nm (standard error, 14–117 nm; four experiments). Isoproterenol-stimulated cAMP accumulation was not enhanced in C6-S193A or C6-S194A cells at any concentration of butaclamol tested, demonstrating a lack of inverse agonist activity at these mutant receptors.

Concentration-dependent potentiation of isoproterenol-stimulated cAMP accumulation by butaclamol. Data are from a single experiment representative of four or five independent experiments, each carried out in triplicate, and are expressed as the percentage of isoproterenol-stimulated cAMP accumulation in the absence of butaclamol (D2S, 142 pmol/well; S193A, 15 ± 9 pmol/well; S194A, 65 ± 20 pmol/well; S197A, 73 ± 15 pmol/well). No curve is shown for the S193A and S194A mutants because there was no detectable potentiation by butaclamol at these receptors.
The potentiation of isoproterenol-stimulated cAMP accumulation by inverse agonists at D2S and S197A receptors was prevented by pretreatment with PTX (data not shown). In addition, PTX treatment revealed the constitutive activity of these receptors in that the toxin increased isoproterenol-stimulated cAMP levels in C6-D2S or C6-S197A cells by 50–100% (Fig.7). In contrast, PTX treatment had no effect on cAMP accumulation in untransfected C6 cells or in C6-S193A and C6-S194A cells.

Effects of PTX on isoproterenol-stimulated cAMP accumulation in untransfected C6 cells and cells expressing D2S or mutant receptors. Intact cells were treated with PTX (25 ng/ml) for 18–24 hr before incubation for 15 min at 37° with 1 μm isoproterenol. Data are expressed as the percentage of isoproterenol-stimulated cAMP accumulation in cells not treated with PTX (C6, 232 ± 48 pmol/well; D2S, 128 ± 11 pmol/well; S193A, 23 ± 6 pmol/well; S194A, 84 ± 14 pmol/well; S197A, 61 ± 21 pmol/well) and are the mean ± standard error of four to six independent experiments. ∗,p < 0.05 compared with cells not treated with PTX (Student’s t test).
Inverse agonist-inhibited binding of [35S]GTPγS.
To confirm the lack of constitutive activity observed for the inhibition of cAMP accumulation by S193A and S194A, we measured [35S]GTPγS binding in the presence of the inverse agonist butaclamol. To maximize basal [35S]GTPγS binding, assays were carried out in the absence of GDP and NaCl. Butaclamol (100 μm) reduced basal [35S]GTPγS binding from 90 to 65 fmol/mg in membranes prepared from C6-D2Scells and from 75 to 55 fmol/mg in membranes from C6-S197A cells (Fig.8), and this reduction was blocked by overnight incubation of the cells with 200 ng/ml PTX (data not shown). Basal [35S]GTPγS binding in untransfected C6 cells (100 fmol/mg) or in C6-S193A (91 fmol/mg) and C6-S194A (99 fmol/mg) cells was not affected by butaclamol, again indicating a lack of inverse agonism (Fig. 8).

Butaclamol inhibition of [35S]GTPγS binding to membranes expressing D2S or mutant receptors. The data are expressed as the percent change from the binding observed in the absence of butaclamol for each receptor. Bars, mean ± standard error of three to six independent experiments, carried out in triplicate. ∗∗, p < 0.01 compared with binding in the absence of butaclamol (Dunnett’spost hoc repeated-measures analysis of variance).
Discussion
Serine residues in TM5 of catecholamine receptors contribute to both binding of agonists and receptor activation (Strader et al., 1989; Wang et al., 1991; Mansour et al., 1992; Pollock et al., 1992; Hwa and Perez, 1996;Woodward et al., 1996). Our prior characterization of mutant D2S receptors, in which each of the serine residues in TM5 was converted to an alanine, demonstrated the unique result that the S194A mutant displays an agonist-dependent loss of inhibition of adenylate cyclase (Cox et al., 1992). Here, we confirmed that dopamine does not inhibit cAMP accumulation via S194A, although the efficacy of several other agonists (NPA, 7-OH-DPAT, and quinpirole) at S194A was similar to that of the wild-type D2S receptor. We also demonstrated that mutation of Ser194 selectively reduced the efficacy of dopamine for activation of Kir3 channels and stimulation of [35S]GTPγS binding to PTX-sensitive G proteins. Furthermore, mutation of either Ser194 or Ser193 abolished constitutive activity of the D2S receptor and inhibition of cAMP accumulation by the agonist DHX, suggesting that Ser193 also may contribute to efficacy in an agonist-dependent manner. Our data are consistent with models of G protein-coupled receptor activation in which the assumption of two states (active and inactive) is expanded to include two or more active conformations, with a given agonist selecting among the possible conformations based on its structure (Gardner, 1995; Kenakin, 1996; Perez et al., 1996;Krumins and Barber, 1997; Leff et al., 1997).
Agonist-selective loss of efficacy for cAMP accumulation.
The loss of efficacy for inhibition of cAMP accumulation by dopamine via S194A occurred despite only modest changes in the affinity of the mutant for dopamine, determined in radioligand binding assays in the presence of GTP. Because an antagonist is a drug with binding affinity but no efficacy (Kenakin, 1996), we determined that dopamine acted as an antagonist at S194A, shifting the dose-response curve for 7-OH-DPAT rightward in an apparently competitive manner. In addition to dopamine and para-tyramine (Cox et al., 1992), the current results show that DHX was unable to inhibit cAMP accumulation via S194A. The mutation-induced loss of efficacy does not seem to be related to the efficacy of the agonists at the wild-type receptor because both full (dopamine) and partial (para-tyramine) agonists lost efficacy at S194A, whereas other full (quinpirole) and partial (meta-tyramine) agonists retained efficacy at the mutant receptor (current results and Cox et al., 1992).
Agonist-selective loss of efficacy for activation of Kir3 channels.
Alterations in signaling by the S194A mutant were not confined to one signaling pathway. Many G protein-coupled receptors, including D2 dopamine receptors, activate inwardly rectifying potassium channels (GIRK or Kir3) viathe release of G protein βγ subunits (Einhorn and Oxford, 1993;Wickman et al., 1994; Werner et al., 1996). Here, we used an oocyte expression system to show that the ability of dopamine to activate the Kir3.1/Kir3.2 heteromultimer was dramatically reduced by mutation of Ser194, whereas quinpirole and 7-OH-DPAT were as active at S194A as at the wild-type D2S receptor. Thus, the pattern of agonist-dependent efficacy via S194A was similar for both G protein α and βγ subunit-regulated pathways.
Relationship between changes in efficacy and high affinity binding of agonists.
We observed decreases in the high affinity binding of some agonists to S193A and S194A similar to those observed for TM5 serine mutants in studies of the human and rat D2L dopamine receptors (Mansour et al., 1992; Woodward et al., 1996). The percentage of high affinity binding sites, which is thought to represent the G protein-coupled state of the receptor, was not affected by the serine mutations and constituted approximately one third of the total number of binding sites for all experiments in which two classes of binding sites were detectable. The ratioKL /KH , on the other hand, was changed by the serine mutations in an agonist-selective manner, being greatly decreased as a result of larger mutation-induced reductions in high affinity compared with low affinity binding for dopamine at S194A, NPA at S193A and S194A, quinpirole at S193A, and S197A, and 7-OH-DPAT at S194A (Table 3). These results are consistent with the prediction thatKL /KH correlates with agonist efficacy (Wreggett and De Léan, 1984;Lahti et al., 1992; Harley et al., 1995) insofar as both the greatest decreases inKL /KH and the loss of efficacy for inhibition of cAMP accumulation were observed for dopamine at S194A and DHX at S193A and S194A. Still, it is surprising that substantial reductions inKL /KH were observed, such as the decrease for NPA from a ratio of 1000 at D2S to 100 at S194A, without any detectable effect on the efficacy of the agonist.
Agonist-selective loss of efficacy for activation of G proteins.
G protein-coupled receptors stimulate the binding of [35S]GTPγS to G proteins by accelerating the dissociation of bound GDP. Consistent with the selective loss of efficacy by dopamine at S194A, the ability of dopamine to stimulate the binding of [35S]GTPγS was significantly decreased via S194A while maximal stimulation of binding by 7-OH-DPAT and quinpirole was not significantly decreased. Although there was a tendency for the binding stimulated by the latter two drugs to be decreased at S194A, the effect of the mutation on the efficacy of dopamine was qualitatively different because the residual dopamine-stimulated binding of [35S]GTPγS via S194A was completely insensitive to treatment with PTX. It is possible that mutation of Ser194 decreases dopamine-induced coupling to PTX-sensitive G proteins while concurrently increasing coupling to a PTX-insensitive G protein. Dissociation of βγ subunits from this G protein may contribute to the small dopamine-activated potassium current mediated by S194A.
Constitutive activity of the D2S receptor.
Receptor activation by an agonist is hypothesized to involve induction or selection of an active receptor conformation because of the higher affinity of the active conformation for the agonist (Kenakin, 1996). A mutation-induced loss of agonist efficacy could be due to a failure of the active conformation to provide a high affinity binding site for the agonist, perhaps because of the loss of an interaction between the agonist and a residue that is specifically involved in high affinity binding. Thus, the agonist-selective loss of efficacy that we observed would be a special example of the frequent observation that the binding of a given drug to a receptor is due to interactions with a set of binding determinants that is unique to the drug, although the set may overlap substantially with determinants for other drugs (Marulloet al., 1990; Cox et al., 1992). Alternatively, a mutation-induced loss of efficacy could be due to a decrease in the ability of the mutant receptor to adopt an active conformation. In this case, our finding of a selective loss of efficacy for some agonists at S194A, S193A, or both would support the hypothesis that a receptor can assume multiple active conformations and that the chemical structure of the agonist determines which form predominates (Gardner, 1995; Kenakin, 1995, 1996; Krumins and Barber, 1997).
To determine whether the loss of efficacy was due to a loss of determinants for high affinity binding or to a decrease in the ability of the receptor to adopt an active conformation, we characterized the constitutive activity of serine mutant receptors as an indirect means for assessing changes in receptor conformation. As described by the extended allosteric ternary complex model of G protein-coupled receptor activation (Samama et al., 1993), receptors spontaneously isomerize between active and inactive conformations, so a receptor can modulate signaling pathways in the absence of agonist. Significant constitutive activity of recombinant D2 receptors expressed in mammalian cells has been described previously (Nilssonet al., 1996; Hall and Strange, 1997; Kozell and Neve, 1997). We confirmed that the antagonists butaclamol, clozapine, epidepride, haloperidol, and spiperone potentiate isoproterenol-stimulated cAMP via D2S and via S197A. The inverse agonist activity of butaclamol was concentration dependent and also could be detected as inhibition of basal [35S]GTPγS binding. Furthermore, the response to all of the antagonists was specific to the D2Sreceptor and the Gi/Gofamily of G proteins in that it did not occur in untransfected C6 cells and was blocked by PTX. None of the antagonists tested potentiated cAMP responses to isoproterenol in C6-S193A and C6-S194A cells. To verify that the loss of the ability to detect inverse agonism via S193A and S194A was the result of mutation-induced decreases in the constitutive activity of the receptors rather than an effect of the mutations on the inverse agonism of the drugs, we determined that treatment with PTX enhanced isoproterenol-stimulated cAMP accumulation in C6-D2S and C6-S197A cells but not in C6-S193A or C6-S194A cells. The demonstration that S193A and S194A were not constitutively active supports the hypothesis that the inability of these mutants to respond to certain agonists of the wild-type D2S receptor reflected their decreased ability to adopt an active conformation, in agreement with results of experiments involving the α1A-adrenergic receptor (Hwa and Perez, 1996).
Conclusions.
The ability of D2S to adopt a conformation capable of constitutive activity was dependent on the presence of both Ser193 and Ser194, as was inhibition of adenylate cyclase activity by DHX. Dopamine-stimulated activation of PTX-sensitive G proteins, inhibition of adenylate cyclase activity, and activation of Kir3 channels, however, required the presence of Ser194 but not of Ser193. For a number of other agonists, including NPA, quinpirole, and 7-OH-DPAT, the lack of any one of the serine residues had no discernible influence on signal transduction. Thus, just as a receptor adopts different conformations in response to full and partial agonists (Gether et al., 1995; Krumins and Barber, 1997), it also distinguishes among full agonists. Although there are considerable data to suggest that agonists select among distinct receptor conformations to activate multiple signaling pathways (Eason et al., 1994; Gurwitz et al., 1994; Robb et al., 1994; Kenakin, 1995; Perez et al., 1996; Realeet al., 1997), the current results demonstrate that there are multiple active conformations for the regulation of one signaling pathway, inhibition of cAMP accumulation. Two possible explanations for our results are that multiple conformations of D2S activate a single subtype of G protein and that distinct active conformations couple to distinct G protein subtypes that converge on one signaling pathway (Gerhardt and Neubig, 1994).
Acknowledgments
We are grateful to Drs. Val Watts, Greg Wiens, and Aaron Janowsky for careful reading of the manuscript.
Footnotes
- Received January 14, 1998.
- Accepted May 8, 1998.
-
Send reprint requests to: Dr. Brenda L. Wiens, Research Service (R & D-30), Veterans Affairs Medical Center, 3710 S.W. U.S. Veterans Hospital Road, Portland, OR 97201. E-mail:wiensb{at}ohsu.edu
-
This work was supported by the Veterans Affairs Merit Review and Career Scientist Programs and by United States Public Health Service Grant T32-DA07262.
Abbreviations
- TM5
- fifth-transmembrane domain
- 7-OH-DPAT
- 7-hydroxy-2-dipropylaminotetralin
- GTPγS
- guanosine-5′-O-(3-thio)triphosphate
- C6-D2S
- C6-S193A, C6-S194A, or C6-S197A, C6 glioma cells expressing the D2S, S193A, S194A, or S197A receptor, respectively
- DHX
- dihydrexidine
- NPA
- N-n-propylnorapomorphine
- PTX
- pertussis toxin
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}