


Abstract
The human cannabinoid receptors, central cannabinoid receptor (CB1) and peripheral cannabinoid receptor (CB2), share only 44% amino acid identity overall, yet most ligands do not discriminate between receptor subtypes. Site-directed mutagenesis was employed as a means of mapping the ligand recognition site for the human CB2 cannabinoid receptor. A lysine residue in the third transmembrane domain of the CB2receptor (K109), which is conserved between the CB1 and CB2 receptors, was mutated to alanine or arginine to determine the role of this charged amino acid in receptor function. The analogous mutation in the CB1 receptor (K192A) was found to be crucial for recognition of several cannabinoid compounds excluding (R)-(+)-[2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthalenyl)methanone (WIN 55,212–2). In contrast, in human embryonic kidney (HEK)-293 cells expressing the mutant or wild-type CB2 receptors, we found no significant differences in either the binding profile of several cannabinoid ligands nor in inhibition of cAMP accumulation. We identified a high-affinity site for (−)-3-[2-hydroxyl-4-(1,1-dimethylheptyl)phenyl]-4-[3-hydroxyl propyl] cyclohexan-1-ol (CP-55,940) in the region of helices 3, 6, and 7, with S3.31(112), T3.35(116), and N7.49(295) in the K109A mutant using molecular modeling. The serine residue, unique to the CB2 receptor, was then mutated to glycine in the K109A mutant. This double mutant, K109AS112G, retains the ability to bind aminoalkylindoles but loses affinity for classical cannabinoids, as predicted by the molecular model. Distinct cellular localization of the mutant receptors observed with immunofluorescence also suggests differences in receptor function. In summary, we identified amino acid residues in the CB2 receptor that could lead to subtype specificity.
The pharmacological effects of marijuana are largely mediated through cell surface cannabinoid receptors. Two cannabinoid receptors have been identified; central cannabinoid receptor (CB1), which is predominantly expressed in the central nervous system, and peripheral cannabinoid receptor (CB2), which is largely restricted to cells of immune origin. The cannabinoid receptors are members of the G protein-coupled receptor superfamily, and share many of this family’s structural features. Both the CB1 and CB2 receptors inhibit adenylyl cyclase activity via a pertussis toxin-sensitive G protein (Howlett and Fleming, 1984; Matsuda et al., 1990; Felder et al., 1995; Slipetz et al., 1995). Furthermore, in neuronal and transfected cell lines, the potency of a series of cannabinoid analogs to inhibit cAMP accumulation correlates with their ability to displace [3H](−)-3-[2-hydroxyl4-(1,1-dimethylheptyl)phenyl]-4-[3-hydroxyl propyl] cyclohexan-1-ol (CP-55,940) binding (Howlett and Fleming, 1984, Matsuda et al., 1990). However, in the same neuronal cell line, others have demonstrated a G protein-mediated inhibition of Ca2+ channels that was not cAMP dependent (Mackie and Hille, 1992; Felder et al., 1993). Furthermore, AtT20 pituitary cells transfected with CB1 receptor cDNA exhibited cannabinoid-mediated inhibition of Q-type Ca2+ channels and activation of an inwardly rectifying potassium channel, as well as inhibition of adenylyl cyclase (Mackie et al., 1995). In contrast, when the CB2receptor was transfected into AtT20 cells, cannabinoid-mediated inhibition of adenylyl cyclase activity was conferred but not modulation of calcium or potassium channels (Felder et al., 1995).
The human CB1 and CB2receptors share only 44% overall identity, which rises to 68% shared identity in the transmembrane domains (Munro et al., 1993). However, most cannabinoid receptor agonists do not discriminate between the receptor subtypes (Felder et al., 1995; Slipetz et al., 1995; Showalter et al., 1996). Site-directed mutagenesis of receptor cDNAs followed by expression in mammalian cells to examine alterations in function provides an approach to mapping the ligand recognition site of the cannabinoid receptors.
Recently, K192 in the third transmembrane domain of the human CB1 receptor was shown to be critical for receptor recognition by several cannabinoid receptor agonists [CP-55,940, (−)-11-hydroxy-Δ8-tetrahydrocannabinol-dimethylheptyl (HU-210), and anandamide] but not for (R)- (+)-[2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthalenyl)methanone (WIN 55,212–2) (Song and Bonner, 1996). These findings were confirmed and expanded upon by Chin et al. (1998) who reported similar results for mutation of K192 in CB1 to glutamine, leucine, and glutamate. These authors also found that only the conservative mutation K192R retained binding affinity for CP-55,940, whereas all four mutant receptors displayed similar binding affinities for WIN-55,212–2. From these results has come the hypothesis that the CB1 binding site for WIN-55,212–2 is distinct from, but partially overlapping with, the binding site for other structural classes of cannabinoid receptor ligands and that K192 is a key interaction site in the latter binding pocket only (Bramblett et al., 1995; Reggio et al., 1997).
Studies using a combination of receptor chimeras between CB1 and CB2 as well as site-directed mutagenesis have also suggested differences in the ligand recognition sites of these receptors (Shire et al., 1996). Alignment of the primary amino acid sequences of the CB1 and CB2 cannabinoid receptors indicates a conserved lysine in the third transmembrane domain that corresponds to K109 in the CB2 receptor. To determine whether this analogous lysine residue was involved in ligand recognition for the CB2 receptor, we changed K109 to alanine (K109A) or arginine (K109R).
We report here that, in agreement with the K192R mutation results of Chin et al. (1998), the binding and signal transduction properties of the corresponding K109R mutant receptor are similar to those of the wild-type CB2 receptor. However, in contrast to the K192A mutation, which had profound results on the affinity and efficacy of several structural classes of cannabinoid receptor agonists, the corresponding mutation, K109A, exhibits binding and signal transduction properties similar to the wild-type receptor for ligands from each structural class tested. The possible origins of the profound differences between the lysine to alanine mutations in the CB1 and CB2 receptors were explored in this study using molecular modeling. S3.31(112) was identified as a key amino acid residue for the binding of CP-55,940 in CB2. Expression of the double mutant, K109AS112G, in accordance with the model, results in a loss of binding activity for several cannabinoid receptor agonists excluding WIN 55,212–2 and a related aminoalkylindole.
Experimental Procedures
Materials
[3H]CP-55,940 and [3H]WIN 55,212–2 were purchased from DuPont-NEN (Wilmington, DE). (−)-Δ9-tetrahydrocannabinol (Δ9-THC) and anandamide were obtained from the National Institutes on Drug Abuse (Rockville, MD). CP-55,940 was synthesized by Dr. Larry Melvin (Pfizer Inc., Groton, CT). WIN-55,212–2 was purchased from Research Biochemicals Inc/ (Natick, MA). Anandamide was synthesized and provided by Dr. Raj Razdan (Organix, Inc., Woburn MA). 2-Methyl-3-napthoyl-N-propylindole (JWH-015) was synthesized and provided by Dr. John Huffman (Clemson Univ., SC). Dr. Sean Munro (MRC Laboratories, Cambridge, England) generously provided the human CB2 cDNA clone.
Mutagenesis
Mutations of the CB2 receptor were introduced with the QuikChange site-directed mutagenesis kit (Stratagene, LaJolla, CA; Papworth et al., 1996). This method allows mutagenesis to be performed in any vector, hence we used human CB2 subcloned into pcDNA3 (Invitrogen, San Diego, CA; Showalter et al., 1996). Oligonucleotide primers, each complementary to opposite strands of the sequence to be altered, were annealed and extended during 12 to 18 cycles of temperature cycling by means of Pfu DNA polymerase (which replicates both strands with high fidelity and without displacing the mutant oligonucleotide primers). The product was treated with DpnI, which digests methylated and hemi-methylated DNA (the parental, nonmutated DNA), then the remainder (containing nicked, double-stranded mutant DNA) transformed into Escherichia coli. The DNAs were sequenced to confirm mutation in the desired regions only. To make the K109A mutation, the primers T GTC TTC CTG CTG GCG ATT GGC AGC GT (forward) and AC GCT GCC AAT CGC CAG CAG GAA GAC A (reverse) containing the desired mutation (AAG to GCG) were used. To make the K109R mutation, the primers TTC CTG CTG AGG ATT GGC A (forward) and CT GCC AAT CCT CAG CAG GAA (reverse) containing the desired mutation (AAG to AGG) were used. To make the K109AS112G mutation, primers containing the desired mutation (AGC to GGC, S to G) were constructed to hybridize to the K109A cDNA.
Cell Culture and Transfection
Human embryonic kidney (HEK)-293 cells obtained from American Type Culture Collection (Rockville, MD) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal clone II (Hyclone Labs., Logan, UT) and 5% CO2 at 37°C in a Forma incubator (Forma Scientific, Marietta, OH). Cell lines were created by transfection of wild-type or mutant CB2 into HEK-293 cells by the lipofectamine reagent (Life Technologies, Gaithersburg, MD). Stable transformants were selected in growth medium containing geneticin (G418, 1 mg/ml). Colonies of about 500 cells were picked (about 2 weeks post-transfection) and allowed to expand, then tested for expression of receptor mRNA by Northern blot analysis. Cell lines containing moderate to high levels of receptor mRNA were tested for receptor binding properties. Transfected cell lines were maintained in DMEM with 10% fetal clone II (Hyclone) plus 0.3 to 0.5 mg/ml G418 and 5% CO2 at 37°C in a Forma incubator.
Cannabinoid Receptor Radioligand Binding Determinations
The assay has been previously described (Tao and Abood, 1998). Briefly, cells were harvested in PBS containing 1 mM EDTA and centrifuged at 500g. The cell pellet was homogenized and centrifuged three times at 1600g (10 min). The combined supernatants were centrifuged at 100,000g (60 min). The (P2 membrane) pellet was resuspended in 3 ml of buffer B (50 mM Tris-HCl, 1 mM EDTA, and 3 mM MgCl2, pH 7.4) to yield a protein concentration of approximately 1 mg/ml. The tissue preparation was divided into equal aliquots, frozen on dry ice, and stored at −70°C. Binding was initiated by the addition of 25 to 75 μg membrane protein to silanized tubes containing [3H]CP-55,940 (102.9 Ci/mmol) or [3H]WIN 55,212–2 (43 Ci/mmol) and a sufficient volume of buffer C [50 mM Tris-HCl, 1 mM EDTA, 3 mM MgCl2, and 5 mg/ml fatty acid free bovine serum albumin (BSA), pH 7.4] to bring the total volume to 0.5 ml. The addition of 1 μM unlabeled CP-55,940 or WIN 55,212–2 was used to assess nonspecific binding. Specific binding averaged >50% of total binding at 1 nM [3H]CP-55,940 in all cell lines used in the analysis, except for K109AS112G. Following incubation (30°C for 1 h), binding was terminated by the addition of 2 ml of ice-cold buffer D (50 mM Tris-HCl, pH 7.4, plus 1 mg/ml BSA) and rapid vacuum filtration through Whatman GF/C filters [pretreated with polyethyleneimine (0.1%) for at least 2 h; Whatman Inc., Clifton, NJ]. Before radioactivity was quantitated by liquid scintillation spectrometry, filters were shaken for 1 h in 5 ml of scintillation fluid. CP-55,940 and all cannabinoid receptor analogs were prepared by suspension in assay buffer from a 1 mg/ml ethanolic stock without evaporation of the ethanol (final concentration of no more than 0.4%). When anandamide was used as a displacing ligand, experiments were performed in the presence of phenylmethylsulfonyl fluoride (50 μM). Saturation experiments were conducted with seven concentrations of [3H]CP-55,940 or [3H]WIN 55, 212–2 ranging from 250 pM to 10 nM. Competition assays were conducted with 0.5 nM [3H]CP-55,940 or 3 nM [3H]WIN 55,212–2 and six concentrations (0.1 nM to 10 μM displacing ligands).
The Bmax andKd values obtained from Scatchard analysis of saturation binding curves (Scatchard, 1951; Rosenthal, 1967) were determined by the KELL package of binding analysis programs for the Macintosh computer (Biosoft, Milltown, NJ). Displacement IC50 values were originally determined by unweighted least-squares linear regression of log concentration-percent displacement data and then converted toKi values using the method of (Cheng and Prusoff, 1973).
cAMP Accumulation Assay
Intracellular cAMP levels were measured (Abood and Tao, 1995) with a competitive protein binding assay (Diagnostic Products, Inc., Los Angeles, CA). Cells were harvested at 70 to 90% confluency in PBS containing 1 mM EDTA and counted with a hemacytometer. After pelleting at 500g, the cell pellet was resuspended at a concentration of 1 × 106 cells/ml in DMEM containing 20 mM HEPES, pH 7.3, 0.1 mM RO-20–1724 and 1 mM isobutylmethylxanthine and incubated for 30 min at 37°C. Aliquots of cells (90 μl) were added to polypropylene microfuge tubes containing 1.0 μM forskolin ± compound + 1 mg/ml fatty acid free BSA, in a final volume of 100 μl and incubated for 5 min at 37°C. Because the compounds tested were dissolved in ethanol, all tubes contained an equivalent amount of ethanol (0.5%). The reactions were terminated by boiling for 4 min, followed by centrifugation and removal of 50 μl of the supernatant, which was assayed for cAMP levels. The results are expressed as percentage of inhibition of forskolin-stimulated cAMP accumulation. EC50 curves were generated with the use of the GraphPad Prism program (GraphPad, San Diego, CA).
Statistical Analyses
The Ki and EC50 values in the mutant versus wild-type cell lines were compared using ANOVA. Bonferroni-Dunn post hoc analyses were conducted when appropriate. P < .05 defined statistical significance.
Molecular Modeling
Residue Identifications.
To facilitate comparisons between homologous positions in the CB1 and CB2 receptor sequences, the generalized numbering scheme suggested by Ballesteros and Weinstein (1995) was employed. In this scheme, each amino acid position in a sequence is given a number that begins with the helix number followed by a two-digit decimal. The most highly conserved residue in each helix is assigned a value of 0.50 and all other residues in the helix are numbered relative to this highly conserved residue. This number may be followed by the sequence number in parentheses.
Ligand Conformational Analysis.
The focus of the modeling studies was upon the K to A mutation at position 3.28 and its effect upon the binding of one cannabinoid ligand, CP-55,940 in both the CB1 and CB2 receptors. The numbering system employed for CP-55,940 (Compton et al. 1992) is provided with the drawing of the CP-55,940 structure (Fig.1). References to each hydroxyl throughout this paper will use a nomenclature suggested by Makriyannis (Tius et al., 1995). In this nomenclature, the C1-hydroxyl group is termed the northern aliphatic hydroxyl (NAH) group; the hydroxyl of the C-4 side chain is termed the southern aliphatic hydroxyl (SAH) group; and the hydroxyl at C2′ is termed the phenolic hydroxyl group (or phenol).

Cannabinoid receptor agonists used in the study. Numbering system commonly used for bicyclic cannabinoids such as CP-55,940 is illustrated in ii (Compton et al., 1992).
A complete conformational analysis of CP-55,940 was performed using the method of Molecular Mechanics as encoded in the MM3 program (Molecular Design, Ltd., San Leandro, CA). This analysis included Dihedral Driver studies in 10-degree increments for all rotateable bonds in CP-55,940.
Model Construction.
Computer models of the K3.28(192)A CB1 and K3.28(109)A CB2mutant receptors were constructed by mutating the residue of interest, K3.28 (192) in CB1 and K3.28(109) in CB2, using the Chem Protein module within the Chem-X molecular modeling package (Chemical Design Ltd., Chipping Norton, U.K.). Mutations were performed on previously constructed models of the cannabinoid CB1 and CB2 receptor transmembrane bundles (see Bramblett et al., 1995; Huffman et al., 1996) with supplementary materials therein for cannabinoid receptor model building details). Very recently the helix tilts within each CB1 and CB2 bundle were adjusted to be consistent with the 7.5 Å projection structure of frog rhodopsin (Unger et al., 1997).
Ligand Docking.
CP-55,940 was initially docked such that its C-4′ alkyl side chain could interact with a hypothesized hydrophobic binding pocket in the Hx6-Hx7 region. Using this hydrophobic binding pocket as an anchor point, each receptor bundle was probed for additional interactions available to the ligand first using interactive computer graphics. In these docking studies, all exocyclic torsion angles in CP-55,940 were allowed to vary to alleviate all Van der Waals overlaps.
Each ligand-receptor complex identified by this initial screen was subjected to energy minimization using the All Atom force field in Amber (Pearlman et al., 1991). The protocol used for the eight-step minimization was as follows. In the first step, the backbones of the helices were held constant with all atoms of the ligand and all side chain atoms free to move. The complex was subjected to 100 cycles steepest descent (SD) followed by 900 cycles of conjugate gradient (CG). A distance dependent dielectric, nonbonded cut-off of 20 Å and nonbonded update each 500 cycles were employed. The second step included 50 cycles SD followed by 1000 cycles CG. In this step, all atoms were free to move and a nonbonded update each 200 cycles was employed. The third and fifth steps were repeats of step one with 20 cycles (SD) and 1980 cycles (CG). The fourth and sixth steps were repeats of step 2. In the last step, the helix backbones were again frozen, and CG cycles were run until the system reached a gradient of 0.001 kcal/mol Å.
Analysis of Ligand/Receptor Interactions.
Each resultant complex was probed for ligand-receptor hydrogen bonding interactions using the Calculate H-Bond facility in the Chem-X molecular modeling suite of programs (Chemical Design Ltd.). A hydrogen bond was considered present if the heteroatom-heteroatom distance was between 2.5 Å and 3.2 Å and the heteroatom-H-heteroatom angle was between 90 and 180 degrees (Jeffrey, 1997). Hydrogen bonds with angles approaching linearity (i.e., close to 180 degrees) were considered to be stronger, as were hydrogen bonds that were shorter (i.e., closer to 2.5 Å).
Immunofluorescence
Cells were grown and maintained in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (Biowhitaker, Walkersville, MD) and 0.5 mg/ml G418. For immunofluorescence, cells (2 × 105/ml) were seeded into 60-mm culture dishes containing sterile glass coverslips and allowed to incubate overnight at 37°C in an humidified atmosphere containing 5% CO2. Coverslips were drained of excess medium, washed in PBS at room temperature three times, air dried (30 min), fixed in absolute acetone (5 min, room temperature), and air dried (30 min). Fixed coverslip cultures were rehydrated in PBS (20 min), blocked (1 h, room temperature) in SuperBlock in Blocking Buffer in PBS (Pierce, Rockford, IL), and incubated (1 h) with an affinity-purified rabbit antibody (1:10 dilution in SuperBlock) directed against an immunodominant carboxy terminal domain of the human CB2 receptor. This antibody, designated anti-hCB2.CV, was elicited in New Zealand White rabbits using a keyhole limpet hemocyanin-human CB2 peptide (amino acids 320–336) fusion as immunogen and was assessed for specificity and antibody titer as described previously (Nowell et al., 1998). Slides were then washed in PBS (3×), incubated with FITC-labeled goat anti-rabbit immunoglobulin G (60 min, 1:32 dilution in PBS, 2.5% Evans Blue, H + L; Cappel, West Chester, PA) at room temperature, rinsed in PBS three times, rinsed in distilled H20 (5 min), and mounted in Aquamount (Lerner Laboratories, New Haven, CT). Slides were examined with an Olympus BHA Microscope equipped with a model BH2RFl reflected fluorescence attachment and a model PM-10AD photomicrographic system (Olympus Corp., Lake Success, NY).
Results
Ligand Binding and Signal Transduction Properties of CB2 Wild-Type and K109 Mutant Receptors.
To assess the role of K109 in ligand binding and signal transduction at the CB2 cannabinoid receptor, we replaced the lysine residue with an alanine (K109A) or an arginine (K109R). These mutations led to removal or extension of the positively charged amine, respectively. Stable cell lines were established that expressed the human CB2 (wild-type), K109A or K109R receptors in HEK-293 cells. No specific [3H]CP-55,940 binding to HEK-293 cells was found before transfection (data not shown). In the HEK-293 cell line stably expressing the CB2 receptor, Kdand Bmax values of 0.88 ± 0.09 nM and 1.55 ± 0.39 pmol/mg protein were obtained (Table1). These values are comparable with a CB2-expressing Chinese hamster ovary (CHO) cell line previously described (Kd = 0.61 ± 0.14 nM and Bmax = 3.1 ± 0.9 pmol/mg protein; Showalter et al., 1996). In contrast to what had been found with the analogous lysine mutation in the CB1 receptor, [3H]CP-55,940 binding was unaffected in the K109A mutant cell line; both the affinity and the absolute levels of expression were comparable with the wild-type CB2receptor cell line (Kd = 0.82 ± 0.23 nM and Bmax = 1.60 ± 0.21 pmol/mg protein, Table 1). Similarly, the K109R mutant cell line exhibited wild-type [3H]CP-55,940 binding (Kd =1.68 ± 0.29 nM andBmax = 1.00 ± 0.21 pmol/mg protein, Table 1). Specific binding averaged >50% of total binding at 1 nM [3H]CP-55,940 in all cell lines used in these analyses.
Scatchard analysis of wild-type and mutant cell lines
Several representative cannabinoid receptor agonists were chosen to test for comparison with the mutant receptors. TheKi values, shown in Table2, demonstrate that the binding profile using these compounds in the K109A mutant cell lines is similar to that in wild-type CB2-transfected cells.
Binding profile of wild-type CB2 and K109A and K109AS112G mutant cell lines
The CB2- transfected HEK-293 cells demonstrated functional coupling to G proteins, as measured by inhibition of forskolin-stimulated cAMP accumulation (Fig.2). Inhibition of forskolin-stimulated cAMP accumulation was maximal at 73% with 1 μM WIN 55,212–2 or CP-55,940 (Fig. 2). No cannabinoid receptor agonist inhibition of forskolin-stimulated cAMP accumulation was observed in untransfected HEK-293 cells (data not shown). That the K109A receptor is fully functional is demonstrated by the potencies and maximal inhibition of forskolin-stimulated cAMP with WIN 55,212–2 (2.5 ± 1.7 nM, 95% inhibition at 1 μM) and CP-55,940 (6.9 ± 1.6 nM, 95% inhibition at 1 μM; Fig. 2). Similarly, the K109R cell line exhibited wild-type EC50s and inhibition of forskolin-stimulated cAMP accumulation with WIN 55,212–2 (4.05 ± 1.4 nM, 63% inhibition at 10 μM) and CP-55,940 (97.2 ± 26.9 nM, 73% inhibition at 10 μM; Fig. 2).
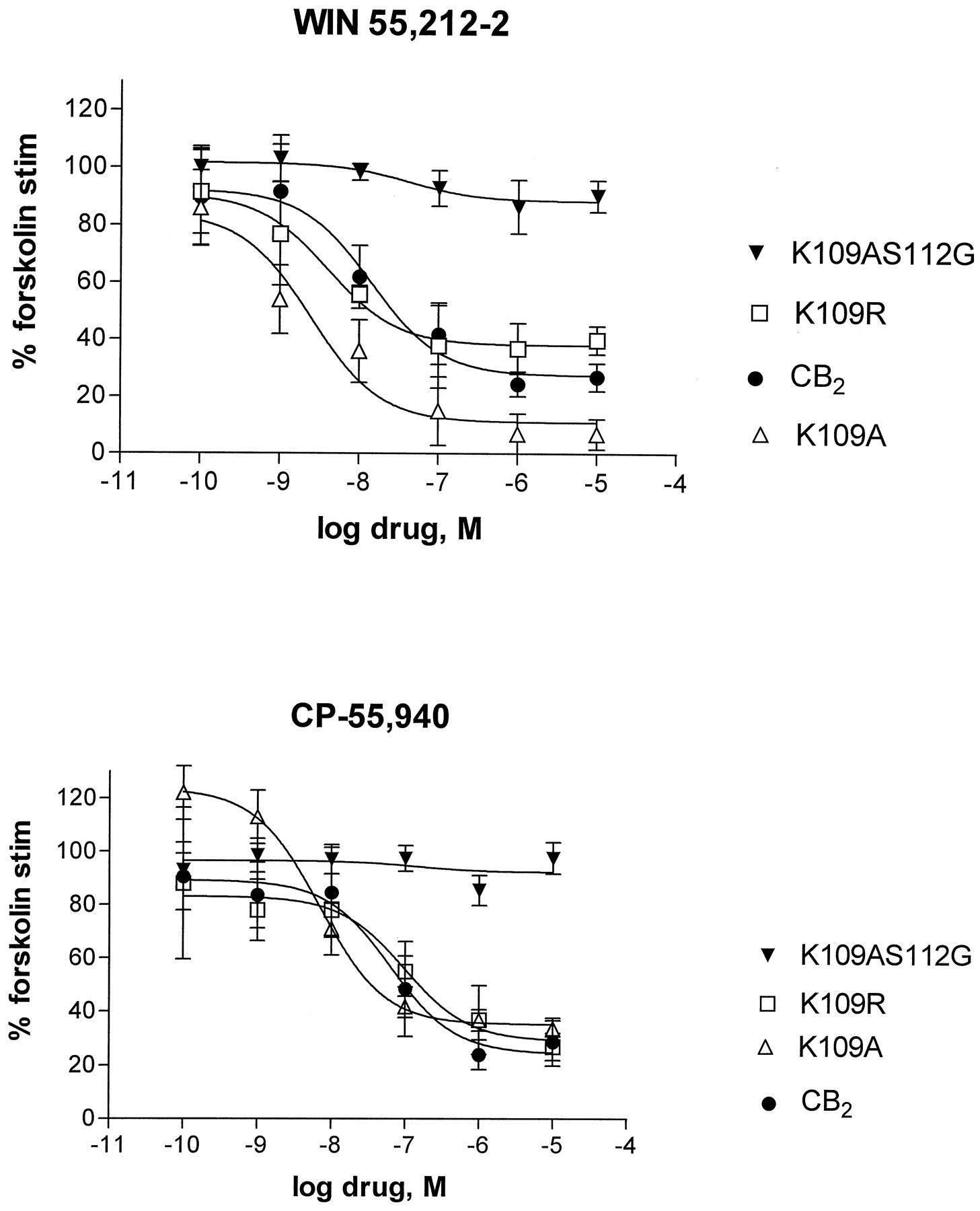
Comparison between wild-type and mutant CB2 receptors for agonist-induced inhibition of cAMP accumulation by WIN 55,212–2 (top) or CP-55,940 (bottom). cAMP accumulation assays were performed on HEK-293 cells stably expressing wild-type CB2 or K109A or K109R or K109AS112G mutant receptor cDNAs. Points indicate mean ± S.E.M. of three or more independent experiments performed in triplicate. Curves were generated as described under Experimental Procedures. EC50 values were 14.2 ± 4.3 nM for WIN-55,212–2 and 63.1 ± 18.8 nM for CP-55,940 in the wild-type CB2receptor; 2.5 ± 1.7 nM (WIN) and 6.9 ± 1.6 nM (CP) for K109A; 4.1 ± 1.4 nM (WIN) and 97.2 ± 26.9 nM (CP) for K109R. Levels of forskolin-stimulated cAMP accumulation (expressed in pmol/106 cells/min) for the cell lines tested were: 8.55 ± 2.78 (CB2); 7.28 ± 0.97 (K109A); 7.97 ± 2.13 (K109R); and 8.39 ± 1.82 (K109AS112G).
Molecular Modeling.
A molecular modeling study with CP-55,940 was undertaken to examine whether an alternate high-affinity binding mode may be achieved in the K109A CB2 mutant in contrast to K192A CB1. Conformational analysis of CP-55,940 revealed the following. One major point of flexibility in CP-55,940 is the C1′-C3 bond. Rotation about this bond controls the placement of the C1 hydroxyl (NAH) and the C4 side chain hydroxyl (SAH) in space relative to the C2′ phenolic hydroxyl. MM3 conformational analysis revealed that there are two minimum energy conformers associated with rotation about the C1′-C3 bond in CP-55,940. The global minimum occurs at C2-C3-C1′-C2" = −116 degrees. The second minimum at 64 degrees is 0.91 kcal/mol higher in steric energy than the global minimum. These results are consistent with reported NMR solution and molecular dynamics studies of CP-55,940 (Xie et al., 1996). These investigators reported two minima for the C2-C3-C1"-C2" torsion angle, 60 degrees and −120 degrees.
Another major point of flexibility in the CP-55,940 structure is the C4′ to C1" bond. Rotation about this bond controls the placement in space of the dimethylheptyl (DMH) side chain relative to the phenyl ring. The MM3 conformational analysis revealed four low-energy minima that occurred at C3′-C4′-C1′-C2" values of −73, 67, 107, and −113 degrees, which are within 0.03 kcal/mol of each other. Xie et al. (1996) reported four minima (60, −60, 120, and −120 degrees) for the C3′-C4′-C1"-C2" torsion angle.
One underlying assumption in all receptor docking studies reported here is that ligand binding to the cannabinoid receptors occurs within the pore formed by the transmembrane helix bundle. A second hypothesis underlying the studies is that the large hydrophobic cluster of amino acids on helices (Hxs) 6 and 7 form the hydrophobic pocket with which the DMH side chain of CP-55,940 interacts.
Receptor docking studies revealed that in CB1versus CB2, CP-55,940 is oriented differently in the binding pocket. In CB1, a largely hydrophobic cluster of residues surround the DMH sidechain of CP-55,940. These are V6.43(351), C6.47(355), L6.51(359), L7.41(385), and L7.44(388). In CB2, this cluster is composed of V6.43(253), C6.47(257), V6.51(261), L7.41(287), and I7.44(290). The binding sites for CP-55,940 in CB1 and CB2 then diverge from each other.
In CB1, the phenyl ring of CP-55,940 is near Hx 3, whereas the NAH group of the cyclohexyl ring points toward Hx 5. In this orientation, CP-55,940 exists in nearly its global minimum C2-C3-C1′-C2′ conformation (−118 degrees compared with global minimum at −116 degrees). The major hydrogen bonding interactions identified for CP-55,940 in CB1 involve the phenolic hydroxyl with K3.28(192), the NAH group with W5.43(279), and the SAH group with N7.45(389). The binding site is illustrated in Fig.3.
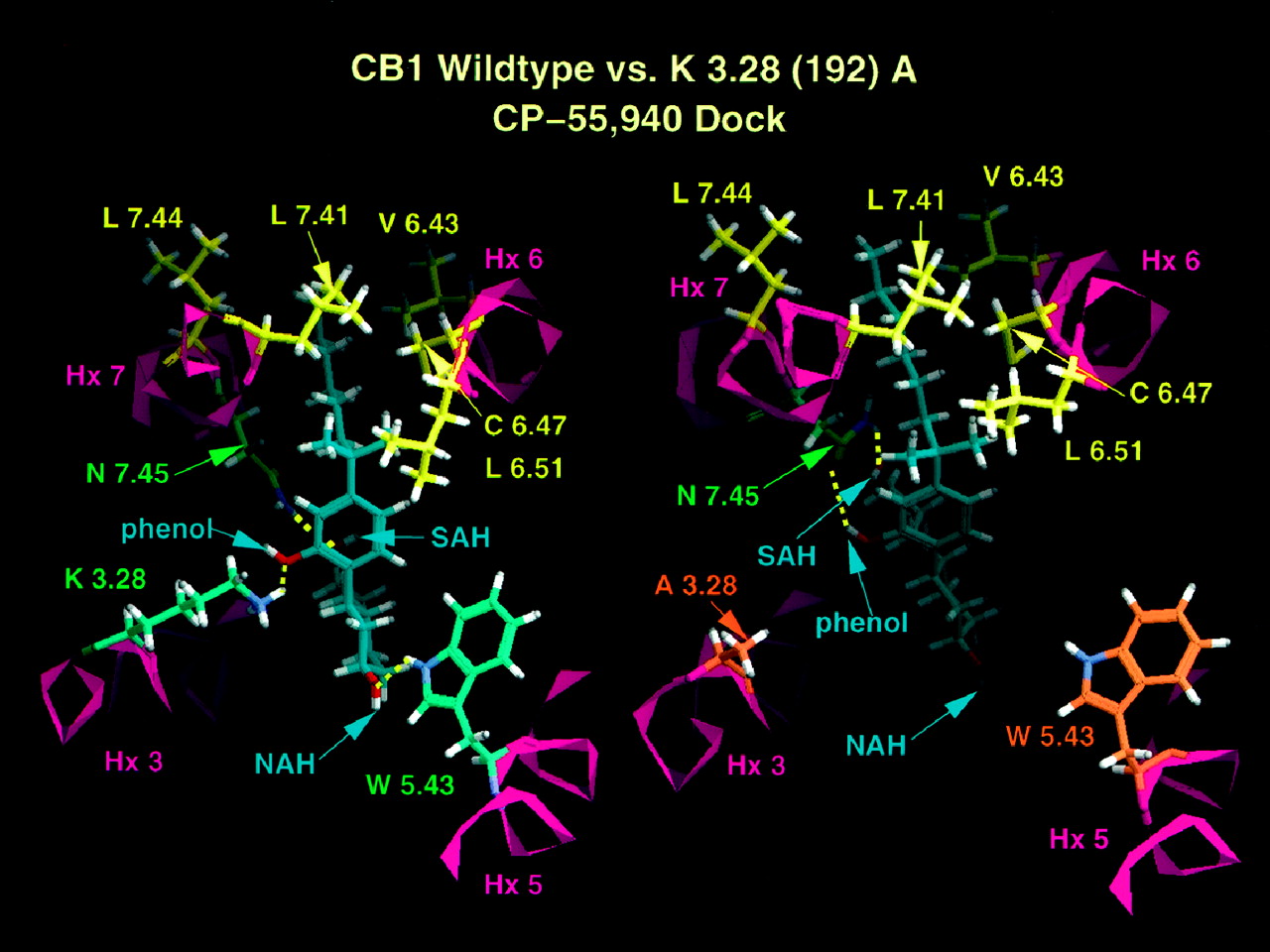
Extracellular views of CP-55,940 docked in a model of the transmembrane helix bundle of CB1 wild type (to the left) and K3.28(192)A (to the right). Residues that form largely hydrophobic binding pocket for the 1", 1"-DMH side chain are shown in yellow. Residues that form hydrogen bonding interactions with the ligand are shown in green. Residues that are no longer able to form hydrogen bonds with CP-55,940 as a consequence of this mutation are shown in orange. C-1 hydroxyl group of CP-55,940 is labeled NAH. Hydroxyl of C-4 side chain of CP-55,940 is labeled SAH. The C-2′ phenolic hydroxyl of CP-55,940 is labeled phenol.
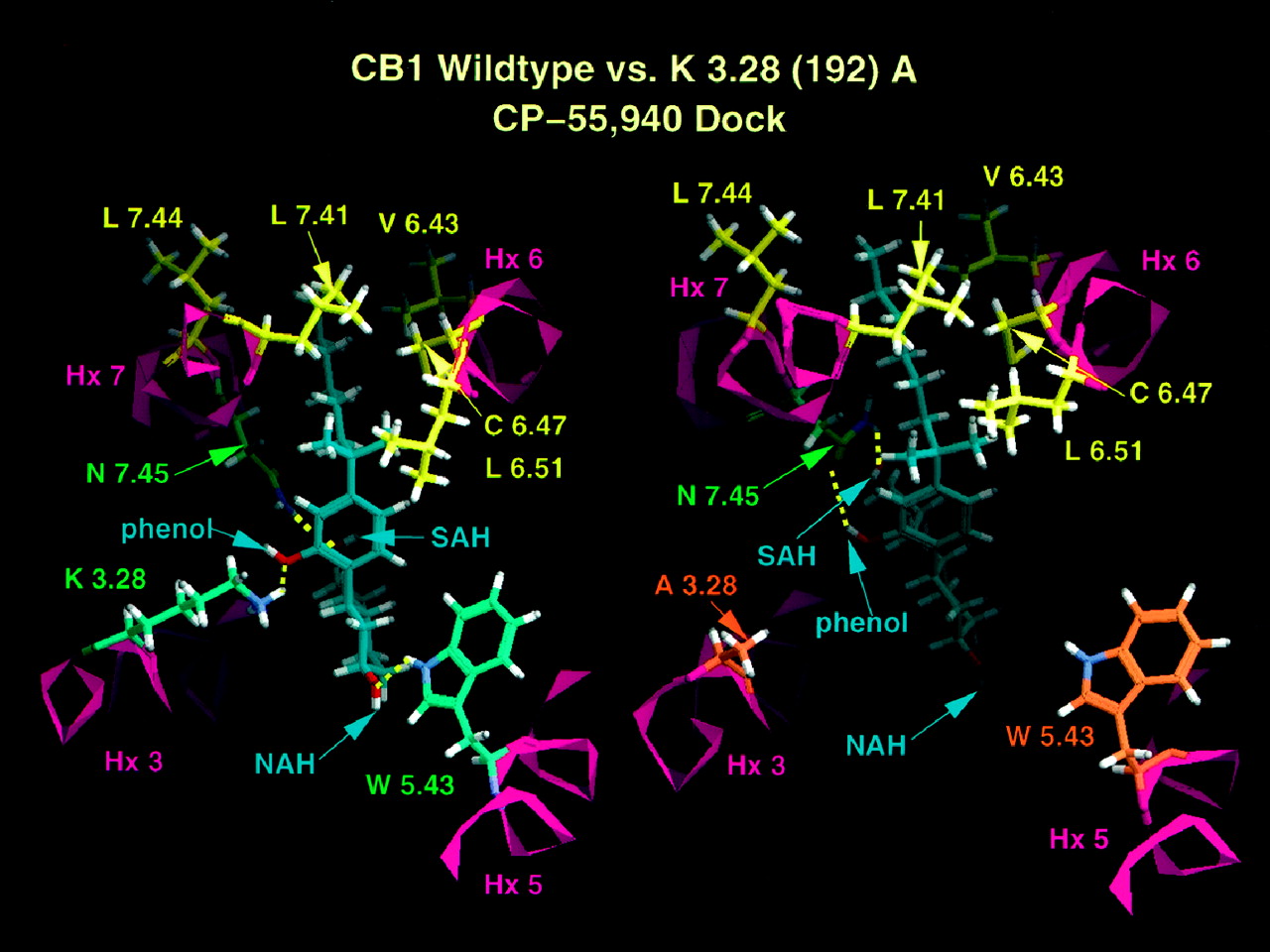
Modeling studies indicate that in CB2, the phenyl ring is near Hx 7, whereas the cyclohexyl ring is between Hxs 3 and 7 pointing toward Hx 2. Hydrogen bonding interactions involve the SAH group with K3.28(109), the phenolic hydroxyl with N7.45(291), and the NAH group with both S3.31(112) and T3.35(116), shown in Fig.4. A unique feature identified in the CP-55,940/CB2 binding site is the hydrogen bonding cluster formed by S3.31(112) and T3.35(116). In its interaction with these two residues, the oxygen of the NAH hydroxyl group acts as a hydrogen bond acceptor in a hydrogen bond with T3.35(116) and the NAH group acts as a hydrogen bond donor to S3.31(112). This hydrogen bonding cluster exists only in CB2. In CB1, residue 3.31 is a G(195) and residue 3.35 is a S(199).

Interactions for CP-55,940 in CB2wild-type and K3.28(109)A receptors. Left top, hydrogen bonding interactions of CP-55,940 in CB2. Hydrogen bonding residues are shown in green. Labels for key functional groups are as explained in Fig. 3. View is from Hx 2 looking toward Hx 5. Left bottom, largely hydrophobic binding pocket for DMH side chain of CP-55,940 in CB2. Residues in this pocket are shown in yellow. View is from lipid looking between Hxs 6 and 7 and into bundle. Right top, hydrogen bonding interactions of CP-55,940 in K3.28 (109)A mutant. Hydrogen bonding residues are shown in green. View is from Hx 2 in membrane looking toward Hx 5. See Fig. 3 for further details. Right bottom, largely hydrophobic binding pocket for DMH side chain of CP-55,940 in the CB2 K3.28 (109)A mutant. Residues that comprise this pocket are shown in yellow. View is from lipid looking between Hxs 6 and 7 into the transmembrane helix bundle.
As indicated in Fig. 4, the K3.28(109)A mutation in CB2 results in the loss of the interaction between CP-55,940 and residue 3.28(109), whereas three other nearly linear hydrogen bonds with S3.31(112), T3.35(116), and N7.45(291) are retained. The ligand C2-C3-C1′-C2′ angle is 55 degrees, which is 1.09 kcal/mol above the global minimum at −116 degrees, but above the second energy minimum at 64 degrees by only 0.18 kcal/mol. Figure 4reveals that there is a slight change in the depth of penetration of CP-55,940 within the receptor pore. This is most evident in the hydrophobic binding pocket. In CB2, the DMH side chain is closer to L7.41(287), which forms the upper portion of the hydrophobic cluster, than in K3.28A(109)CB2. This difference does not appear to have consequences of an overall significance.
In contrast to the K3.28(109)A CB2 results, modeling indicates that the K3.28(192)A mutation in CB1 would have a more deleterious effect upon the binding of CP-55,940. Figure 3 reveals that loss of the K3.28(192) interaction causes the cyclohexyl ring portion of the molecule to angle deeper into the pocket, resulting in the loss of the NAH/W5.43(279) hydrogen bond that exists in the CB1 WT receptor. In the K3.28(192)A CB1 mutant, only one amino acid, N7.45(389), interacts with CP-55,940, producing a hydrogen bond with the phenolic hydroxyl and with the SAH group.
Alternate binding modes were sought in the K3.28(192)A CB1 mutant. Changing the ligand orientation to mimic the orientation of CP-55,940 in the CB2receptor was not found to improve interactions for CP-55,940. This appears to be due to the fact that the hydrogen bonding cluster does not exist in CB1. Although a serine does exist at 3.35 and could potentially serve as a hydrogen bonding partner, Amber studies revealed that the ligand is not drawn close enough to S3.35(199) for a hydrogen bond to form (heteroatom-heteroatom distance = 4.2 Å). Only a single hydrogen bond occurs for CP-55,940 docked in this position. This hydrogen bond is between the phenolic hydroxyl and N7.45(389). This alternate binding site, therefore, does not result in improved ligand/receptor interactions relative to those presented above.
Ligand Binding and Signal Transduction Properties of K109AS112G Double Mutant.
To test the hypothesis that S3.31(112) provides critical hydrogen bonding stability in the K109A mutant, we substituted S3.31(112) with a glycine, creating the K109AS112G double mutant. None of several cell lines tested showed appreciable specific binding using [3H]CP-55,940 as a radioligand (data not shown). On the other hand, specific binding of [3H]WIN 55,212–2 was observed (Table 1). It should be noted that analysis of several stable cell lines showed lowered expression of this receptor mutant; a cell line with aBmax of 0.484 ± 0.01 pmol/mg was chosen for further characterization. Competition experiments using [3H]WIN 55,212–2 as a radioligand confirmed the absence of CP-55,940 binding in the K109AS112G mutant (Table 2). Similarly, neither Δ9-THC, cannabinol, nor anandamide displaced [3H]WIN 55,212–2. Only JWH-015, another aminoalkylindole-like [3H]WIN 55,212–2 (see Fig. 1), competed for the binding site. When inhibition of forskolin-stimulated cAMP accumulation was examined in the K109AS112G cell line, some inhibition was observed with WIN 55,212–2 (10% at 10 μM, EC50 value of 42.3 ± 2.6 nM) and no effect was found with CP-55,940 (Fig. 2).
Immunofluorescence Studies.
In an effort to understand the binding and cAMP results found with the mutant receptors, we employed the technique of immunofluorescent microscopy to localize receptor proteins. Immunofluorescence studies were performed using an antibody directed against an immunodominant carboxy terminal domain of the human CB2 receptor to define the intracellular localization of the expressed mutant CB2 proteins (Fig. 5). In HEK-293 cells expressing the wild-type CB2 receptor, approximately 90% were positive for fluorescence. Approximately 10% of these cells exhibited a punctate pattern of fluorescence within the cytoplasm. For the majority of cells, however, staining was within the cytoplasmic compartment in a diffuse pattern and apparently delineating the outer periphery consistent with localization at the cell surface.
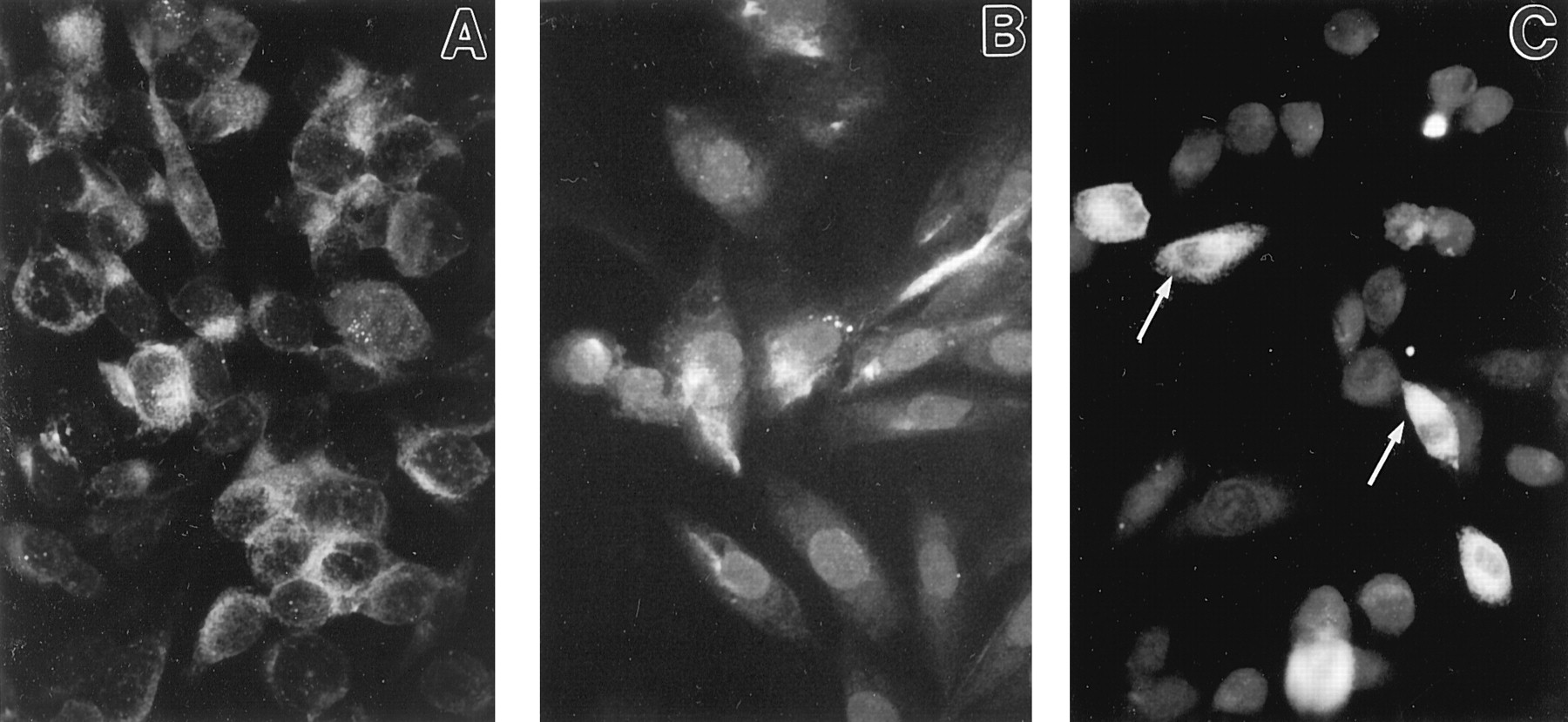
Immunofluorescence staining of HEK-293 cells expressing human CB2 mutant cannabinoid receptors. A, cells expressing wild-type CB2 exhibit immunofluorescence throughout cytoplasm with characteristic punctations in approximately 10% of positive cells. B, cells transfected with the K109A mutant exhibit a punctate pattern of intracytoplasmic staining and accumulation of immunoreactive product at margin of the cytoplasmic compartment. C, cells transfected with K109AS112G double mutant exhibit an entrapment of immunoreactive product within perinuclear region (arrows) and absence of immmunoreactive product from distal cellular extensions. Original magnification, 350×.
Approximately 50% of cells transfected with the K109A mutant were positive for fluorescence. Intense fluorescence, indicative of relatively high level expression of CB2 receptor, was observed in approximately 10% of cells. The pattern of immunofluorescence was characterized by distinct punctations in the cytoplasmic compartment as well as a delineated pattern of staining at the outer margin of the cytoplasm consistent with localization of the receptor on the cell surface.
In contrast, cells transfected to express the K109AS112G double mutant exhibited a pattern of receptor protein expression distinctive from that of cells transfected to express the wild-type or K109A mutant. A smaller percentage (approximately 30%) of positively stained cells was observed when compared with wild-type and K109A mutant-transfected cells. In the majority (i.e., approximately 70%) of cells shown to be positive for fluorescence, a distinctive perinuclear accumulation of fluorescence was noted. This perinuclear accumulation often encompassed the greater part of the cytoplasm. Untransfected HEK-293 cells, CB2, and K109A transfected cells incubated with normal rabbit immunoglobulin G exhibited minimal fluorescence.
Discussion
The lysine residue in the third transmembrane domain of the CB2 receptor, which is conserved between the CB1 and CB2 cannabinoid receptors, appears to mediate different functional roles in the receptor subtypes. K192 of the CB1 receptor is critically important for ligand recognition for several agonists excluding WIN 55,212–2 (Song and Bonner, 1996; Chin et al., 1998). Mutation of the analogous residue in the CB2receptor (K109) to alanine or arginine resulted in fully functional CB2 receptors with all ligands tested.
Because the K109R mutation preserves the charge at position 3.28(109) within the CB2 transmembrane bundle, the lack of an effect on ligand binding and efficacy may not be surprising. However, the fact that the K109A mutation in CB2produces results very different from those of the corresponding CB1 mutation is an unexpected, intriguing, finding. There are two possible explanations for this result. First, lysine 3.28(109) in the wild-type CB2 receptor is not an interaction site for the cannabinoid receptor agonists tested. Mutation of this residue to alanine, therefore, would not affect ligand binding or the ensuing activation of the receptor. Second, lysine 3.28(109) is a ligand interaction site in wild-type CB2. However, in the absence of this interaction site in the K109A mutant, an alternate binding mode with other residues within the pore of the transmembrane bundle is possible. This alternate binding mode provides sufficient ligand interaction to maintain high-affinity binding and activation of the receptor.
Modeling studies revealed that CP-55,940 adopts a different orientation in CB1 versus CB2. In CB1, K3.28(192) appears to be a key residue for CP-55,940 binding. Mutation of this residue to alanine results in altered ligand depth in the binding pocket and loss of two ligand interaction sites [K3.28(192) and W5.43(279)]. An alternate binding orientation producing greater ligand interaction in the K3.28(192)A mutant was not found. Even though a hydrogen bonding residue [S3.35(199)] exists in CB1, no hydrogen bonding interaction between ligand and S3.35(199) was possible.
The loss of the K3.28(192) and W5.43(279) interactions should result in a significant drop in affinity of CP-55,940 for the K3.28(192)A mutant, because only the N7.45(389) and the hydrophobic binding pocket interactions are retained. However, it is important to note that the model does indicate some residual interactions available to CP-55,940. In their K3.28(192)A study of the CB1 receptor,Song and Bonner (1996) reported no displacement of [3H]WIN-55,212–2 by CP-55,940, HU-210, or anandamide. However, CP-55,940, HU-210, and anandamide were found to inhibit cAMP accumulation at concentrations in the micromolar range. Thus, some interaction between the K3.28(192)A mutant and CP-55,940 must occur, albeit at higher concentrations of agonist.
In contrast to results for CB1, K3.28 was not found to be key to the binding of CP-55,940 in CB2. Instead, the hydrogen bonding cluster (S3.31(112)/T3.35(116)), a feature unique to CB2, appears important. The retention of three hydrogen bonding sites and the hydrophobic binding pocket interaction for CP-55,940 in the K3.28(109)A mutant is consistent with the retention of high-affinity binding (with a slight decrease relative to CB2) exhibited by CP-55,940 in the K3.28(109)A CB2mutant. Furthermore, as the presence of a hydrogen bonding residue only at S3.35(199) in CB1 did not result in ligand interaction, modeling studies suggest that amino acid position 3.31 may be key to the binding affinity of CP-55,940 at CB2.
To test the hypothesis that Ser3.31(112) provides a critical hydrogen-bonding interaction in K109A, we constructed the K109AS112G double mutant. The K109AS112G mutants lost the ability to bind CP-55,940, Δ9-THC, and anandamide, and retained affinity for WIN 55,212–2 as well as a related aminoalkylindole, JWH-015. These data support the molecular model. However, although the K109AS112G double mutant retained binding affinity for WIN 55,212–2, receptor activation by this compound was drastically reduced. This suggested that the K109AS112G mutants were not properly coupled. Furthermore, the number of receptor binding sites was reduced in the K109AS112G cell lines compared with the other mutants. Therefore, immunofluorescence studies were conducted to define the pattern of CB2 receptor expression in cells transfected with the K109A mutant and the K109AS112G double mutant.
The immunofluorescence data indicated that there was a differential pattern of CB2 expression within HEK-293 cells that was dependent on the presence of the introduced point mutations. Wild-type CB2 and K109A-transfected cells exhibited a pattern of immunostaining consistent with membrane-associated protein expression on the cell surface. A characteristic of these cells was a punctate cytoplasmic staining pattern consistent with vesicular transport of receptor from the perinucleus to the cell surface and their contextual association with membranes at those sites. These findings are supported pharmacologically by the ligand binding and cAMP data that revealed comparable affinity and absolute levels of receptors for the K109A mutant when compared with the wild-type CB2-expressing cell line. Collectively, these data suggest that the K109A mutant CB2 receptors are processed, transported, and compartmentalized in a normal fashion consistent with functional coupling. On the other hand, cells transfected with the K109AS112G double mutant demonstrated an accumulation of immunostaining in a perinuclear arrangement. This pattern of localization suggests that the double-mutant receptor may undergo abortive transport from the Golgi apparatus or the endoplasmic reticulum. Such abortive translocation could be a consequence of a protein that is improperly folded and/or inappropriately integrated into cellular membranes. Binding data support this hypothesis as the double mutant was unable to bind CP-55,940, anandamide, and Δ9-THC and exhibited less than 10% inhibition of cAMP. Nonetheless, binding of WIN 55,212–2 and a related indole was retained. The single versus the double mutations did not result in apparent differential processing of CB2, because the anti-CB2 antibody directed against carboxy terminal amino acid residues (320–336) elicited immunoreactive product in both types of transfected cells. Thus, both mutants allowed for expression of a mostly mature, if not fully mature protein. These results suggest that the major difference in expression between the single and double mutants is at the level of receptor translocation and/or compartmentation.
The overall aim of this mutagenesis research is to elucidate important molecular components of the cannabinoid pharmacophore. This knowledge may lead to the design of more specific cannabinoid receptor ligands, which could offer increased therapeutic activity and decreased side effects. Mutation of the conserved lysine residue in the third transmembrane domain of the CB2 receptor revealed discrimination between receptor subtypes, and provided new insight into the molecular structure of the cannabinoid receptors.
Footnotes
-
Send reprint requests to: Dr. Mary Abood, P.O. Box 980524, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond VA 23298-0524. E-mail:mabood{at}hsc.vcu.edu
-
This Council for Tobacco Research Award was supported by the following grants: DA-05274 and DA-09978 and the CTR 4482 (to M.E.A.); DA-07027 (to S.D.M.); DA-05832, DA-05274 and DA 09158 (to G.A.C); and DA-03934 (to P.H.R.).
- Abbreviations:
- CB1
- central cannabinoid receptor
- CB2
- peripheral cannabinoid receptor
- Δ9-THC
- (−)-Δ9-tetrahydrocannabinol
- CP-55
- 940, (−)-3-[2-hydroxyl-4-(1,1-dimethylheptyl)phenyl]-4-[3-hydroxyl propyl] cyclohexan-1-ol
- WIN-55
- 212–2, (R)- (+)-[2,3-dihydro-5-methyl-3-[(4-morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl](1-naphthalenyl)methanone
- JWH-015
- 2-methyl-3-napthoyl-N-propylindole
- HU-210
- (−)-11-hydroxy-Δ8-tetrahydrocannabinol-dimethylheptyl
- BSA
- bovine serum albumin
- DMH
- dimethylheptyl
- NAH
- northern aliphatic hydroxyl
- SAH
- southern aliphatic hydroxyl
- Hx
- helix
- SD
- steepest descent
- CG
- conjugate gradient
- Received May 14, 1998.
- Accepted December 9, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}