


Abstract
Signaling of G protein-coupled receptors is terminated by phosphorylation of intracellular serine and threonine residues. Resensitization of these receptors requires internalization and subsequent dephosphorylation. We have recently shown that the resensitization rate of the rat μ opioid receptor (MOR) isoforms MOR1 and MOR1B is mainly determined by the amino acid composition of their alternatively spliced C-terminal tails. Upon agonist stimulation, MOR1B passes through an accelerated cycle of receptor endocytosis and reactivation, which in turn promotes a greater resistance to agonist-induced desensitization, as compared with MOR1. Given the fact that MOR1B lacks only one putative phosphorylation site (T394 of MOR1), we replaced this threonine by an alanine and stably expressed the wild-type MOR1 and its T394A mutant in mouse neuroblastoma Neuro2a cells. We show that during prolonged [d-Ala2, MePhe4, Gly5-ol]enkephalin exposure (5 h), the T394A receptor mutant desensitized at a slower rate than MOR1. In contrast, T394A is more rapidly removed from the cell surface than MOR1, as determined by flow cytometry using epitope-tagged receptors. This fast internalization was followed by immediate resensitization of T394A during 20 min of agonist removal while the wild-type MOR1 remained inactive. Similar to MOR1B, rapid internalization and reactivation of T394A may explain its delayed desensitization. These findings suggest that T394 represents a negative regulatory signal for MOR1 internalization. Furthermore, phosphorylation of this threonine residue may influence the time course of μ opioid receptor resensitization.
It is well established that prolonged exposure to agonists reduces responsiveness of G protein-coupled receptors to a second stimulus (Lohse, 1993; Chakrabarti et al., 1995; Freedman and Lefkowitz, 1996). This phenomenon, termed receptor desensitization, is mostly initiated by kinase-catalyzed phosphorylation of serine and/or threonine residues within the third intracellular loop and the C terminus. The phosphorylated receptor is then internalized via clathrin-coated pits and vesicles into early endosomes. Within the acidic environment of the endosomes, the ligand is effectively separated from the receptor, followed by dephosphorylation and recycling of the receptor protein to the cell surface.
For the μ opioid receptor, several agonist-independent second messenger kinases, such a Ca++ calmodulin-dependent protein kinase and protein kinases A and C, but also agonist-dependent G protein receptor kinases (GRKs) have been implicated in receptor desensitization (Mestek et al., 1995; Pak et al., 1997; Chakrabarti et al., 1998). In addition, the serine residues S261/S266 within the third intracellular loop, which are putative Ca++/calmodulin-dependent protein kinase II phosphorylation sites, have been shown to be involved in agonist-induced desensitization of the μ opioid receptor (Koch et al., 1997a).
We have previously shown that the μ opioid receptor is alternatively spliced into two C-terminal isoforms, rat μ opioid receptor (MOR)1 and MOR1B (Zimprich et al., 1995). Both variants differ from amino acid 387 to the C terminus (MOR1,387LENLEAETAPLP398; MOR1B,387KIDLF391). MOR1B, which lacks only one putative phosphorylation site, T394, is more resistant to agonist-induced desensitization than MOR1 (Zimprich et al., 1995;Koch et al., 1998). Interestingly, site-directed mutagenesis of T394 to alanine has been shown to result in a slow desensitizing MOR1 mutant (Koch et al., 1997b; Pak et al., 1997). However, despite its slow desensitization, MOR1B is internalized faster than MOR1. This fast internalization permits a rapid recycling and resensitization of the MOR1B receptor (Koch et al., 1998).
In the present study, we investigated the role of T394 in the internalization and resensitization process of the μ opioid receptor. We provide evidence that the slow desensitizing T394A receptor mutant is subject to rapid endocytosis and reactivation.
Materials and Methods
Epitope Tagging and In Vitro Mutagenesis of the μ Opioid Receptor.
The wild-type μ opioid receptor was tagged at the N terminal tail with the epitope tag sequence MASMTGGQQMGKL using the polymerase chain reaction. Amino acids KL were coded by nucleotide sequence AAGCTT representing a HindIII cloning site. The MOR1 cDNA subcloned into the pRc/CMV expression vector was kindly provided by Dr. Lei Yu (Indianapolis, IN). The HindIII restriction site upstream of the ATG start codon as well as the point mutation of T394 to alanine were generated by polymerase chain reaction mutagenesis using MOR1/HindIII-primer 5′-TCCGCAGCAAAGCTTCACACCATG-3′ and 1A-T394A mutagenesis primer 5′-TTAGGGCAATGGAGCAGCTTCTGC-3′(MWG-Biotech, Ebersberg, Germany). Mutagenesis primer MOR1/HindIII corresponds to 24 nucleotides from −21 up to base position 3, whereas mutagenesis primer 1A-T394A corresponds to 24 nucleotides 1174 to 1197 of the coding sequence of MOR1. Mismatch at position 1180 led to the replacement of threonine by alanine at amino acid position 394. Mutation was confirmed by double-strand DNA sequencing.
Generation of Cell Lines Expressing Rat μ Opioid Receptors and Drug Treatments.
Transfection of mouse neuroblastoma cell line (Neuro2a) cells was performed by the calcium phosphate precipitation method as described by Chen and Okayama (1988). Approximately 1.5 × 106 cells were transfected with 20 μg of plasmid DNA. Cells were selected in the presence of 500 μg/ml G418 (Gibco/BRL, Eggenstein, Germany), and the whole pool of resistant cells was grown in the presence of 400 μg/ml G418 without selection of individual clones.
Measurements of cAMP Levels.
One and one-half × 105 cells were seeded in 22-mm 12-well dishes with Dulbecco′s modified Eagle’s medium Nut-F12 medium containing 10% fetal calf serum. On the day of assay, media were removed from individual wells and replaced by 0.5 ml of serum-free medium containing 20 μM 3-isobutyl-1-methylxanthine and 25 μM forskolin or a mixture of 25 μM forskolin, 20 μM 3-isobutyl-1-methylxanthine, and 1 μM [d-Ala2, Me Phe4, Glyol5]enkephalin (DAMGO). The cells were then incubated at 37°C for 15 min. The reaction was terminated by removing the medium and sonicating the cells in 1 ml of ice-cold HCl/EtOH (1 volume of 1 N HCl/100 volume of EtOH). After centrifugation, the supernatant was evaporated, the residue was dissolved in Tris-EDTA buffer, and the cAMP content was measured using a commercial radioimmunoassay (Amersham, Braunschweig, Germany).
Radioligand-Binding Assay.
Binding studies were performed on membranes prepared from cells using a modified method described bySimantov (1989). All assays were carried out at least in triplicate. Specific binding was calculated by subtracting nonspecific binding, defined as that seen in the presence of 2.5 nM [3H]DAMGO plus 1 μM unlabeled DAMGO, from total binding obtained with 2.5 nM [3H]DAMGO alone. Data were calculated as femtomole-bound radioligand per milligram of protein.
Confocal Microscopy.
Neuro2a cells stably expressing either MOR1 or T394A receptor mutant were grown on poly-l-lysine-treated coverslips overnight. Cells were then exposed to 1 μM DAMGO for 0, 10, 30, or 50 min. Cells were fixed with 4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer, pH 6.9, for 40 min at room temperature and subsequently washed several times in 10 mM Tris, 10 mM phosphate buffer, 137 mM NaCl, and 0.05% thimerosal, pH 7.4 (TPBS). Cells were then incubated for 3 min in 50% methanol and for 3 min in 100% methanol and subsequently washed several times in TPBS. After 1-h preincubation in TPBS containing 3% normal goat serum (NGS), cells were incubated with mouse monoclonal anti-T7-tag antibody (Novagen, Madison, WI) at a dilution of 1:1000 in TPBS containing 1% NGS overnight at room temperature. This antibody was generated against the sequence MASMTGGQQMGKL. Bound primary antibody was detected using biotinylated goat anti-mouse IgG (1:200; Vector, Burlingame, CA) followed by streptavidine-cyanine 3.18 (1:400; Amersham). Cells were then dehydrated, cleared in xylol, and permanently mounted in DPX (Fluka, Neu-Ulm, Germany). Specimens were examined using a Leica TCS-NT laser scanning confocal microscope. Cyanine 3.18 was imaged with 568-nm excitation and 570- to 630-nm bandpass emission filters. Confocal micrographs were taken by a person blinded to the treatments who was instructed to randomly select one colony of 4 to 12 cells per coverslip.
Flow Cytometry.
For quantitative analysis of agonist-induced receptor internalization, 106 cells were plated into 6-well plates. After 24 h, the cells were treated with Dulbecco’s modified Eagle’s medium alone or with 1 μM DAMGO for various times at 37°C. Cells were then immediately chilled to 4°C, washed with PBS, and incubated with 0.5 μg/ml anti-T7-tag antibody in TPBS containing 3% NGS for 1 h at 4°C. The cells were washed twice with TPBS and incubated with 2.5 μg/ml phycoerythrin-labeled goat anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA) for 1 h at 4°C. After washing twice with TPBS, cells were fixed with 1% paraformaldehyde and then collected from the wells with 5 mM EDTA and analyzed on a FACScan flow cytometer (Becton Dickinson Immunocytometry Systems, San Jose, CA). Five thousand cells were acquired for each time point. Mean fluorescence of all cells minus mean fluorescence of cells stained only with phycoerythrin-conjugated secondary antibody was used for calculation.
Results
Expression of μ Opioid Receptors in Neuro2a Cells.
Neuro2a cells were stably transfected with cDNAs encoding rat μ opioid receptors MOR1 or the T394A mutant. Both receptor types revealed similar specific binding of [3H]DAMGO. Displacement-binding experiments of radioactive DAMGO indicated that MOR1 and T394A mutant did not differ significantly with respect to their binding affinity (IC50 = 8–10 nM). Surface expression of MOR1 (413 ± 70 fmol/mg protein) in Neuro2a cells was comparable to that of T394A (522 ± 64 fmol/mg protein) as measured in the ligand-binding assay. Moreover, no difference was observed between the individual receptor subtypes with regard to their abilities to inhibit adenylate cyclase activity in Neuro2a cells. The reduction of forskolin-stimulated cAMP levels by 1 μM DAMGO was 56% ±7 and 53% ±5 for MOR1 and T394A, respectively (mean ± S.E.M.,n = 4–6).
Agonist-Induced Desensitization of μ Opioid Receptors Expressed in Neuro2a Cells.
Desensitization was measured as the decreasing ability of the agonist DAMGO to inhibit forskolin-stimulated adenylate cyclase after agonist pretreatment. Therefore, receptor-expressing Neuro2a cells were pretreated with 1 μM DAMGO for various time intervals, followed by the determination of the DAMGO-induced inhibition of forskolin-stimulated adenylate cyclase. Forskolin treatment resulted in a 5- to 8-fold increase of cAMP levels (up to 4 pmol) as compared with untreated Neuro2a cells. DAMGO inhibited forskolin-stimulated cAMP accumulation by about 50% in Neuro2a cells expressing either MOR1 or T394A mutant. In Neuro2a cells lacking opioid receptors, DAMGO did not inhibit the forskolin-stimulated increase in cAMP levels (data not shown). The time course of agonist-dependent loss of receptor activity is illustrated in Fig.1. For each receptor type, maximum agonist-induced inhibition of cAMP accumulation (without DAMGO preincubation) has been defined as 100%. After 1 h of DAMGO administration, the maximum DAMGO-induced cAMP inhibition of MOR1-expressing cells dropped to nearly 30%, whereas T394A mutant showed only a decrease of maximum cAMP inhibition to approximately 90%. After 5 h of DAMGO preincubation, both receptor types were completely desensitized. These data demonstrated that MOR1-expressing cells desensitize faster than those expressing the T394A mutant receptor.

Time course of agonist-induced desensitization of MOR1 wild-type and the T394A receptor mutant. Transfected Neuro2a cells were preincubated at 37°C with 1 μM DAMGO for the indicated time intervals. After removal of the preincubation medium, cells were treated with forskolin (25 μM) or forskolin (25 μM) plus DAMGO (1 μM) and cAMP levels were determined as described above (seeMaterials and Methods). Values represent means ± S.E.M. from six separate measurements. Asterisk indicates significant difference (p <.05) between MOR1 and T394A as determined using analysis of variance followed by Bonferroni test.
Agonist-Induced Loss of Receptors from Cell Surface.
Flow cytometry was used to quantify the internalization of epitope-tagged MORs in intact, nonpermeabilized cells detected by immunostaining with T7-tag antibody. DAMGO triggered a rapid loss of opioid receptors from the plasma membrane during 50 min of drug treatment (Fig.2). Replacement of T394 by alanine led to a facilitated receptor internalization. After 2 min of agonist treatment, only 15% of MOR1 receptor but nearly 30% of the T394A receptor were internalized. After 50 min of agonist treatment, 40% of MOR1 and 60% of T394A receptor mutant were internalized.

Flow cytometric analysis of MOR1 and T394A internalization. Neuro2a cells expressing each receptor type were treated for the times shown with 1 μM DAMGO. After drug treatment, cells were chilled to 0°C to arrest further trafficking and stained with anti-T7-tag antibody. The cells were then analyzed on a FACScan flow cytometer. Values are the mean ± S.E. of four experiments.
Direct Observation of Agonist-Induced Endocytosis of μ Opioid Receptors.
To test whether the T394A mutation of the μ opioid receptor has an effect on agonist-induced receptor endocytosis, MOR1- and T394A-expressing Neuro2a cells were exposed to 1 μM DAMGO for 0, 10, 30, and 50 min. These cells were subsequently fixed, permeabilized, and fluorescently labeled with anti-T7-tag-antibodies. The subcellular distribution of the receptor proteins was then analyzed by confocal microscopy. The results, depicted in Fig.3, show that in the absence of DAMGO, both receptor types were mostly present at the level of the plasma membrane and to a lesser extent in the cytoplasm. Although agonist-induced internalization of MOR1 was complete not before 50 min, in most cells internalization of T394A was already complete after 30 min. The results clearly show that T394 internalization proceeds at a faster rate than that of MOR1.

Comparison of agonist-induced endocytosis of MOR1 and T394A mutant in Neuro2a cells. MOR1 (top) and T394A receptor mutant- (bottom) expressing Neuro2a cells were exposed to 1 μM DAMGO for the indicated time intervals. Cells were subsequently fixed, fluorescently labeled with anti-T7-tag antibody, and examined by confocal microscopy. Shown are representative results from one of three independent experiments performed in duplicate.
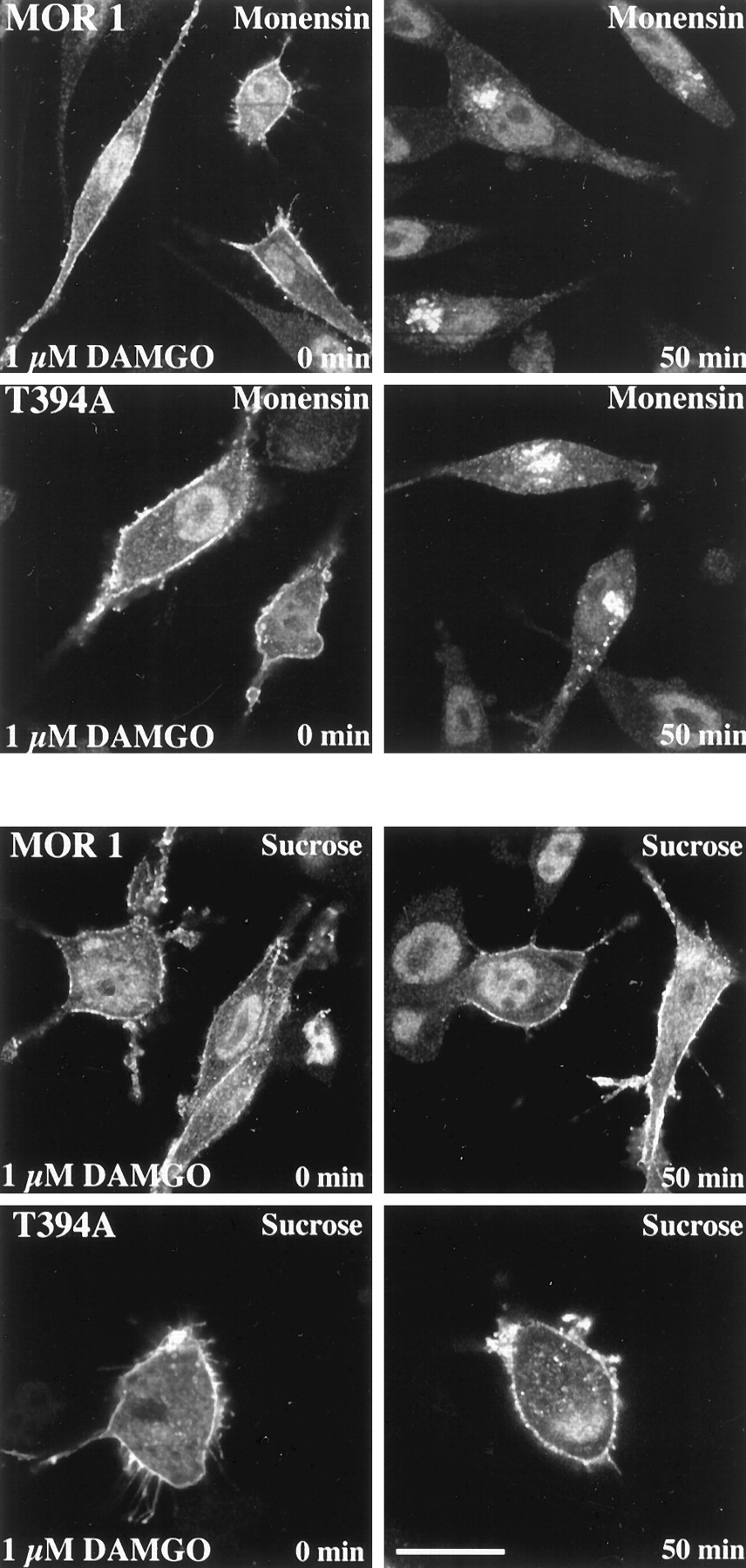
To test the possibility that T394A mutant is constitutively internalized, we compared the internalization extent of MOR1 and T394A mutant in the presence of monensin, which blocks receptor recycling to the cell membrane. Figure 4 (top) shows that in the absence of agonist, neither MOR1 nor T394A receptor mutant is internalized, indicating that T394A mutant does not undergo constitutive internalization.

Comparison of agonist-induced endocytosis of MOR1 and T394A mutant in Neuro2a cells in the presence of recycling blocker monensin (top) or sucrose (bottom). MOR1 and T394A receptor mutant-expressing Neuro2a cells were exposed to 1 μM DAMGO for the indicated time intervals. Cells were subsequently fixed, fluorescently labeled with anti-T7-tag antibody, and examined by confocal microscopy. Shown are representative results from one of three independent experiments performed in duplicate.
The agonist-induced internalization of MOR1 and T394A mutant was blockable by sucrose pretreatment (Fig. 4, bottom).
Resensitization of MOR1 and T394A in Neuro2a Cells.
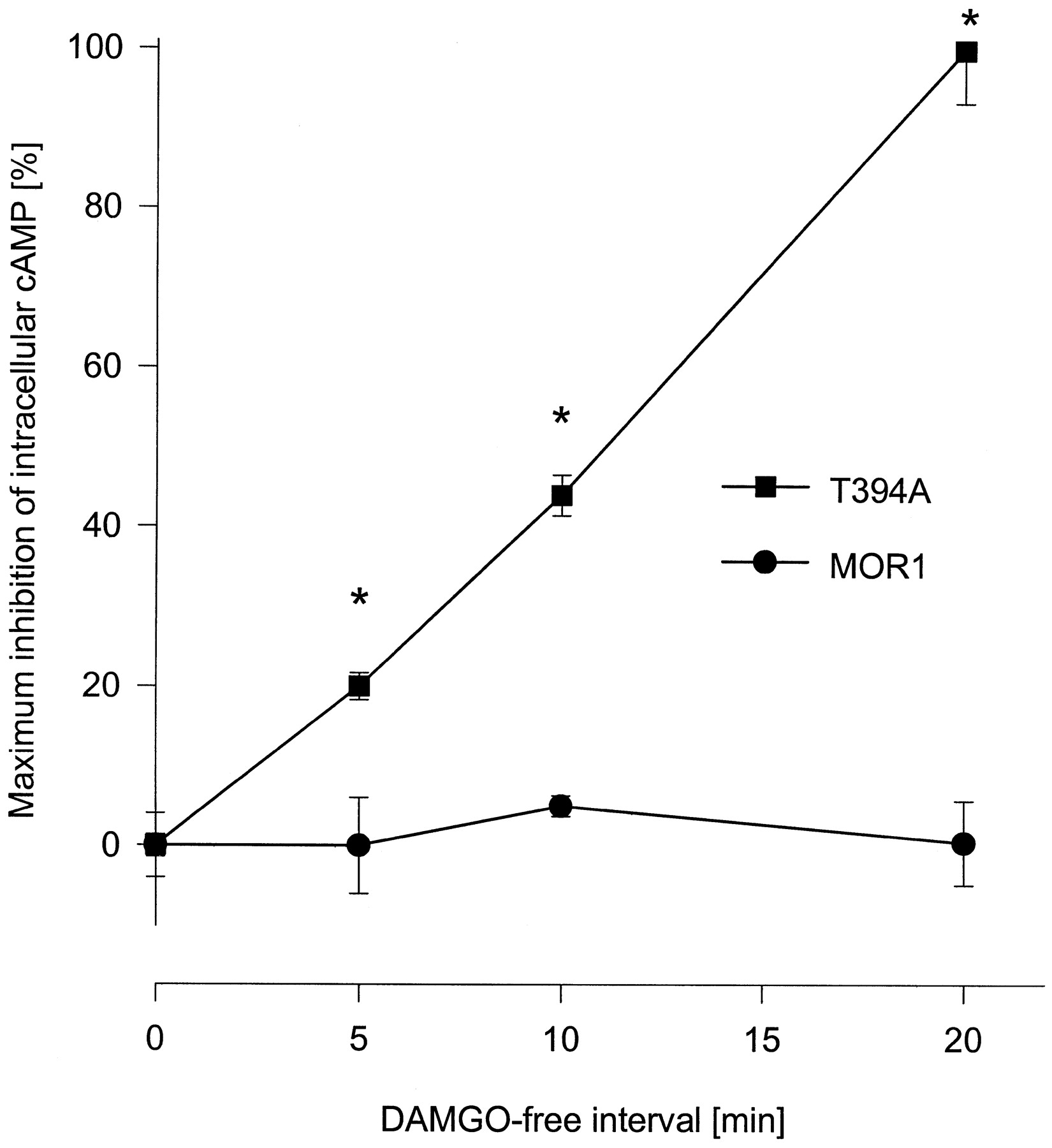
To investigate whether MOR1 and T394A receptor mutants differ in their resensitization kinetics after complete desensitization, Neuro2a cells expressing either MOR1 or T394A were treated for 5 h with 1 μM DAMGO. Cells were subsequently washed followed by an additional agonist-free incubation interval of either 0, 5, 10, or 20 min and determination of cAMP accumulation. Figure5 shows that after complete receptor desensitization, only T394A but not MOR1 resensitized during the 20 min of DAMGO withdrawal. Resensitized T394A receptors dropped forskolin-stimulated cAMP accumulation from 4 pmol to 2.5 pmol, whereas the cAMP level of MOR1 receptor-expressing cells remained unchanged at 4 pmol after agonist withdrawal. These results suggest that the faster rate of internalization of T394A is associated with accelerated receptor resensitization and redistribution of receptor proteins to the cell surface.

Resensitization of MOR1 and T394A. Transfected Neuro2a cells were cultured in the presence of 1 μM DAMGO for 5 h, which results in complete desensitization of both receptor types. Medium was removed and cells were washed three times with PBS followed by an additional incubation for 0, 5, 10, or 20 min in agonist-free medium. Cells were then treated with 25 μM forskolin or 25 μM forskolin plus 1 μM DAMGO for 15 min and cAMP levels were determined using a radioimmunoassay. Values represent means ± S.E.M. of triplicate determinations from two independent experiments. Asterisk indicates significant difference (p <.05) between MOR1 and T394A mutant as determined using analysis of variance followed by the Bonferroni test.
Discussion
It is commonly held that agonist-induced receptor desensitization involves phosphorylation of the agonist-bound form of the receptor by GRKs. Binding of arrestin or arrestin-like proteins to the phosphorylated receptor leads to uncoupling of the activated receptor from its G protein (Hausdorff et al., 1990; Lefkowitz et al., 1993;Lohse, 1993). The C-terminal tail and the third intracellular loop of various G protein-coupled receptors contain numerous serine and threonine residues that are potential phosphorylation sites for GRKs. For the δ opioid receptor, the C terminus, especially T353, has been shown to be important for receptor desensitization and internalization (Cvejic et al., 1996; Trapaidze et al., 1996). Also for the μ opioid receptor and platelet-activating factor receptor C-terminal serine and threonine residues have been identified to be involved in receptor desensitization (Takano et al., 1994; Segredo et al., 1997; Pak et al., 1997).
As shown previously, C-terminal splicing of the rat μ opioid receptor modulates agonist-induced desensitization (Zimprich et al., 1995). The splice variant MOR1B, which is more resistant to agonist-induced desensitization, lacks only one putative phosphorylation site (T394) compared with MOR1. In view of the fact that mutation of this T394 into an alanine resulted in a μ opioid receptor mutant that is also more slowly desensitized than MOR1 (Pak et al., 1997; Koch et al., 1997), it is not unreasonable to speculate that due to the loss of this phosphorylation site, the T394A mutant may become a poor substrate for phosphorylation by GRKs and, hence, more resistant to agonist-induced desensitization.
However, we have recently demonstrated an alternative mechanism for the observed greater resistance of the splice variant MOR1B to agonist-induced desensitization. Using confocal microscopy we showed that the C-terminal sequence of MOR1B facilitates clathrin-coated endocytosis which, in turn, promotes rapid receptor reactivation. In the present study, we provide evidence that also the T394A receptor mutant, in a manner similar to that seen with MOR1B, passes through an accelerated cycle of endocytosis and receptor resensitization which may explain its slow desensitization. This indicates that the T394 in the MOR1 receptor may represent a negative regulatory signal for receptor endocytosis.
In the present work we also show that, unlike MOR1B, the T394A receptor mutant does not undergo constitutive internalization, suggesting that the C terminus of MOR1B not only lacks the T394 but appears to contain sequence information that facilitates targeting of the receptor protein to the endocytotic machinery leading to constitutive endocytosis.
Our model that fast receptor internalization may be an important mechanism for receptor reactivation/recycling is supported by recent studies implicating endocytosis in the resensitization of G protein-coupled receptors (for review, Yu et al., 1993; Pippig et al., 1995; Krupnick and Benovic, 1998). For several receptors such as the δ opioid (Trapaidze et al., 1996), somatostatin (Roth et al., 1997), transferrin (Johnson et al., 1996), platelet-activating factor (Ishii et al., 1998), and human insulin receptors (Vogt et al., 1991), the C terminus seems to be involved in the process of internalization, but a common endocytotic motif has not been identified. For the μ opioid receptor, constitutive receptor internalization has been shown to follow from the truncation of the C-terminal threonine/serine region354ThrSerSerThr357 (Segredo et al., 1997). In addition to this threonine/serine region, our findings confirm a role of C-terminal T394 as inhibitor of μ opioid receptor endocytosis.
In summary, we have established that rate and extent of MOR1 internalization and resensitization is largely determined by a single phosphorylation site (T394) within the C-terminal tail. Exchange of this threonine residue leads to facilitated endocytosis via clathrin-coated pits and vesicles. This rapid internalization permits enhanced resensitization and thus counteracts agonist-induced desensitization.
Acknowledgments
We thank Dana Wiborny and Inge Schwarz for excellent technical assistance.
Footnotes
- Received July 30, 1998.
- Accepted October 15, 1998.
-
Send reprint requests to: Dr. Volker Höllt, Department of Pharmacology and Toxicology, Otto-von-Guericke University, 39120 Magdeburg, Leipziger Strasse 44, Germany. E-mail:volker.hoellt{at}medizin.uni-magdeburg.de
-
↵1 Both authors contributed equally to this publication.
-
This study was supported by Grants 1895A/0025 (to T.K.) and 1908A/0025 (to S.S.) from the Land Sachsen-Anhalt, Grant SCHU 924/4–1 (to S.S.) from the Deutsche Forschungsgemeinschaft, Grant SFB 426 TPA2, and a grant from Fonds der Chemischen Industrie (to V.H.).
Abbreviations
- DAMGO
- [d-Ala2, MePhe4, Gly5-ol]enkephalin
- Neuro2a
- mouse neuroblastoma cell line
- MOR
- rat μ opioid receptor
- GRK
- G protein receptor kinase
- TPBS
- 10 mM Tris, 10 mM phosphate buffer, 137 mM NaCl, 0.05% thimerosal
- NGS
- normal goat serum
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}