


Abstract
The recently identified γ-aminobutyric acid type B receptors (GABABRs) share low sequence similarity with the metabotropic glutamate (mGlu) receptors. Like the mGlu receptors, the N-terminal extracellular domain (NTED) of GABABRs is proposed to be related to bacterial periplasmic binding proteins (PBPs). However, in contrast to the mGlu receptors, the GABABRs lack a cysteine-rich region that links the PBP-like domain to the first transmembrane domain. This cysteine-rich region is necessary for the PBP-like domain of mGlu receptors to bind glutamate. To delimit the ligand-binding domain of GABABRs, we constructed a series of chimeric GABABR1/mGluR1 and truncated GABABR1 receptor mutants. We provide evidence that despite the lack of a cysteine-rich region, the NTED of GABABRs contains all of the structural information that is necessary and sufficient for ligand binding. Moreover, a soluble protein corresponding to the NTED of GABABRs reproduces the binding pharmacology of wild-type receptors. This demonstrates that the ligand-binding domain of the GABABRs can correctly fold when dissociated from the transmembrane domains.
The diverse actions of γ-aminobutyric acid (GABA) neurotransmission are mediated by a variety of receptors. They can be classified into ionotropic GABAA/C and metabotropic GABAB receptors (GABABRs) on the basis of physiological, pharmacological, and molecular criteria. GABAA and GABAC receptors comprise an intrinsic Cl− channel and are responsible for fast inhibition (Bormann and Feigenspan, 1995; Smith and Olsen, 1995). GABABRs act on a slower time scale and inhibit neuronal activity through second messenger systems that regulate transmitter release and the activity of Ca2+ and K+ channels (Kerr and Ong, 1995; Bettler et al., 1998).
The cloning of GABABRs revealed a topological organization typical of G protein-coupled receptors, with seven transmembrane domains (TMDs), an N-terminus facing the extracellular space and a C-terminus protruding into the cell (Kaupmann et al., 1997). GABABRs share low sequence similarity with mGlu, the Ca2+-sensing (CaS) receptor, and a family of vomeronasal and putative taste receptors but not with other G protein-coupled receptors (Kaupmann et al., 1997; Bettler et al., 1998;Hoon et al., 1999). The overall sequence identity between GABABRs and CaS, mGlu, vomeronasal, and taste receptors is between 18 and 23%. A striking difference between GABABRs and the other members of this gene family is the lack of the highly conserved cysteine-rich region in the N-terminal extracellular domain (NTED).
The two first GABABRs isolated are the splice variants GABABR1a (R1a) and GABABR1b (R1b) with a molecular mass of 130 and 100 kDa, respectively. They differ at the N-terminus where R1a consists of a tandem pair of consensus sequences for the complement protein (CP) module [also called short consensus repeat (SCR) and Sushi domain] that is missing in R1b (Bettler et al., 1998;Hawrot et al., 1998). A second structurally related GABABR, GABABR2 (R2), has been identified (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998; Kuner et al., 1999; Ng et al., 1999). The R2 receptor does not bind available GABABR antagonists with measurable affinity and assembles with R1a and R1b into functional heteromeric complexes. The R1a and R1b variants share agonist- and antagonist-binding properties, and strikingly, the NTEDs of the smaller R1b and R2 receptors are limited to a region with remote sequence similarity to a family of bacterial periplasmic binding proteins (PBPs;O’Hara et al., 1993; Kaupmann et al., 1997). Standard sequence comparison tools reveal that the similarity with prokaryotic PBPs is more pronounced for GABABRs than for mGlu receptors, where a homology with PBPs has been proposed previously (O’Hara et al., 1993).
Consistent with a structural relationship between the NTED of mGlu receptors and PBP (O’Hara et al., 1993; Costantino and Pellicciari, 1996), it was shown that in chimeric mGlu receptors, the PBP-like domain confers selectivity for agonists (Takahashi et al., 1993). More recent experiments with truncated versions of mGluR1 underscored that this domain is necessary for agonist binding (Okamoto et al., 1998). The binding of mGluR1 ligands to the PBP-like domain requires the cysteine-rich region, a hallmark of the mGlu/CaS/vomeronasal/taste receptor gene family, which is conspicuously absent in GABABRs. The NTED is responsible for the covalent, disulfide-linked homodimerization of mGlu and CaS receptors (Bai et al., 1998, 1999; Fan et al., 1998; Okamoto et al., 1998; Ward et al., 1998). The significance of this dimerization process for ligand binding and receptor activation is unclear. Although functional GABABRs form dimeric complexes as well, these heteromeric complexes are not linked through disulfide bridges but instead assemble via C-terminal coiled-coil structures. The low sequence similarity and the structural differences between GABAB and mGlu/CaS receptors indicate that their NTEDs do not necessarily fulfill related functions.
We wanted to examine the role of the NTED of the GABABR1 receptor in ligand binding and therefore constructed a series of chimeric and truncated GABABR mutants to map the ligand-binding domain. We eventually designed a secreted miniprotein that retains wild-type (WT) agonist- and antagonist-binding properties. The availability of a soluble GABABR miniprotein that binds high-affinity antagonists should enable a crystallization of the binding domain, similar to that recently demonstrated for the ionotropic glutamate receptors (Armstrong et al., 1998). A first account of these studies has been published in abstract form (Klix et al., 1998).
Experimental Procedures
Materials.
All GABABR-selective ligands were synthesized in-house. 125I-labeled CGP64213 and 125I-labeled CGP71872 were labeled to a specific radioactivity of >2000 Ci/mmol, and [3H]CGP54626A was labeled to 40 to 60 Ci/mmol (ANAWA AG, Wangen, Switzerland).
Cell Culture, Transfection of Mammalian Cells, Ca2+Imaging, and Electrophysiology.
Spodoptera frugiperda(Sf21) cells were propagated at 27°C in ExCell 401 medium (JRH Biosciences) supplemented with 5% FCS and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin). Trichopulsanicells (High Five/TN5 cells; InVitrogen, Carlsbad, CA) were propagated at 27°C in suspension cultures (with shaking at 120 rpm) in Sf900-II SFM medium (Life Technologies, Basel, Switzerland) supplemented with antibiotics as above. COS1 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% FCS and antibiotics as above. Transient transfections with cDNA constructs were carried out as described previously (Kaupmann et al., 1997). The cells were washed with PBS and fed with fresh medium 15 h after transfection. Cells were harvested for membrane preparation 36 to 40 h after transfection. Electrophysiology usingXenopus oocytes and two electrode voltage-clamp and Ca2+ imaging were as described previously (Mosbacher et al., 1994; Tones et al., 1995).
Antibodies and Immunoblotting.
The C-terminal mGluR1b-specific serum (αmGluR1b) was generated as described previously (Kaupmann et al., 1998). To obtain an antiserum against the N-terminus of GABABRs, we isolated a cDNA fragment encoding amino acids 485 to 523 of R1a (Kaupmann et al., 1997). This fragment is common to R1a and R1b and was amplified by polymerase chain reaction (PCR) using primers that add flankingBamHI (5′) and EcoRI (3′) sites. The PCR product was inserted downstream of the glutathione-S-transferase gene encoded by pGEX-2T (Pharmacia, Freiburg, Germany). Expression and purification of the fusion protein were done as described previously (Malitschek et al., 1998). Polyclonal antibodies against the fusion protein were raised in New Zealand White rabbits. The resulting antiserum, antibody (Ab)176a, was used for immunoblotting as described previously (Malitschek et al., 1998).
Chimeric and Truncated Receptor cDNAs.
We assembled chimeric R1a/mGluR1b as well as truncated R1a and R1b receptors using gene splicing by overlap extension (Horton et al., 1989). Truncated receptor mutants were generated in a single PCR that adds flankingEcoRI (5′) and NotI (3′) sites. In addition, a novel stop codon was added at the 3′ end of truncated cDNA constructs. All constructs were inserted into pcDNAI (InVitrogen). The boundaries of the three chimeric receptors are as follows: aN530, N-terminal 530 residues of R1a spliced to mGluR1b at position 475 (G530/D475); aN550, R1a/GluR1b (G550/T500); and aN587, R1a/mGluR1b (S587/I592). In truncated R1a receptor mutants, the following deletions were introduced: aN1C, amino acid residues 614 to 854; aN7C, residues 589 to 829; and aN1, residues 614 to 960. In truncated R1b mutants, the deletions were bN7C, residues 458 to 700; and bN1, residues 485 to 831. The numbering of amino acid residues is as described previously (Kaupmann et al., 1997). All constructs were verified by sequencing.
Transfer Vectors for Expression in Insect Cells.
PCR using primers that add a stop codon at the 3′ end and includeBamHI (5′) and NotI (3′) sites amplified a fragment encoding the NTED of R1a. The PCR product was inserted into pBacPak8 (Clontech, Palo Alto, CA). The final construct, pBakPak8-sNa, encodes the NTED of R1a (residues 1–588), including its signal peptide. pAcGP67B-sNb encodes the NTED of the mature R1b protein (residues 30–459) preceded by the signal peptide of gp67, a baculovirus secretion signal derived from pAcGP67B (PharMingen, San Diego, CA).
Generation of Recombinant Baculovirus.
Sf21 cells were transfected with BacPAK6 (Clontech) and pAcGP67B-sNb or pBakPAK8-sNa using Bacfectin reagent (Clontech) according to the manufacturer’s protocol. The cell culture supernatant of the transfected cells was harvested 5 days after transfection and used for a plaque assay at a 1:10 and 1:100 dilution. For each construct, we analyzed 10 plaques for production of recombinant protein using immunoblotting. Two subsequent rounds of plaque purification produced clonal viruses. For protein production, High Five cells were infected at a multiplicity of infection of 2. Cell culture supernatants were harvested 65 h after transfection. The supernatant was cleared by centrifugation at 5000g for 10 min, sterilized by filtration, and used for protein purification.
Protein Purification.
Supernatants of infected TN5 cells were concentrated by ultrafiltration in a stirred cell (Amicon, Grace & Co., Beverly, MA) with a cellulose membrane (Amicon YM10 membrane, cut-off >10 kDa). Simultaneous diafiltration with Krebs-Henseleit buffer (118 mM NaCl, 20 mM Tris-Cl, 5.6 mM glucose, 1.2 mM KH2PO4, 1.2 mM MgSO4, 4.7 mM KCl, 1.8 mM CaCl2, pH 7.4) kept the pH in the solution constant. Proteins in the concentrated supernatant were adsorbed to Concanavalin A Sepharose (Pharmacia). The beads (1.5 ml; 1:1 slurry in a buffer containing 20 mM Tris · HCl, 0.5 M NaCl, pH 7.4) were incubated with 50 ml of the supernatant at 4°C for 4 h under continuous shaking and then washed twice with Krebs-Henseleit buffer. Protein was eluted from the beads overnight at 4°C with 1 M of α-d-methylmannoside. The affinity purified material was concentrated in half of the initial volume of Krebs-Henseleit buffer and used for photoaffinity cross-linking and radioligand-binding experiments.
Photoaffinity Labeling and Radioligand Binding Assays.
Radioligand-binding experiments with membrane-anchored mutants and125I-labeled CGP71872 photoaffinity labeling were carried out as described previously (Kaupmann et al., 1997). For the soluble secreted proteins, the binding assay was performed in a 96-well filtration plate with a mixed cellulose-ester membrane at the bottom of the well (MHAB N45; Millipore, Bedford, MA).125I-labeled CGP64213 (0.1 nM) and competitor compounds were added to the protein solution and incubated at room temperature for 1 h. Control experiments were done in the presence of unlabeled GABAB antagonist CGP54626 (Kaupmann et al., 1997). Unbound radioactivity was removed under vacuum using the MultiScreen manifold (Millipore). Proteins retained by the nitrocellulose membrane were washed twice with 400 μl of ice-cold Krebs-Henseleit buffer. After drying the membranes at 50°C for 5 min, scintillation liquid was added, and the plates and the radioactivity were counted. All assays were performed in triplicate.
Results
Characterization of a GABABR1 Antiserum Directed toward N-Terminal Epitopes.
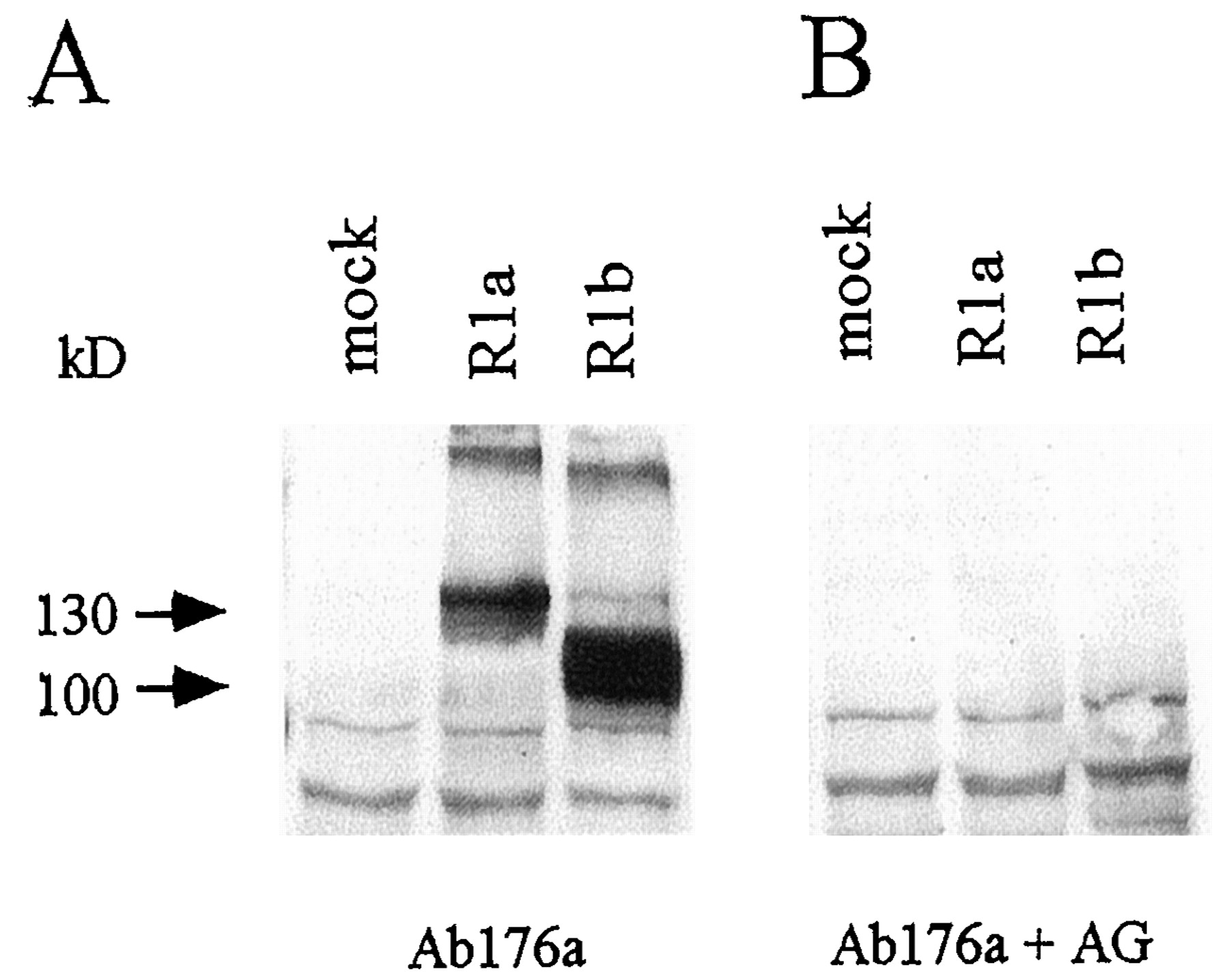
To be able to analyze the expression of the mutant GABABRs, we produced Ab176a, a polyclonal antiserum directed against a N-terminal epitope shared between R1a and R1b (residues 485–542 of R1a). The antiserum was characterized on immunoblots of membranes prepared from COS1 cells expressing GABABRs (Fig. 1A). As expected (Kaupmann et al., 1997), the antiserum detects proteins with an apparent molecular mass of 130 and 100 kDa in cells expressing R1a and R1b, respectively. These proteins are not detected in membranes prepared from mock-transfected COS1 cells. No immunoreactivity is observed after preincubation of the antiserum with purified GST/ GABABR1 fusion protein, which is the immunizing antigen (Fig. 1B). Additionally, Ab176a detects high-molecular-weight bands of approximately 200 and 185 kDa in R1a- and R1b-expressing cells, respectively (Fig. 1A). The high-molecular-weight protein species are recognized not only by the N-terminal antiserum Ab176a but also by the C-terminal antiserum Ab174.1 (Malitschek et al., 1998; data not shown). This clearly indicates that the high-molecular-weight material relates to R1a and R1b protein. For several reasons, it is unlikely that the high-molecular-weight proteins represent disulfide-linked homodimers. Recent reports demonstrate that GABAB receptors do not homodimerize (White et al., 1998, Kuner et al., 1999). Consistent with these findings, the high-molecular-weight proteins run on SDS-polyacrylamide gel electrophoresis (PAGE) significantly below the molecular mass of expected receptor homodimers [(200 kDa (R1a) and 185 kDa (R1b) instead of 260 and 200 kDa, respectively]. Moreover, the high-molecular-weight proteins are also detectable in the presence of 100 mM dithiotreitol or β-mercaptoethanol (data not shown). Significantly, we observe only the high-molecular-weight material when using heterologous expression systems and never observe them when analyzing neuronal GABAB receptors. The high-molecular-weight proteins are not labeled by125I-labeled CGP71872, suggesting that they form aggregates that are inaccessible to the photoaffinity label. We expect that the high-molecular-weight forms represent SDS-resistant receptor aggregates, resulting from the high expression level of recombinant protein in COS1 cells.

Characterization of Ab176a. The antiserum that is directed to an N-terminal epitope common to R1a and R1b (Ab176a) was characterized on Western blots. Cell lysates of COS1 cells transiently transfected with vector DNA (mock) or R1a or R1b expression plasmids were subjected to SDS-PAGE (6% gel), blotted, and immunostained. A, Ab176a detects R1a and R1b as proteins of 130 and 100 kDa, respectively. No cross-reactivity is observed in mock-transfected cells. B, when the Ab176a is preadsorbed to the immunizing antigen (AG), immunostaining of the 130- and 100-kDa proteins is prevented.
Construction, Expression, and Pharmacology of Chimeric mGluR1/GABABR1 Receptor Mutants.
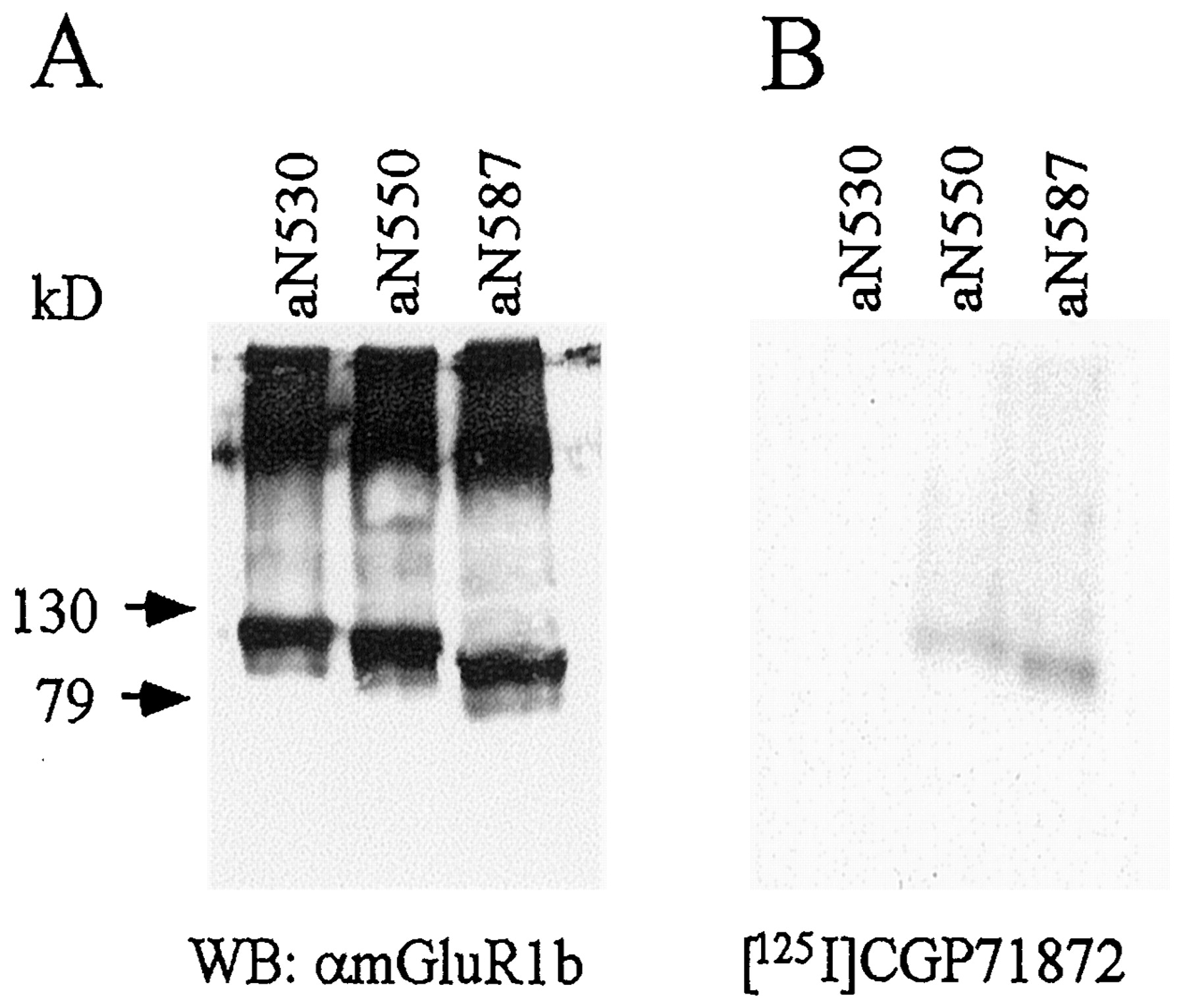
To investigate whether the NTED of GABABRs is involved in agonist binding, we constructed chimeric receptors in which the NTED of mGluR1 was replaced with the corresponding region of R1a (Fig.2A). We expected the chimeric receptors to be activated by GABA and to couple to the signal transduction pathway of mGluR1 because intracellular regions determine the coupling preference of chimeric mGlu receptors (Takahashi et al., 1993; Tones et al., 1995; Parmentier et al., 1998). Because of the low sequence similarity between GABABRs and mGluR1, the localization of suitable boundaries is difficult. We therefore made three constructs in which the chimeric receptors contain residues 1 to 530 (aN530), 1 to 550 (aN550), and 1 to 587 (aN587) of the NTED of R1a; the C-terminal part is derived from downstream mGluR1 sequences (Fig.2A). We tested these chimeric receptors in assay systems normally used to study mGluR1 function. All the chimeric receptors do not activate Ca2+-activated Cl−channels in Xenopus oocytes and do not increase the intracellular Ca2+ concentration in transfected COS1 cells (data not shown). This precluded a pharmacological analysis of the chimeras using functional read-outs. The chimeric receptors are correctly expressed as shown on immunoblots with antibodies specific for the C terminus of mGluR1b (Fig. 3A). Moreover, aN550 and aN587 are able to bind the photoaffinity ligand125I-labeled CGP71872 (Fig. 3B), whereas aN530 is devoid of binding activity. This demonstrates that aN550 and aN587 contain the molecular determinants necessary for antagonist binding. For aN587, we measured the IC50 value for GABA and the antagonist CGP64213 using the standard125I-labeled CGP64213-binding assay (Kaupmann et al., 1997). Significantly, the IC50 value of CGP64213 at WT receptors (2.3 nM ± 0.1, n = 3; Table 1) and aN587 (1.7 nM ± 0.1,n = 3) is virtually identical. However, the chimeric receptor exhibits a slightly increased affinity for GABA (IC50 = 22.9 ± 2.4 μM for WT,n = 3, Table 1; IC50 = 4.7 ± 0.4 μM for aN587, n = 3).
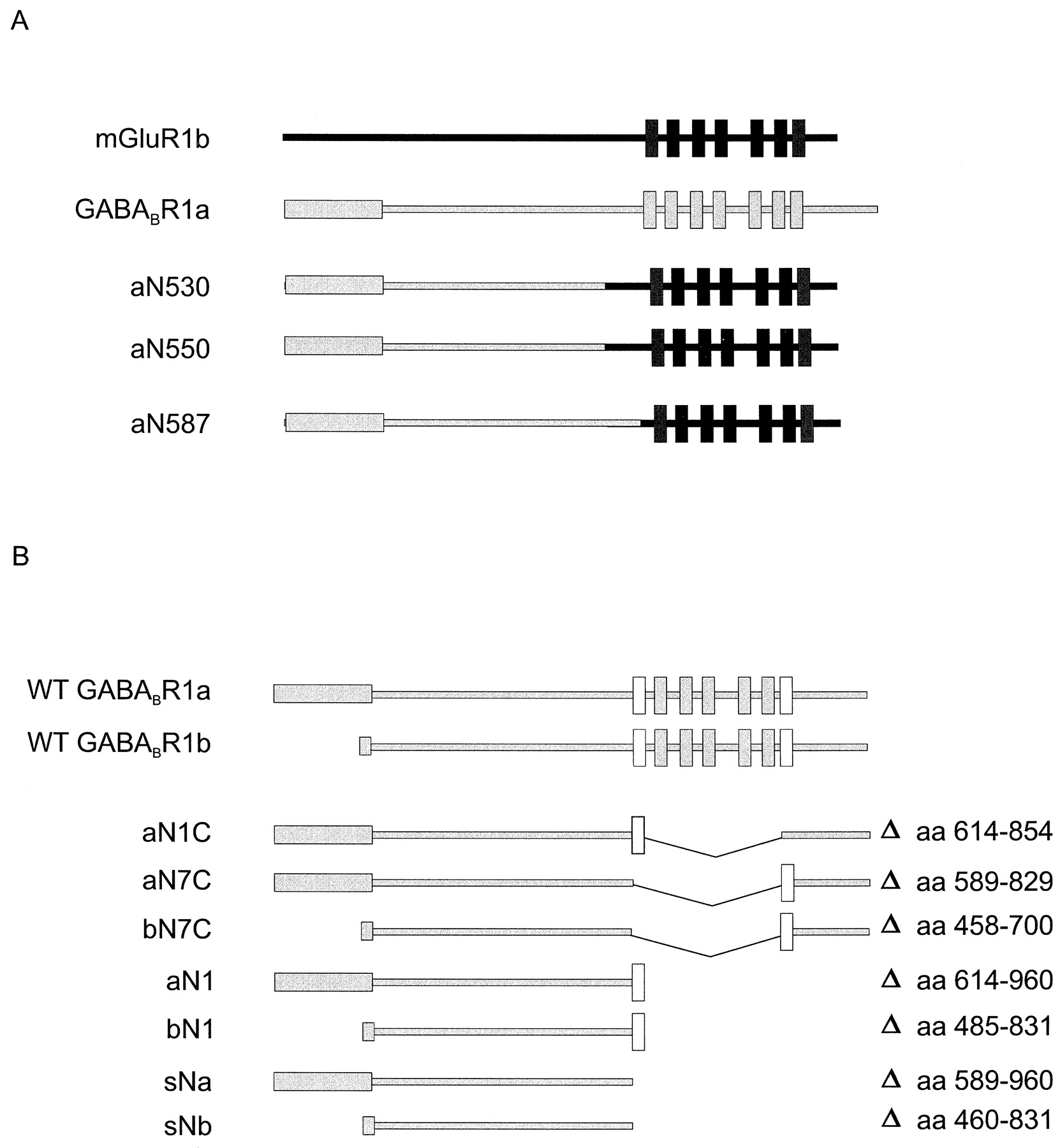
Schematic representation of WT and mutant receptors. A, schematic representation of chimeric GABABR1/mGluR1 receptors. Amino acids 1 to 530 (aN530), 1 to 550 (aN559), and 1 to 587 (aN587) of R1a were fused to the corresponding amino acids of mGluR1b. B, schematic representation of membrane-anchored and soluble truncated R1a and R1b receptors. Deleted (thin lines) amino acid residues are indicated; white boxes represent TMD1 and TMD7.

Expression and antagonist binding of chimeric R1a/mGluR1b receptors. A, COS1 cells were transiently transfected with the expression constructs for the chimeric GABABR1/mGluR1 receptors aN530, aN550, and aN587. Membrane proteins were separated on 6% SDS gels, blotted, and immunostained with an antibody directed against mGluR1b (αmGluR1b). B, membranes of transfected cells were photoaffinity labeled with 125I-labeled CGP71872 and subjected to SDS-PAGE on a 6% gel. Labeled proteins were detected by autoradiography. In agreement with the above data, specific125I-labeled CGP64213 binding was detected with chimeric receptors aN550 and aN585 but not with aN530.
Inhibition of [125I]-labeled CGP64213 binding to chimeric and truncated GABAB receptors by agonists and antagonists
Construction, Expression, and Pharmacology of Membrane-Anchored GABABR Mutants.
We analyzed a possible influence of the transmembrane and C-terminal domains of GABABRs on agonist and antagonist binding by constructing several truncation/deletion mutants (Fig. 2B). These constructs correspond to the NTED of R1a or R1b preceding either TMD1 (aN1C, aN1, bN1) or TMD7 (aN7C, bN7C), with (aN1C, aN7C, bN7C) or without (aN1, bN1) the intracellular C-terminal tail (Fig. 2B). As shown with immunoblots, all receptor proteins are expressed in the membrane fraction of transiently transfected COS1 cells (Fig.4A). Significantly, deletion of large parts of the WT protein does not impair stability of the polypeptides. We analyzed the-binding properties of the mutant receptors using the photoaffinity ligand 125I-labeled CGP71872 (Fig.4B). All truncated receptors bind to 125I-labeled CGP71872, and photaffinity labeling is, as expected, prevented in the presence of the unlabeled GABABR antagonist CGP54626A (Fig. 4B). Mutants aN1C and aN7C show that the substitution of TMD1 for TMD7 does not significantly alter recombinant protein expression levels and 125I-labeled CGP71872-binding properties. Similarly, deletion of the large C-terminal domain in aN1 and bN1 does not seem to influence125I-labeled CGP71872-binding activity.

Expression and photoaffinity ligand binding of truncated GABABRs. A, COS1 cells were transiently transfected with the expression constructs for the WT and truncated, membrane-anchored R1a (aN1C, aN7C, and aN1) and R1b (b7C and bN1) receptors. Membrane proteins were separated on 6% SDS gels, blotted, and immunostained with Ab176a. B, membranes of transfected cells were photoaffinity labeled with 125I-labeled CGP71872 in the absence (−) or presence (+) of 1 μM unlabeled CGP54626A and subjected to SDS-PAGE on a 6% gel. Labeled proteins were detected by autoradiography. For recombinant GABAB proteins, the main band seen on immunoblots does not always closely agree in molecular mass with the main band seen in photoaffinity labeling experiments. It appears that with the crude membrane preparation used in these studies, the photoaffinity ligand labels a minor fraction of protein that can be detected with antibodies. Most likely, this is due to the fact that antibodies will also detect the bulk of incompletely processed proteins present on internal membranes. Not fully maturated proteins are unlikely to adapt a structure that will allow the binding of the photoaffinity ligand. Alternatively, photoaffinity labeling may change the migration properties of proteins on SDS-PAGE. Proteins at various maturation stages may also provide an explanation for the multiple bands on immunoblot seen with some of the mutants (e.g., bN1).
The antagonist-binding affinities of the truncated receptor proteins match closely the affinities reported for WT receptors (Fig.5, Table 1). In contrast, the binding affinity for agonists is increased by a factor of 10 when comparing mutant with WT receptors (Fig. 5, Table 1). No significant differences in the IC50 values are observed between different mutant proteins. Clearly, all molecular determinants necessary for ligand binding reside within the NTED of the receptor. The C-terminal part of the molecule can be removed, and TMD1 and TMD7 can be exchanged without significantly influencing binding properties. In accordance with earlier observations with native (Malitschek et al., 1998) and recombinant (Kaupmann et al., 1997) receptors, the unique N-terminal sequences of R1a and R1b do not measurably alter the pharmacological properties of the mutant proteins.

Binding pharmacology of the aN1 (A) and bN1 (B) mutants transiently expressed in COS1 cells. Inhibition of125I-labeled CGP64213 binding to aN1 and bN1 by the agonists GABA, l-baclofen, and 3-aminopropylphosphinic acid and the antagonists CGP64213 and SCH50911. Results are shown from typical experiments performed in triplicate. Bars indicate S.E.M. The curves were fitted using nonlinear regression (Prism; GraphPAD Software, San Diego, CA).
Construction, Expression, and Pharmacology of a Soluble Ligand-Binding Domain.
The previous experiments show that the NTED of GABABRs is responsible for ligand binding. To analyze whether the NTED can fold independently of any TMD, we produced soluble R1a (sNa) and R1b (sNb) miniproteins in insect cells using recombinant baculovirus (Fig. 6A). The sNb construct using the viral secretion signal was more efficient in releasing recombinant protein into the medium, although the mammalian R1a signal peptide in sNa was also effectively used as a secretion signal in insect cells (Fig. 6A). Glycosylation most likely accounts for the difference between the calculated molecular mass of the sNa (63 kDa) and sNb (45 kDa) proteins and the apparent molecular mass in SDS-PAGE of 80 and 60 kDa, respectively (Fig. 6A). We took advantage of the extensive glycosylation of the sNa and sNb proteins to affinity purify them from the medium using Concanavalin A Sepharose. No further enrichment was necessary to perform 125I-labeled CGP64213- and 125I-labeled CGP71872-binding studies with sNa and sNb. Clearly, the sNa and sNb proteins bind to125I-labeled CGP71872 (Fig. 6B). The agonist- and antagonist-binding affinities of the soluble sNa and sNb proteins are similar to those of the membrane-anchored truncated receptors (Table1). Similar to the membrane-anchored receptors, the agonist-binding affinities of the soluble proteins are slightly increased compared with the WT receptors. It is concluded that the NTED of GABABRs contains a strong intrinsic fold that confers binding and ligand recognition selectivity in the absence of integral membranous domains.

Expression and antagonist binding of soluble N-terminal ligand-binding domains. Supernatants of TN5 insect cells infected with recombinant baculovirus were concentrated and purified as described (see Experimental Procedures). A, the enriched soluble proteins were separated on 7.5% SDS gels, blotted, and immunostained with Ab176a. B, purified sNa and sNb were photoaffinity labeled with125I-labeled CGP71872 in the absence (−) or presence (+) 1 μM unlabeled CGP54626A, precipitated with trichloroacetic acid, and subjected to SDS-PAGE on a 7.5% gel. Proteins labeled with125I-labeled CGP71872 were detected by autoradiography. The radiolabeled sNa and sNb proteins do not correspond to major bands on Coomassie blue-stained SDS-PAGE.
Discussion
We found that the NTED of GABABRs is both necessary and sufficient for agonist and antagonist binding. Our study thus reaches the same conclusions as recent studies on the structure of mGlu receptors (O’Hara et al., 1993; Takahashi et al., 1993; Okamoto et al., 1998) and demonstrates that the molecular determinants of GABA and l-glutamate binding reside in similar domains. This further strengthens an evolutionary relationship between mGlu and GABAB receptors, despite the lack of absolutely compelling sequence homology. As already suggested for the mGlu receptors (O’Hara et al., 1993), the binding domain of GABABRs also shares a remote sequence similarity with a family of PBPs. Structural models for the PBPs thus may apply not only to mGlu receptors but also to GABABRs. Accordingly, it is expected that GABA is accommodated in a groove between two globular domains that presumably fold like the lobes of PBP. The sequence similarity maps the PBP-like domain of R1a to amino acids 164 to 550. This hypothesis has been validated in a separate study using molecular homology modeling and site-directed mutagenesis (Galvez et al., 1999). Strikingly, chimera aN530 with a boundary located within the PBP-like domain (at amino acid G530 of R1a) does not bind 125I-labeled CGP71872. This provides further experimental support for the PBP-like structure of the binding site, as the disruption of the fine architecture of the domain is likely to be responsible for the loss of radioligand-binding affinity.
Nine cysteine residues are closely spaced in the NTED of the mGlu, CaS, vomeronasal, and taste receptors, a part of the protein referred to as the cysteine-rich region. This cysteine-rich region links the leucine-binding protein-like domain to the first TMD. Strikingly, such a cysteine-rich region is missing in the GABABRs. The role of the cysteine-rich region in receptor function is not fully understood. Studies to define the role of specific cysteines in the CaS receptor have indicated that all nine cysteines are essential for proper receptor trafficking and function (Fan et al., 1998). Similarly, it has been shown that the leucine-binding protein-like domain of mGluR1 binds [3H]quisqualate only when produced as a soluble protein that encompasses the cysteine-rich region (Okamoto et al., 1998). This indicates a critical role of the cysteine-rich region for proper folding and/or surface translocation. Interestingly, the nine cysteines are not implicated in the formation of intermolecular disulfide bridges that stabilize the mGlu receptor homodimers (Fan et al., 1998). In contrast to the results obtained with CaS and mGlu receptors, the PBP-like core of GABABRs can fold correctly and retains ligand-binding properties in the absence of any additional sequence elements. The lack of a cysteine-rich region in the soluble GABAB miniprotein should aid the production and refolding of a bacterial protein to crystallize the binding domain.
It is remarkable that none of the chimeric R1a/mGluR1 receptors are functional. Clearly, the chimeric proteins aN550 and aN587 bind the photoaffinity ligand, and the failure in responding to GABA is not due to impaired binding. Competition experiments showed that aN587 has almost WT affinities for GABA and CGP64213 (Table 1). Similar functional defects were observed for some chimeric mGluR2/mGluR1 (Takahashi et al., 1993) and mGluR1/DmGluRA receptors (Parmentier et al., 1998). Assembly of R1 with R2 has recently been shown to be a prerequisite for effective transport of GABABRs to the cell surface and for a robust coupling to the G protein (Couve et al., 1998; Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998; Kuner et al., 1999; Ng et al., 1999). The heteromerization of GABABRs is mediated by the C termini of R1 and R2 (White et al., 1998; Kuner et al., 1999). Instead, some mGlu receptors form homodimers via disulfide bridges within their NTED (Romano et al., 1996; Okamoto et al., 1998). The lack of function of the chimeric GABAB/mGluR1 receptors may therefore reflect an inability to form complexes that efficiently target to the effector system. In this respect, it is interesting to note that a coexpression of the chimeric R1/mGluR1 receptors with R2 does not rescue function (data not shown). Nevertheless, our study indicates that the NTED of R1 contains all the structural information that is needed to bind GABAB ligands with a rank order that is identical to that of WT receptors.
It is notable that although there are no differences in the rank order of agonist-binding affinities at WT and mutant receptors, there is a significant increase in agonist affinity at all mutant receptors (Fig.5, Table 1). Significantly, this also is a feature of the soluble sNa and sNb proteins. An increased agonist affinity compared with the WT receptor has also been reported for the soluble mGluR1-binding domain (Okamoto et al., 1998). It is apparent that although the NTED determines the rank order of agonist potency, other receptor regions (i.e., the TMDs) also influence agonist affinity. We assume that the high-affinity agonist site observed in the mutant proteins represents an alternate conformational state of the low-affinity site present in WT R1a and R1b receptors. A possible reason for the increase in agonist affinity is that the binding domain is locked in a high-affinity state due to the structural constraints imposed by the mutation. In a simple model, the GABA-binding site can assume distinct affinity states reflecting the conformational changes associated with receptor activation. This causes the allosteric coupling of a spatially separated regulatory G protein at the cytoplasmic side. Alternatively, as has been suggested for mGlu receptors (Pin and Bockaert, 1995), GABA binding may induce a conformation that allows the NTED to interact with a binding pocket situated within the TMDs. Such an interaction may result in a decrease in agonist affinity. According to such a model, and in agreement with the data herein, the TMDs do not influence the affinity of antagonists because antagonists are not expected to promote the interaction between the binding and the membrane-spanning domains. Crystallization of the GABA-binding site with and without ligand would certainly facilitate the understanding of the conformational changes involved in intramolecular signaling. With our demonstration that a soluble form of the receptor is still able to bind GABAergic ligands with high affinity, X-ray crystallography of the binding domain now appears feasible.
Footnotes
- Received April 1, 1999.
- Accepted May 7, 1999.
-
Send reprint requests to: Dr. Bernhard Bettler, Novartis Pharma AG, K-125.6.08, Nervous System Research, CH-4002 Basel, Switzerland. E-mail:bernhard.bettler{at}pharma.novartis.com
Abbreviations
- GABA
- γ-aminobutyric acid
- GABABR
- γ-aminobutyric acidB receptor
- R1a
- γ-aminobutyric acidB receptor 1a
- R1b
- γ-aminobutyric acidB receptor 1b
- R2
- γ-aminobutyric acidBreceptor 2
- mGlu
- metabotropic glutamate
- CaS
- calcium sensing
- PCR
- polymerase chain reaction
- PAGE
- polyacrylamide gel electrophoresis
- Ab
- antibody
- TMD
- transmembrane domain
- NTED
- N-terminal extracellular domain
- PBP
- periplasmic binding proteins
- WT
- wild-type
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}