


Abstract
Etoposide (VP-16) is extensively used to treat cancer, yet its efficacy is calamitously associated with an increased risk of secondary acute myelogenous leukemia. The mechanisms for the extremely high susceptibility of myeloid stem cells to the leukemogenic effects of etoposide have not been elucidated. We propose a mechanism to account for the etoposide-induced secondary acute myelogenous leukemia and nutritional strategies to prevent this complication of etoposide therapy. We hypothesize that etoposide phenoxyl radicals (etoposide-O⋅) formed from etoposide by myeloperoxidase are responsible for its genotoxic effects in bone marrow progenitor cells, which contain constitutively high myeloperoxidase activity. Here, we used purified human myeloperoxidase, as well as human leukemia HL60 cells with high myeloperoxidase activity and provide evidence of the following. 1) Etoposide undergoes one-electron oxidation to etoposide-O⋅ catalyzed by both purified myeloperoxidase and myeloperoxidase activity in HL60 cells; formation of etoposide-O⋅radicals is completely blocked by myeloperoxidase inhibitors, cyanide and azide. 2) Intracellular reductants, GSH and protein sulfhydryls (but not phospholipids), are involved in myeloperoxidase-catalyzed etoposide redox-cycling that oxidizes endogenous thiols; pretreatment of HL60 cells with a maleimide thiol reagent, ThioGlo1, prevents redox-cycling of etoposide-O⋅radicals and permits their direct electron paramagnetic resonance detection in cell homogenates. VP-16 redox-cycling by purified myeloperoxidase (in the presence of GSH) or by myeloperoxidase activity in HL60 cells is accompanied by generation of thiyl radicals, GS⋅, determined by HPLC assay of 5,5-dimethyl-1-pyrroline glytathionyl N-oxide glytathionyl nitrone adducts. 3) Ascorbate directly reduces etoposide-O⋅, thus competitively inhibiting etoposide-O⋅-induced thiol oxidation. Ascorbate also diminishes etoposide-induced topo II-DNA complex formation in myeloperoxidase-rich HL60 cells (but not in HL60 cells with myeloperoxidase activity depleted by pretreatment with succinyl acetone). 4) A vitamin E homolog, 2,2,5,7,8-pentamethyl-6-hydroxychromane, a hindered phenolic compound whose phenoxyl radicals do not oxidize endogenous thiols, effectively competes with etoposide as a substrate for myeloperoxidase, thus preventing etoposide-O⋅-induced redox-cycling. We conclude that nutritional antioxidant strategies can be targeted at minimizing etoposide conversion to etoposide-O⋅, thus minimizing the genotoxic effects of the radicals in bone marrow myelogenous progenitor cells, i.e., chemoprevention of etoposide-induced acute myelogenous leukemia.
Etoposide (VP-16), a semisynthetic epipodophyllotoxin, has become one of the most widely used anticancer drugs in the United States since its introduction in 1971 (Slevin, 1991). The tumoricidal effect of etoposide and related epipodophyllotoxins is considered to be dependent on a dual mechanism of DNA strand cleavage via direct inhibition of DNA topoisomerase II (topo II), as well as direct or indirect DNA modification (Corbett and Osheroff, 1993). It is frequently used as a first-line drug for treating small cell lung cancer, germ cell tumors, lymphomas, and, more recently, Kaposi’s sarcoma associated with AIDS. It is also used to treat a variety of leukemias including acute lymphocytic leukemia. Etoposide is used in combination with other antitumor drugs (Slevin, 1991), as well as irradiation (Goss et al., 1993).
In the past 10 years, however, numerous groups have reported that treatment schedules associated with the impressive efficacy of etoposide are also associated with an increased risk of secondary acute myeloid leukemia (AML). This has prompted the removal of this highly effective agent from some treatment regimens (Chen et al., 1996).
Although some studies report that the rate of treatment-related AML and myelodysplastic syndrome is within the range previously reported for alkylator-based regimens that did not include etoposide (Smith et al., 1993), a large majority of data indicates that patients receiving epipodophyllotoxins are at risk for developing secondary leukemia that has features distinct from the syndrome of secondary leukemia associated with alkylating agents (Pui et al., 1991, 1995; Whitlock et al., 1991; Horibe et al., 1993; Sugita et al., 1993; Bokemeyer et al., 1995). In a study of children with acute lymphoblastic leukemia treated with etoposide without alkylating agent therapy or irradiation, overall event-free survival at 4 years was only 79.3 ± 5.1%, with a risk of secondary AML at 4 years of 5.9 ± 3.2% (Winick et al., 1993). Another study evaluated 734 children with acute lymphoblastic leukemia who attained complete remission and received maintenance treatment according to different schedules of epipodophyllotoxin administration. Whereas the overall cumulative risk of AML at 6 years was 3.8%, the subgroups treated twice weekly or weekly had risks of 12.3 and 12.4%, respectively. In the subgroups not treated with epipodophyllotoxins or treated with them only during remission induction or every 2 weeks during continuation treatment, the highest cumulative risk was 1.6% (Pui et al., 1991). Thus, the causative link between treatment of cancer with etoposide and the development of secondary AML in children and adults has been firmly established. Yet, the biochemical mechanisms to explain the high susceptibility of myeloid stem cells to the leukemogenic effects of etoposide has not been elucidated.
Etoposide contains a hindered phenolic ring (E-ring), a critical structural prerequisite for its antitumor activity (Usui and Sinha, 1990). It has been suggested that metabolic activation of etoposide is essential for its cytotoxicity (Sinha and Trush, 1983;Haim et al., 1987; Usui and Sinha, 1990). In particular, cytochrome P-450-dependent monooxygenases, peroxidases, prostaglandin synthetase, and tyrosinase may be involved in the metabolic activation of etoposide. Usui and Sinha (1990) demonstrated that etoposide was significantly more cytotoxic to B-16/F-10 melanoma cells expressing high tyrosinase activity as compared with MCF-7 breast tumor cells with low tyrosinase activity. Phenylthiocarbamide, an inhibitor of tyrosinase activity, selectively decreased etoposide toxicity only in melanoma cells. Based on this data it was suggested that the etoposide oxidation product formed by tyrosinase-catalyzed reaction (via intermediate formation of an etoposide phenoxyl radical) confers an enhanced cytotoxicity.
Our previous studies demonstrated that the phenoxyl radical, the primary one-electron oxidation intermediate of phenol formed by myeloperoxidase in the presence of the cosubstrate, hydrogen peroxide, may contribute significantly to phenol-induced oxidative stress and genotoxicity (Stoyanovsky et al., 1996). Recent studies have demonstrated inhibition of human topo I and II by high concentrations of quinoid metabolites of benzene; however, a remarkably greater sensitivity of the enzymes was detected with phenolic metabolites of benzene in the course of their bioactivation using a peroxidase/H2O2 system. This suggests that free radical intermediates of phenolic oxidation (formed in the presence of peroxidase activity) may contribute to the clastogenic and carcinogenic effects of phenolic compounds through inhibition of topoisomerases (Chen and Eastmond, 1995). We propose a mechanism to account for the secondary AML induced by etoposide in myeloid cells, and, as a direct consequence of this mechanism, nutritional strategies for preventing or ameliorating this complication of etoposide therapy. We hypothesize that the one-electron free radical intermediates (phenoxyl radicals) formed from etoposide by myeloperoxidase-catalyzed oxidative metabolism are primarily responsible for its genotoxic effects and carcinogenicity. This hypothesis explains why genotoxic effects of etoposide are especially great in myeloid progenitor cells, which contain constitutively high myeloperoxidase activity.
According to recent crystallographic data (Fenna et al., 1995) the cleft leading to the active site of myeloperoxidase is small and highly constrained and, hence, could limit the accessibility of large, bulky phenolic compounds to the catalytic site (Day et al., 1999). Hence, a bulky etoposide molecule, with its hindered phenolic E-ring, condensed aromatic part, and sugar moiety, could be a poor substrate for myeloperoxidase. Therefore, in the present work, we initially tested whether etoposide could undergo one-electron oxidation catalyzed by purified myeloperoxidase. Our results demonstrated that etoposide could indeed be used as a substrate by myeloperoxidase. This set the stage for further experiments in which we used human leukemia HL60 cells with high endogenous activity of myeloperoxidase to prove that: 1) etoposide is a substrate of myeloperoxidase that undergoes one-electron oxidation to form the etoposide phenoxyl radical; 2) some intracellular reductants, GSH and protein sulfhydryls (but not phospholipids), undergo oxidative modification during myeloperoxidase-catalyzed recycling of etoposide; 3) ascorbate can compete with thiols for the reduction of the etoposide phenoxyl radical (i.e., can protect thiols against oxidation); and 4) a vitamin E homolog, another hindered phenol whose phenoxyl radicals do not oxidize endogenous thiols, can compete with etoposide as a substrate for myeloperoxidase. Based on our hypothesis and the data presented, strategies can be developed to decrease the carcinogenic potential of etoposide in myeloid cells using nutritional antioxidants.
Materials and Methods
Reagents.
Etoposide, guaiacol, phenol, sodium ascorbate, 5,5-dimethyl-1-pyrroline N-oxide (DMPO), 3-amino-1,2,4-triazole, phenylmethyl-sulphonyl fluoride (PMSF), cetyltrimethylammonium bromide, methanol, diethylentriaminepentaacetic acid (DTPA), deferoxamine mesylate [′-demethylepipodophyllotoxin-9-(4,6-O-ethylidene-β-d-glucopyranosine)] (DFO), hydrogen peroxide, GSH, glucose, HEPES, sodium chloride, magnesium chloride, sodium phosphate, potassium phosphate, dithiothreitol, fatty acid-free human serum albumin, polyoxiethylenesorbitan monolaurate (Tween 20), sodium molibdate, malachite green base, butylated hydroxytoluene, horseradish peroxidase (type VI-A), catalase (from bovine liver, EC 1.11.1.6), myeloperoxidase (from human leukocytes, EC 1.11.1.7), and fetal bovine serum (FBS) were purchased from Sigma Chemical Co. (St. Louis, MO). Chloroform, hexane (HPLC grade), and 2-propanol (HPLC grade) were Purchased from Aldrich Chemical Co. (Milwaukee, WI). ThioGlo1 maleimide reagent was obtained from Covalent Associates, Inc. (Woburn, MA) and dimethyl sulfoxide (DMSO) was obtained from Fisher Scientific Company (Pittsburgh, PA).cis-Parinaric acid (PnA) was purchased from Molecular Probes, Inc. (Eugene, OR). The purity of each lot of PnA was determined by UV spectrometry using the molar extinction ε304 = 80 mM−1cm−1 in ethanol. RPMI 1640 medium was purchased from Life Technologies Laboratories (Grand Island, NY). Iscove’s media was purchased from Irvine Scientific (Santa Ana, CA). 2,2,5,7,8-Pentamethyl-6-hydroxy-chromane (PMC) was a generous gift from Eisai Co. (Tokyo, Japan). PMSF was dissolved in 2-propanol and etoposide was dissolved in DMSO.
Cells and Cell Culture.
HL60 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C under 5% CO2 atmosphere. Cells from passages 25 to 40 were used for the experiments. The density of cells at collection time was 0.5 × 106 cells/ml.
Preparation of Cell Homogenates.
Cells were centrifuged at 1000 rpm for 15 min and pellet was washed with L1210 buffer (10 mM glucose, 115 mM NaCl, 25 mM HEPES, 5 mM KCl, 1 mM MgCl2, and 5 mM NaH2PO4, pH 7.4) three times. The homogenate was prepared by freezing the cells at 77°K and subsequent thawing and resuspension. PMSF (100 μM) and DTPA (100 μM) were added immediately to the cell homogenate. The homogenate of 2 × 106 cells/ml was used for subsequent treatments and measurements.
Peroxidase Activity.
Activity of purified myeloperoxidase and peroxidase activity of HL60 cells were determined using guaiacol oxidation assay (Pinnix et al., 1994). In the case of the purified enzyme, the incubation medium contained DFO (100 μM), guaiacol (15 mM), H2O2 (1 mM) in 100 mM phosphate buffer, pH 7.4. For assays in homogenates (106 cells/ml), the reaction mixture contained 3-amino-1,2,4-triazole (2.5 mM), 0.03% cetyltrimethylammonium bromide, PMSF (100 μM), DFO (100 μM), guaiacol (15 mM), and H2O2 (1 mM). The activity was monitored on a Shimadzu UV160U spectrophotometer by changes in optical density at 470 nm (ε470 = 26.6 mM−1 cm−1). On average, the myeloperoxidase activity of cell homogenates was about 5 nmol guaiacol/min/106 cells at 20°C.
Samples for EPR Measurements.
In experiments with purified myeloperoxidase (in most experiments 2 U/ml), etoposide (400 μM) was incubated in the phosphate buffer (0.1 M, pH 7.4 at 25°C) in the presence of different concentrations of H2O2 as indicated. Generation of radicals in HL60 cell homogenates (2 × 106 cell/ml) was studied in incubation medium that contained etoposide (10–500 μM), 3-amino-1,2,4-triazole (2.5 mM), PMSF (100 μM), DTPA (100 μM). In some experiments (see figure legends), the homogenates were preincubated for 1 min with ThioGlo1 (400 μM) after which H2O2(20–1,000 μM) was added immediately before the measurement and subsequently at 10-min intervals during the measurements.
EPR Spectroscopy.
EPR measurements were performed on a JEOL-RE1X spectrometer at 25°C in gas-permeable Teflon tubings (0.8 mm i.d., 0.013 mm thickness obtained from Alpha Wire Corp. (Elizabeth, NJ). The tube (approximately 8 cm length) was filled with 50 μl of mixed sample, folded into quarters, and placed in an opened 3.0 mm i.d. EPR quartz tube. Etoposide phenoxyl radical spectra were recorded under following conditions: 335.7 mT, center field; 2 mT, sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 2 min, time scan. PMC phenoxyl radical spectra were recorded under the following conditions: 335.7 mT, center field; 5 mT, sweep width; 0.2 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 2 min, time scan. Spectra of ascorbyl radicals were recorded under the following conditions: 335.5 mT, center field; 0.5 mT, sweep width; 0.05 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 1 min, time scan. Spectra of DMPO-glytathionyl (GS) adducts were recorded under the following conditions: 335.7 mT, center field; 8 mT, sweep width; 0.1 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 1 min, time scan.
The time course of etoposide and ascorbate radical EPR signals was obtained by repeated scanning of the field (0.15 mT, sweep width; 335.7 mT, center field) corresponding to a part of the EPR signal (for etoposide radical) or the entire EPR signal (for ascorbate radical). Other instrumental conditions were: 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 20 s, time scan; internal mode of recording.
Measurements of Peroxidation in Phospholipids Metabolically Labeled with PnA.
PnA was incorporated into HL60 cells by addition of its human serum albumin (hSA) complex as previously described (Ritov et al., 1996). Briefly, cells in log phase of growth were rinsed twice with L1210 buffer and resuspended in this buffer to give a cell density of 106 cells/ml. PnA–SA complex was added to the cell suspension to give a final concentration of 2 μg PnA/ml and cell were incubated at 37°C in the dark under aerobic conditions. At the end of incubation period, the cells were consequently washed twice with L1210 buffer containing fatty acid-free hSA (0.5 mg/ml) and without hSA, respectively.
Treatment of HL60 Cell Homogenates with Etoposide and H2O2.
Homogenates were incubated in the presence or absence of etoposide and H2O2 for 60 min at 37°C in aerobic conditions in the dark. Etoposide (10 or 50 μM) was added 15 min before addition of H2O2. Equal amounts of H2O2 (25 μM) were added every 10 min during incubation. After 60 min of incubation, lipids were immediately extracted using a slightly modified Folch procedure. Extracted lipids were dissolved in 0.2 ml of 2-propanol:hexane/water (4:3:0.16, v/v/v). Lipid phosphorus was determined using a slightly modified method previously described (Chalvardjian and Rudnicki, 1970).
HPLC Analysis of Cell Lipids.
Lipid extracts were separated by HPLC using an ammonium acetate gradient essentially as described previously (Geurts van Kessel et al., 1977). The lipid extract was applied to a 5-μm Supelcosil LC-Si column (4.6 × 250 mm) equilibrated with a mixture of 1 part of solvent A, 2-propanol/hexane/water (57:43:1, v/v/v), and 9 parts of solvent B, 2-propanol/hexane/40 mM aqueous ammonium acetate (57:43:10 v/v/v), pH 6.7. The column was eluted during the first 3 min with a linear gradient from 10% solvent B to 37% solvent B, 3 to 15 min isocratic at 37% solvent B, 15 to 23 min linear gradient to 100% B, and then 23 to 45 min isocratic at 100% solvent B. The solvent flow rate was 1 ml/min. A Shimadzu high-performance liquid chromatograph (LC-600) equipped with fluorescence detector (RF-551) was used. The effluent was monitored by fluorescence of PnA by emission at 420 nm after excitation at 324 nm. Fluorescence data were processed and stored in digital form with Shimadzu EZChrom software.
Assays of Glutathionyl Radical Adducts (DMPO-GS nitrone) in Model System Containing Purified Myeloperoxidase and in HL60 Cell Homogenates.
We used HPLC measurements to detect etoposide phenoxyl radical-induced formation of EPR-silent adducts of a spin trap, DMPO, with glutathionyl radical: DMPO-GS nitrone. In a model system, myeloperoxidase (1 U/ml) was incubated with GSH (25 mM), H2O2, 3-amino-1,2,4-triazole (2.5 mM), DTPA (100 μM), and DMPO (100 mM) in the presence or absence of etoposide (100 and 500 μM) in L1210 buffer, pH 7.4, at 37°C for 1 h. Etoposide was added to the incubation medium before addition of myeloperoxidase and H2O2. H2O2 (200 μM) was added every 10 min during 1 h of incubation. After incubation, DMPO-GS nitrone was determined by HPLC as described earlier (Stoyanovsky et al., 1996).
Similarly, HL60 cells treated with etoposide and H2O2 were assayed for formation of DMPO-GS nitrone using the HPLC procedure (Stoyanovsky et al., 1996). Cells (5 × 106 cells/ml) were preincubated with DMPO (100 mM) for 2 min and then incubated with H2O2 (25 μM H2O2 was added every 10 min in the course of incubation), 3-amino-1,2,4-triazole (2.5 mM), and DTPA (100 μM) in the presence or absence of etoposide (100 μM) for 1 h at 37°C in L1210 buffer in the dark. Etoposide was added 15 min before any other additions were made. After incubation, cells were washed twice with L1210 buffer and DMPO-GS nitrone was determined by HPLC.
Fluorescence Assay of GSH and Protein SH Groups in HL60 Cell Homogenates.
Cell homogenates were incubated at 25°C in L1210 buffer, pH 7.4, containing 3-amino-1,2,4-triazole (2.5 mM), DTPA (100 μM) in the presence or absence of etoposide (500 μM) and H2O2 (100 μM). Aliquots of cell homogenates were then used for GSH and protein SH groups determination. Total protein sulfhydryl concentration and GSH content in cell homogenates was determined using ThioGlo1, a maleimide reagent, producing a highly fluorescent product on its reaction with SH groups (Langmuir et al., 1996). A standard curve was established by addition of GSH (0.2–6.0 μM) to 50 mM phosphate buffer, pH 7.4, containing ThioGlo1 (20 μM; as DMSO solution). GSH content was estimated by an immediate fluorescence response registered on addition of ThioGlo1 to a cell homogenate. Total protein sulfhydryls were determined as an additional fluorescence response after addition of SDS (2 mM) to the same cell homogenate. A Shimadzu spectrofluorimeter RF-5301PC was employed for determination using an excitation slit of 1.5 nm, and an emission slit of 5 nm. The wavelengths employed in the assay were 388 nm (excitation) and 500 nm (emission). The data acquired were exported from the spectrofluorimeter using a RF-5301PC Personal Fluorescence Software (Shimadzu).
Topo II Covalent Complex Formation.
Mid-log cells (1.5–2.0 × 105 cells/ml) were labeled overnight with 0.5 μCi/ml [3H-methyl]thymidine (0.5 Ci/mM) and 0.1 μCi/ml [4C]leucine (318 mCi/mmol) in Iscove’s media containing 15% FBS. Cells were then pelleted and resuspended in fresh Iscove’s/15% FBS and incubated for 1 h at 37°C. Cells (1 × 106 cells/ml) were treated for 2 h with ascorbate (0–500 μM) in Dulbecco’s modified Eagle’s medium (DMEM) containing 1% FBS. Cells were pelleted and resuspended in L1210 buffer at 37°C at a final density of 1.0 × 106 cells/ml and incubated with etoposide (20 μM) for 1 h. Cells were pelleted 90 s at 2400g, washed with ice-cold L1210 buffer, pelleted, lysed, cellular DNA sheared, and protein-DNA complexes precipitated with SDS and KCl as previously described (Zwelling et al., 1989). Topo II–DNA complexes were quantified by scintillation counting, and [3H]DNA was normalized to cell number using coprecipitated 14C-labeled protein as an internal control.
Results
One-Electron Oxidation of Etoposide by Myeloperoxidase.
Previous work demonstrated that one-electron oxidation of etoposide by tyrosinase or horseradish peroxidase yields phenoxyl radical intermediates readily detectable by EPR (Kalyanaraman et al., 1989;Kagan et al., 1994). As shown on Fig. 1c, incubation of myeloperoxidase with cosubstrates, etoposide, and H2O2, produces a characteristic EPR signal of etoposide phenoxyl radical (etoposide-O⋅) with hyperfine couplings aOCH3 H = 1.4 G, aring H = 1.4 G, aβ H = 4.47 G, aγ H = 0.6 G (Kalyanaraman et al., 1989). No detectable EPR signals were obtained when the enzyme was incubated with etoposide in the absence of H2O2 (Fig. 1b), or when H2O2 and etoposide were incubated in the absence of the enzyme (Fig. 1a).
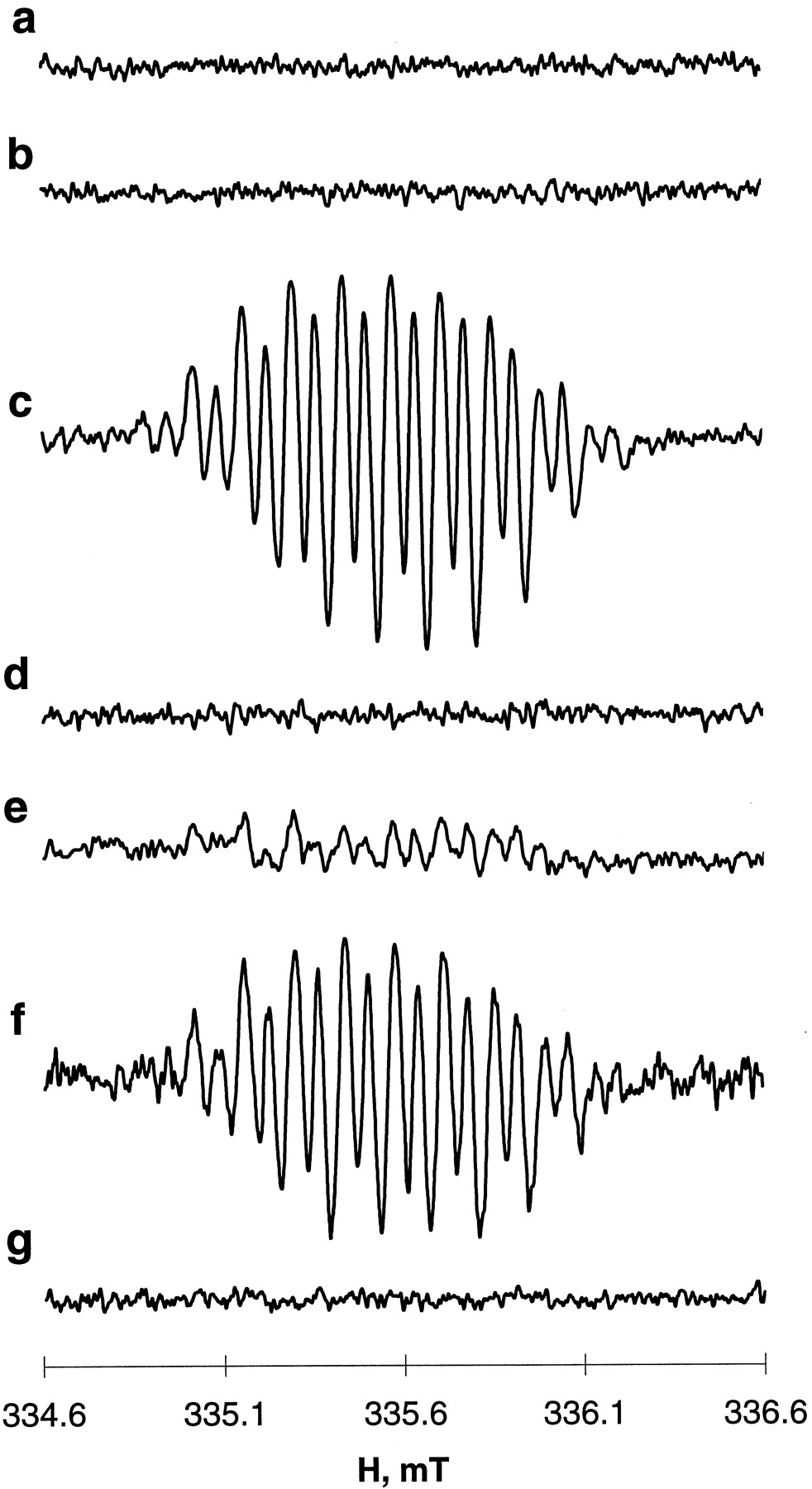
EPR spectra of etoposide phenoxyl radicals generated by myeloperoxidase or myeloperoxidase activity in HL60 cell homogenates in the presence of H2O2. a, etoposide + H2O2; b, myeloperoxidase + etoposide; c, myeloperoxidase + etoposide + H2O2; d, cell homogenate + etoposide + H2O2; e, same as d, but in the presence of ThioGlo1; f, same as e but in the presence of 3-amino-1,2,4-triazole (2.5 mM) and DTPA (100 μM); g, same as f, but in the presence of azide (2.0 mM). Conditions: Myeloperoxidase (2 U/ml) or HL60 cell homogenates (2 × 106 cells/ml) were incubated with etoposide (400 μM) or/and H2O2(100 μM) in L1210 buffer (pH 7.4) at 25°C. HL60 cell homogenates were pretreated with ThioGlo1 (400 μM) 2 min before additions of the reagents were made. Azide (2.0 mM) was added to HL60 cell homogenates 10 min before other reagents. Spectra of etoposide phenoxyl radicals were recorded at 335.7 mT, center field; 2 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 2-min time scan.
We and others demonstrated that physiologically relevant reductants (e.g., ascorbate, GSH, dihydrolipoate, and metallothioneins) can reduce etoposide phenoxyl radicals generated by tyrosinase or horseradish peroxidase (Haim et al., 1987; Mans et al., 1992; Kagan et al., 1994). To test whether myeloperoxidase-derived etoposide phenoxyl radicals are similarly free to leave the active sites and react with extraneous thiols, we further evaluated whether GSH was able to effectively reduce etoposide-O⋅. Figure 2A shows the time course of etoposide-O⋅ generated by myeloperoxidase/H2O2 in the absence and presence of different concentrations of GSH. In the absence of GSH, the EPR signal of etoposide-O⋅ was immediately detectable, whereas its appearance was delayed in the presence of GSH. The duration of the lag period was proportional to the GSH concentration. The appearance of the etoposide-O⋅ EPR signal coincided in time with the GSH depletion (oxidation) (Fig. 2B).

Interactions of myeloperoxidase/H2O2-induced etoposide phenoxyl radicals with GSH. A, time course of etoposide phenoxyl radicals in the presence of different concentration of GSH. GSH concentrations: ▵, 0; ⋄, 2 μM; ■, 4 μM; ○, 6 μM. B, effect of etoposide on GSH depletion: ○, magnitude of the etoposide phenoxyl radical EPR signal; ▪, GSH content. Incubation conditions: L1210 buffer pH 7.4 contained 100 μM DTPA, 2.5 mM 3-amino-1,2,4-triazole, etoposide (400 μM), H2O2 (50 μM), and GSH (2–6 μM). Spectra of etoposide phenoxyl radicals were obtained by repeated scanning of the magnetic field using the internal mode of recording: at 335.7 mT, center field; 0.15 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 20 s, time scan. Shaded area along x-axis represents magnitude of EPR background noise.
One-Electron Oxidation of Etoposide by Endogenous Myeloperoxidase in HL60 Cells.
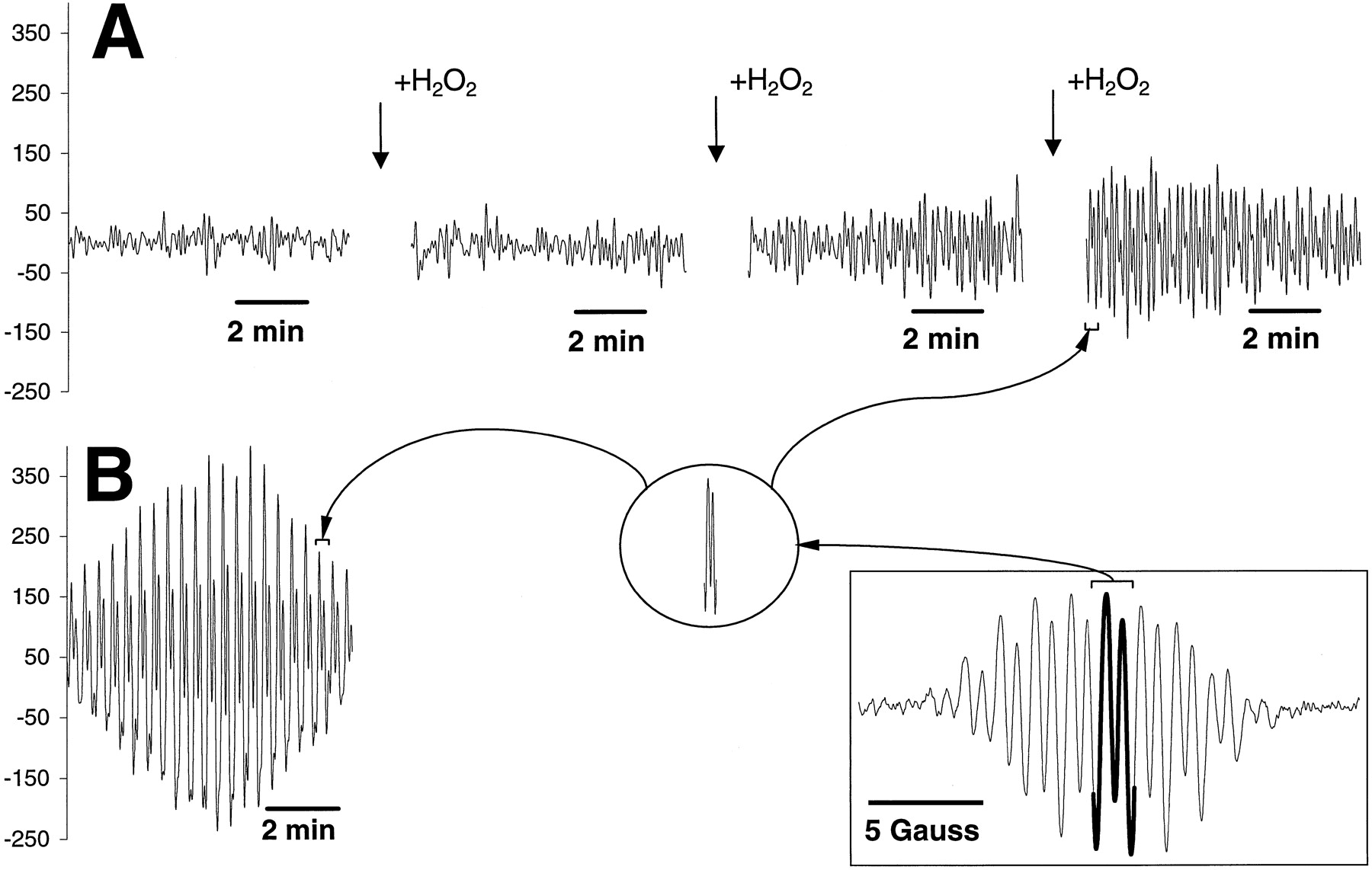
We next tested whether myeloperoxidase activity endogenously present in HL60 cells could effectively catalyze one-electron oxidation of etoposide. In these experiments, we used cell homogenates prepared from 2 × 106 cells in which the myeloperoxidase activity (assayed with guaiacol) was approximately the same as in experiments with the purified enzyme (≈2U/ml). (Note that both the catalase inhibitor, 3-amino-1,2,4-triazole, and an iron-chelator, DTPA, were present in the incubation medium to prevent catalase-mediated degradation of H2O2 and iron-dependent Fenton reactions). When etoposide and H2O2 were added to the homogenate of HL60 cells, no detectable EPR signals were observed for ≈25 to 30 min, after which the characteristic EPR signal of etoposide phenoxyl radical appeared to grow over time. This is shown in Fig.3A where the time course of the etoposide phenoxyl radical is presented. [Note that the recordings represent repeated scans of the field (0.15 mT, sweep width; 335.7 mT, center field) corresponding to a small part of the etoposide-O⋅ radical EPR signal, as demonstrated on the insets (Fig. 3 insets)]. Based on our above results using purified myeloperoxidase and GSH, we postulated that the delay in the EPR detection of etoposide-O⋅ might be caused by rapid reaction with endogenous reductants such as intracellular thiols (GS and protein sulfhydryls), although other factors (e.g., effective decomposition of added H2O2 and inactivation of myeloperoxidase inhibitors) might be contributory. To test our hypothesis, we conducted EPR measurements using cell homogenates after chemical modification of endogenous thiols. To this end, we pretreated the HL60 cell homogenates with a maleimide SH-reagent, ThioGlo1, added in an amount at least 40 times exceeding the level of readily accessible thiols (GSH) and 15 times exceeding the level of total thiols (GSH plus protein SH groups) in the cells (as has been previously titrated using fluorescence yield of ThioGlo1-treated homogenates; see Materials and Methods). This titration out of free SH groups resulted in an immediate appearance of the typical etoposide-O⋅ EPR signal when etoposide and H2O2 were added to the homogenates (Figs. 1f and 3B). The magnitude of the signal increased over time and then declined, mainly due to depletion of H2O2, because repeated addition of H2O2reconstituted the signal (data not shown). This confirmed that endogenous thiols could serve as targets for oxidation by etoposide-O⋅ and presumably mediate recycling of etoposide.

EPR spectrum and time course of etoposide phenoxyl radicals generated by H2O2/myeloperoxidase activity in HL60 cell homogenates in the absence (A) and presence (B) of the maleimide SH-reagent, ThioGlo1. Inset in square, EPR spectrum of etoposide phenoxyl radicals recorded at 335.7 mT, center field; 2 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 2-min time scan. Inset in circle, the fragment of etoposide phenoxyl radical EPR signal that was chosen for repeated scans of the time course (0.15 mT, sweep width; 335.7 mT, center field; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 2-s time scan). Conditions: HL60 cell homogenates (2 × 106 cells/ml) were incubated with etoposide (400 μM) and H2O2 (100 μM) in the presence or absence of ThioGlo1 (400 μM) in L1210 buffer, pH 7.4, containing 100 μM DTPA and 2.5 mM 3-amino-1,2,4-triazole at 25°C. ThioGlo1 was added 2 min before the other additions.
To confirm the involvement of endogenous myeloperoxidase in one-electron oxidation of etoposide, we studied effects of myeloperoxidase inhibitors, azide, and cyanide on the EPR signal of etoposide-O⋅ radical in HL60 cell homogenates pretreated with ThioGlo1. Both azide (Fig. 1g) and cyanide (data not shown), at concentrations of 2.0 mM and higher completely quenched etoposide-O⋅ EPR signal. Almost 80% inhibition of etoposide-O⋅ radical in HL60 cell homogenates was produced by 1.0 mM azide. Although azide and cyanide are not highly specific inhibitors of myeloperoxidase but rather block different heme-containing enzymes, these results suggest that H2O2-dependent myeloperoxidase, the major heme-protein of HL60 cells (Traweek et al., 1995), may be involved in the generation of etoposide-O⋅radicals.
To determine whether relatively low (clinically relevant) concentrations of etoposide can produce steady-state levels of etoposide-O⋅ detectable by EPR, we performed measurements after varying concentrations of etoposide. Well-resolved signals of etoposide-O⋅ radicals were reliably detectable in the EPR spectra obtained from HL60 cells starting at drug concentrations as low as 10 μM. The EPR response continuously increased with the increase of etoposide concentration in the range 10 to 500 μM (Fig.4A).

Effect of different concentrations of etoposide (A) and H2O2 (B) on the time course of etoposide phenoxyl radical EPR signal generated by H2O2/myeloperoxidase activity in HL60 cell homogenates. A, H2O2, 100 μM; etoposide concentrations: ▵, 500 μM; ▴, 400 μM; ▪, 300 μM; ○, 200 μM; ⋄, 100 μM; ♦, 50 μM; ■, 20 μM; ●, 10 μM. B, etoposide, 400 μM; H2O2 concentrations: ▴, 20 μM; ■, 100 μM; ●, 400 μM; ⋄, 800 μM. Incubation conditions: HL60 cell homogenates (2 × 106 cells/ml) were incubated with etoposide (10–500 μM), H2O2 (20–800 μM) in the presence of ThioGlo1 (400 μM) in L1210 buffer pH 7.4 containing 100 μM DTPA and 2.5 mM 3-amino-1,2,4-triazole at 25°C. ThioGlo1 was added 2 min before the other additions. Spectra of etoposide phenoxyl radicals were obtained by repeated scanning of the magnetic field using the internal mode of recording: at 335.7 mT, center field; 0.15 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 20 s, time scan. Shaded area along x-axis represents magnitude of EPR background noise.
To develop the conditions for the optimized detection of etoposide-O⋅ EPR signal, we also performed measurements after varying concentrations of H2O2 (Fig. 4B). The effect of H2O2 was biphasic where signal intensity increased up to 200 μM and then began to decline as higher concentrations of H2O2 were applied (most likely due to enzyme inhibition). The time course of etoposide phenoxyl radical formation uniformly demonstrated initial activation of etoposide oxidation followed by a decreased rate of the radical formation, most likely due to H2O2 depletion (data not shown). For practical reasons (solubility of etoposide), 400 μM etoposide and 100 μM H2O2were used in most of the subsequent experiments.
Effects of Etoposide on H2O2-Induced Oxidation of Endogenous Thiols and Phospholipids in HL-60 Cells.
Because endogenous thiols appeared to be targets serving to reduce etoposide phenoxyl radicals in HL60 cell homogenates, we expected that loss of endogenous thiols should occur in the course of exposure to H2O2 plus etoposide. Incubation of HL60 cell homogenates in the presence of H2O2 alone caused a time-dependent decrease in the endogenous levels of both GSH and protein sulfhydryls (Fig. 5). Combination of H2O2 and etoposide, however, significantly (>2-fold) increased the rate of GSH oxidation. Etoposide plus H2O2slightly increased the rate of oxidation of protein SH groups as compared with H2O2 alone, but this effect did not achieve statistical significance. In the absence of H2O2, etoposide did not cause any thiol oxidation. These results indicate that the attack of myeloperoxidase-catalyzed etoposide phenoxyl radicals on thiols was primarily on GSH, whereas oxidation driven by H2O2 alone appeared to include both GSH and protein sulfhydryls.

Oxidation of GSH (A) and protein thiols (B) in HL60 cell homogenates in the presence of etoposide/H2O2. Protective effect of ascorbate and a vitamin E homolog, PMC. ▵, etoposide; ○, H2O2; ●, etoposide + H2O2; ▪, etoposide + H2O2 + PMC; ■, etoposide + H2O2 + ascorbate HL60 cell homogenates (2 × 106 cell/ml) were incubated in the presence and absence of H2O2 (100 μM) and/or etoposide (500 μM) in L1210 buffer, pH 7.4, containing 2.5 mM 3-amino-1,2,4-triazole and 100 μM DTPA at 25°C. PMC (100 μM) or ascorbate (10 μM) were added before addition of etoposide and H2O2. Aliquots of cell homogenates were taken at time points as indicated, and GSH and protein SH groups were determined as described inMaterials and Methods.
Reactive phenoxyl radicals can also potentially initiate lipid peroxidation by abstracting allylic hydrogens from phospholipid polyenoic fatty acid residues. Therefore, we next studied effects of H2O2 and etoposide on phospholipid peroxidation in HL60 cell homogenates. For this we prepared HL60 cells whose phospholipids were metabolically labeled with oxidation-sensitive fluorescent fatty acid, PnA, and the content of fluorescently labeled individual phospholipids assayed by HPLC. H2O2 induced dramatic oxidation of all individual classes of PnA-labeled phospholipids: phosphatidylcholine (by 68.5%), phosphatidylethanolamine (by 68.8%), phosphatidylserine (by 54.4%), phosphatidylinositol (by 67.9%), sphingomyeline (by 74.1%), and diphosphatidylglycerol (by 59.8%) (Table 1). Etoposide alone (10 and 50 μM) did not produce peroxidation of any phospholipid. In marked contrast to its effect on thiols, etoposide protected all phospholipids against H2O2-induced peroxidation. Moreover, it is likely that the hindered dimethoxy-phenolic ring of etoposide can function similar to the hindered phenolic chromanol ring of vitamin E and act as a chain-breaking antioxidant.
Effect of etoposide on oxidation of PnA-labeled phospholipids induced by H2O2 and myeloperoxidase activity of HL60 cell homogenates
Because etoposide phenoxyl radicals readily oxidize thiols (vide supra) this interaction might have prevented the reaction of the phenoxyl radicals with phospholipids in HL60 cells. To experimentally address the issue, we performed experiments with PnA-labeled cells that were pretreated with the SH-reagent ThioGlo1 (Table 1) to eliminate all possible interactions of the etoposide phenoxyl radicals with endogenous thiols. Under these conditions, the effects were essentially the same as in control nonpretreated cells. H2O2 caused a severe oxidation of all major classes of PnA-labeled phospholipids, whereas etoposide acted as an effective antioxidant and protected phospholipids against H2O2-induced oxidation. These results clearly demonstrate that the reactivity of the etoposide phenoxyl radical is not sufficient to induce phospholipid peroxidation as has been reported earlier for other phenolic compounds [e.g., phenol (Ritov et al., 1996)].
Detection of Etoposide Phenoxyl Radical-Induced Generation of Thiyl Radicals in a Model System and HL60 Cell Homogenates.
Oxidation of thiols by phenoxyl radicals proceeds via one-electron intermediate, the thiyl radical, whose formation may be monitored by spin trapping with EPR detection (Mason and Ramakrishna Rao, 1990) or HPLC detection (Stoyanovsky et al., 1996). Initially, we attempted to directly document the formation of thiyl radicals in the system with purified myeloperoxidase (or horseradish peroxidase)/etoposide/H2O2using a spin trap, DMPO. Spin-adducts of DMPO-thiyl radicals formed by horseradish peroxidase-catalyzed or prostaglandin synthase-catalyzed oxidation of phenolic compounds have been characterized (Schreiber et al., 1989). Indeed, when myeloperoxidase was incubated with phenol, H2O2, and GSH in the presence of DMPO, a typical EPR spectrum of DMPO-GS⋅ adduct with aN = 15.4 G, aHβ = 16.2 G was readily detectable and well resolved (Fig.6A, compare lines a and b).

A, EPR signals of DMPO spin-adducts with GS⋅radicals generated by myeloperoxidase/H2O2 in the presence of phenol or etoposide. B, EPR signals of etoposide-O⋅ radicals generated by myeloperoxidase/H2O2 in the presence or in the absence of DMPO. A, reaction mixture contained 0.1 M phosphate buffer (pH 7.4), 100 mM DMPO, GSH (4 mM), and myeloperoxidase (2 U/ml): a, H2O2 (200 μM); b, H2O2 (200 μM) + phenol (200 μM); c, H2O2 (200 μM) + etoposide (400 μM). B, reaction mixture contained 0.1 M phosphate buffer (pH 7.4), myeloperoxidase (2 U/ml), H2O2 (200 μM), etoposide (400 μM): a, in the absence of DMPO; b, +DMPO (100 mM). Spectra of DMPO-GS adducts were recorded at 335.7 mT, center field; 8 mT sweep width; 0.1 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 1 min time scan. Spectra of etoposide-O⋅ were recorded at 335.7 mT, center field; 2 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 2-min time scan.
In contrast, no DMPO-GS⋅ adduct signals were found in the system including myeloperoxidase/H2O2/etoposide/GSH. Instead, the characteristic EPR signal of nitroxide radicals [with aN = 0.72 G, aH = 0.41 G for 5,5-dimethylpyrrolidone-(2)-oxyl-(1)] was observed (Fig. 6, line c). Similarly, the incubation of horseradish peroxidase with H2O2 and phenol in the presence of GSH caused a dramatic accumulation of EPR-detectable DMPO-GS⋅ adducts. Yet, no EPR signals of DMPO-GS⋅ adducts were discernible during horseradish peroxidase-catalyzed oxidation of etoposide (in the presence of H2O2 and GSH). A well-resolved characteristic EPR signal of nitroxide radicals was also identified in the spectrum (the magnitude of the signal was much greater than that produced by myeloperoxidase; data not shown). The data suggest that in this system myeloperoxidase-catalyzed formation of etoposide phenoxyl radicals in the presence of GSH and DMPO does not yield sufficiently high concentration of DMPO-GS⋅ adducts to be detectable by EPR. Moreover, formation of nitroxide radicals in the presence of DMPO and etoposide indicates that myeloperoxidase-catalyzed metabolism of DMPO may compete with etoposide for one-electron oxidation and consequently prevent generation of thiyl radicals (and DMPO-GS⋅ adducts). This suggestion is supported by the data shown in Fig. 6B in which the formation of radicals in the presence and absence of DMPO by myeloperoxidase was compared (in the absence of GSH). EPR signals of etoposide phenoxyl radicals (readily observable in the absence of DMPO; Fig. 6B, line a) disappeared from the spectra on addition of DMPO (Fig. 6B, line b) and were substituted by the signals of nitroxides radicals (Fig. 6A, line c).
Because EPR detection of DMPO-GS⋅ as a marker for myeloperoxidase-catalyzed formation of etoposide-O⋅-induced thiyl radicals was not sensitive enough for our purposes, we decided to use a previously developed HPLC procedure for detection of EPR-silent DMPO-GS adduct (nitrone). This HPLC assay had sensitivity superior to that of EPR detection of DMPO-GS⋅ adducts (Stoyanovsky et al., 1996). Typical HPLC tracings of the adducts formed with myeloperoxidase/H2O2/GSH/phenol and myeloperoxidase/H2O2/GSH/etoposide are shown on Fig. 7. When phenol was substituted for etoposide, the amount of DMPO-GS nitrone formed dramatically decreased (about 10-fold), although the signal was still readily detectable on the HPLC tracing and it was significantly higher than in the absence of etoposide (Fig. 7 and Table2). Moreover, accumulation of DMPO-GS nitrone adduct was dependent on the etoposide concentration.

Typical HPLC tracings of the DMPO-GS nitrone formed in the system myeloperoxidase/H2O2/GSH/phenol or myeloperoxidase/H2O2/GSH/etoposide. Myeloperoxidase (1 U/ml) was incubated with GSH (25 mM), DMPO (100 mM), and H2O2 (200 μM H2O2was added every 10 min during 1 h of incubation at 37°C) in L1210 buffer (pH 7.4) containing 100 μM DTPA, 2.5 mM 3-amino-1,2,4-triazole in the presence of 300 μM phenol, or 300 μM etoposide. DMPO-GS nitrone was determined using characteristic UV-absorbency at 258 nm as described in Materials and Methods. A, myeloperoxidase/H2O2/GSH; b, myeloperoxidase/H2O2/GSH/etoposide; c, myeloperoxidase/H2O2/GSH/phenol; d, DMPO-GS nitrone (standard)
Effect of addition of etoposide and H2O2 on formation of DMPO-GS nitrone by purified myeloperoxidase and myeloperoxidase activity in HL60 cells
An increased accumulation of DMPO-GS nitrone adduct was also observed in HL60 cell homogenates incubated in the presence of H2O2/etoposide as compared with H2O2 alone (Table 2). As with purified myeloperoxidase, a much more pronounced accumulation of DMPO-GS nitrone was detected when HL60 cell homogenates were incubated in the presence of H2O2/phenol (data not shown). Thus, despite the peroxidase-catalyzed metabolism of DMPO and its interference with EPR assay of DMPO-GS⋅ adducts, the etoposide-induced formation of DMPO-GS nitrone is detectable by HPLC. Therefore, we conclude that one-electron oxidation of etoposide to the phenoxyl radical and its subsequent interaction with GSH produces thiyl radicals.
Effects of Ascorbate and Vitamin E Homolog PMC on Etoposide Phenoxyl Radical in HL60 Cells.
We hypothesized that at least two different approaches may be used to suppress production of etoposide phenoxyl radicals and thereby potentially reduce their redox-cycling leading to cyto- and/or genotoxic effects (i.e., secondary malignancies) in myeloperoxidase-rich cells. These include: 1) quenching of the phenoxyl radical by a reductant whose radicals are not cytotoxic, and 2) combination of etoposide with another phenolic compound that may compete with etoposide as the substrate for myeloperoxidase and whose phenoxyl radicals do not readily undergo redox-cycling. We chose two such compounds, vitamin C (ascorbate) and a vitamin E homolog (PMC), with potential to act through mechanisms 1 and 2, respectively.
Effects of Ascorbate.
As shown in Fig.8A, addition of ascorbate to the etoposide phenoxyl radical-generating system (myeloperoxidase/H2O2/etoposide) results in complete disappearance of the etoposide radical EPR signal and appearance of a characteristic ascorbate radical doublet signal (with aH 1.7 G). The time courses of ascorbate and etoposide radicals (Fig. 8A) indicate that complete oxidation of ascorbate (disappearance of ascorbate radical signal) coincides with the reappearance of the etoposide phenoxyl radical EPR signal. This suggests that ascorbate is capable of reducing the myeloperoxidase-induced etoposide phenoxyl radical as has been previously demonstrated for tyrosinase (Kagan et al., 1994).

A and B, effect of ascorbate on the EPR signal of etoposide-O⋅ generated by myeloperoxidase/H2O2/etoposide (A) and by myeloperoxidase activity in HL60 cell homogenates (B) in the presence of H2O2. C, time course of ascorbate radical generated by myeloperoxidase activity in HL60 cell homogenates in the presence of H2O2/etoposide and different concentrations of ascorbate. A, time course of ascorbate and etoposide-O⋅ radicals generated by myeloperoxidase (2 U/ml)/H2O2 (100 μM)/etoposide (400 μM). Inset, typical etoposide phenoxyl and ascorbate radical EPR signals.●, ascorbate radical EPR signal; ■, etoposide-O⋅ EPR signal. Time course of etoposide-O⋅ (B) and ascorbate (C) radical EPR signal at different concentrations of ascorbate: ●, 0; ▵, 0.5 μM; ▪, 0.75 μM; ♦, 1 μM; ▴, 1.25 μM; ■, 1.5 μM; ○, 2 μM. Incubation conditions: A, 0.1 M phosphate buffer, pH 7.4, 100 μM DTPA, myeloperoxidase (2 U/ml), H2O2 (200 μM), etoposide (400 μM), ascorbate (20 μM). B and C, L1210 buffer, pH 7.4, contained 100 μM DTPA, 2.5 mM 3-amino-1,2,4-triazole, ThioGlo1 (400 μM), etoposide (400 μM), H2O2 (100 μM), HL60 cell homogenates (2 × 106 cell/ml). The time course of etoposide and ascorbate radicals were obtained by repeated scanning of the field (using the internal mode of recording 0.15 mT, sweep width; 335.7 mT, center field) corresponding to a part of the ESR signal (for etoposide radical) or the low field component of ascorbate radical ESR signal. Other conditions were: 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 20-s time scan. Shaded area along x-axis represents magnitude of EPR background noise.
Similarly, the etoposide-O⋅ radical EPR signal, immediately observable on addition of H2O2/etoposide to ThioGlo1-pretreated HL60 cells, disappeared and was substituted by the ascorbate radical signal in the presence of ascorbate (Fig. 8, B and C). The effectiveness of ascorbate inhibition of etoposide phenoxyl radical production was concentration dependent. Complete suppression of etoposide phenoxyl radical signal was achieved by ascorbate concentrations as low as 1.0 to 2.0 μM (whereas the etoposide concentration was as high as 400 μM; Fig. 8B). These low concentrations of ascorbate (1.5–2.0 μM) provided for ascorbate radical presence in the spectra for 8 to 10 min under the conditions used (Fig. 8C). A prominent oxidation of GSH and protein sulfhydryls induced by etoposide/H2O2in HL60 cells was effectively prevented by ascorbate (Fig. 5, A and B). Thus, ascorbate acted as a primary reductant for etoposide phenoxyl radical generated by purified myeloperoxidase or by endogenous myeloperoxidase activity in HL60 cells where it was able to preserve endogenous thiols against oxidative attack by etoposide-O⋅.
Effects of PMC.
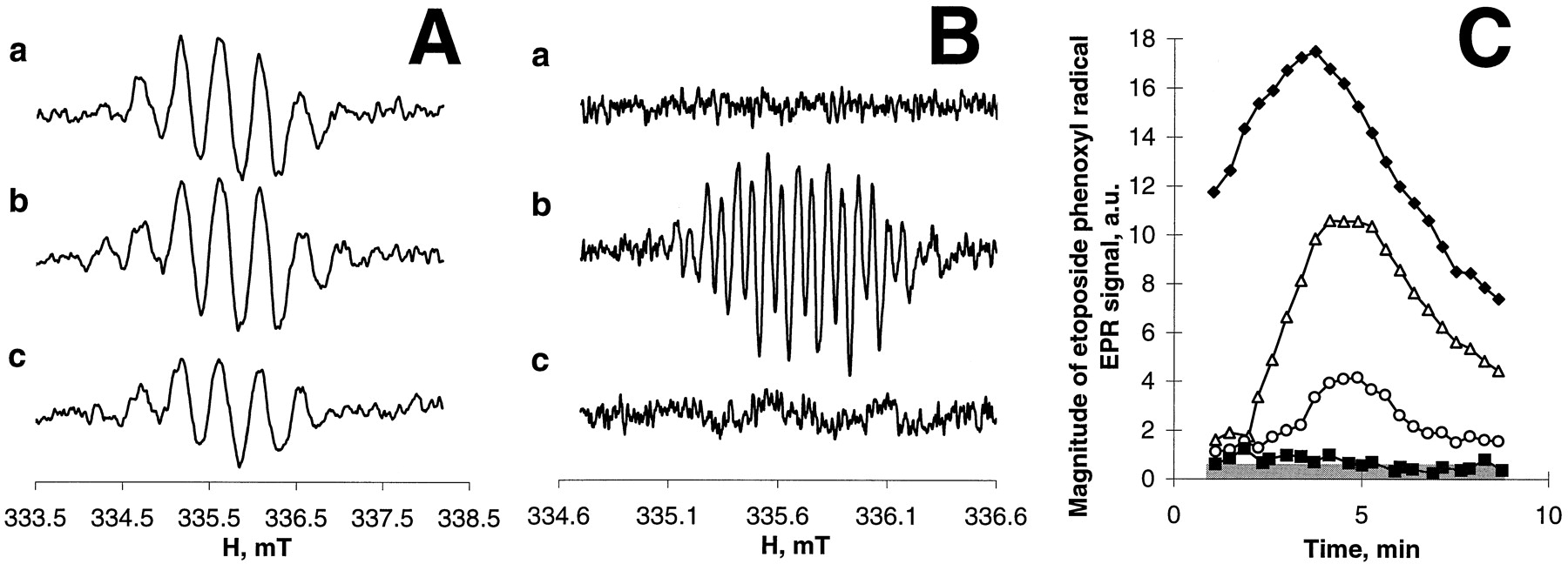
Figure 9demonstrates the EPR spectra of PMC phenoxyl radicals (Fig. 9A) or etoposide phenoxyl radicals (Fig. 9B) generated after addition of H2O2 and the phenolic compound (etoposide or PMC) to HL60 cell homogenates. In untreated cells, a characteristic (but not completely resolved) EPR signal of PMC phenoxyl radical was found after addition of H2O2/PMC (Fig. 9A, line a). No etoposide EPR signals were detectable in the presence of etoposide in homogenates that contained endogenous thiols (Fig. 9B, line a). After pretreatment of the cells with ThioGlo1, both PMC phenoxyl radical signal (Fig. 9A, line b) and etoposide phenoxyl radical signal (Fig. 9B, line b) were immediately apparent in the EPR spectra in the presence of H2O2/PMC or H2O2/etoposide, respectively. Finally, PMC (200 μM) and etoposide (400 μM) were added together to the cell homogenate along with H2O2. In this case, robust PMC radical EPR signal was readily observable in Fig. 9A (line c) under optimized EPR conditions and was discernible even in Fig. 9B (line c) where the EPR conditions were optimized for the registration of etoposide-O⋅ radical signal. [Note that the conditions for the recording of narrow and well-resolved etoposide radical signal and relatively poorly resolved broadened EPR signal of PMC radicals were very different (modulation amplitude, time constant, and sweep width)]. No etoposide-O⋅ EPR signal could be distinguished on either of the recordings (under either of the EPR settings) when the combination of PMC and etoposide was used. These data indicate that the steady-state concentration of PMC phenoxyl radicals was significantly greater than that of etoposide radicals, although the etoposide concentration was 2-fold higher than the concentration of PMC. This suggests that PMC is a much better substrate for myeloperoxidase than etoposide. This is quantitatively supported by the results shown in Fig. 9C, which demonstrates that PMC at concentrations almost two orders of magnitude lower than those of etoposide was able to completely eliminate the etoposide radical signal generated by HL60 cells in the presence of H2O2.

EPR spectra of PMC (A) and etoposide (B) phenoxyl radicals induced by myeloperoxidase activity in HL60 homogenates in the presence of H2O2. Effect of different concentrations of PMC on the etoposide-O⋅ (C). A and B, a, PMC (400 μM) or etoposide (400 μM); b, PMC (400 μM) or etoposide (400 μM) + ThioGlo1; c, PMC (200 μM) + etoposide (400 μM) + ThioGlo1. C, PMC concentrations: ♦, 0; ▵, 2 μM; ○, 4 μM; ▪, 6 μM. Conditions: HL60 cell homogenates (2 × 106 cells/ml) were incubated with PMC (200 or 400 μM) or/and etoposide (400 μM) in the presence or in the absence of ThioGlo1 (400 μM) in L1210 buffer (pH 7.4) containing 100 μM DTPA, 2.5 mM 3-amino-1,2,4-triazole, H2O2 (100 μM) at 25°C. Spectra of etoposide phenoxyl radicals were recorded at 335.7 mT, center field; 2 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.1 s, time constant; 2-min time scan. Spectra of PMC phenoxyl radicals were recorded at 335.7 mT, center field; 5 mT, sweep width; 0.2 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 2 min, time scan. Time course of etoposide-O⋅was obtained by repeated scanning of the magnetic field using the internal mode of recording: 335.7 mT, center field; 0.15 mT sweep width; 0.04 mT, field modulation; 10 mW, microwave power; 0.3 s, time constant; 20 s, time scan. Shaded area alongx-axis (C) represents magnitude of EPR background noise.
In separate experiments, we studied the effect of PMC on thiols in HL60 cells exposed to H2O2/etoposide. We found that PMC significantly protected both GSH and protein sulfhydryls against etoposide/H2O2-induced oxidation (Fig. 5, A and B). Thus, the vitamin E homolog PMC can serve as a competing substrate for myeloperoxidase and thus limit the production of etoposide-O⋅ in myeloperoxidase-rich cells.
Effects of Ascorbate on Etoposide-Induced Topo II-DNA Complex Formation in HL60 Cells.
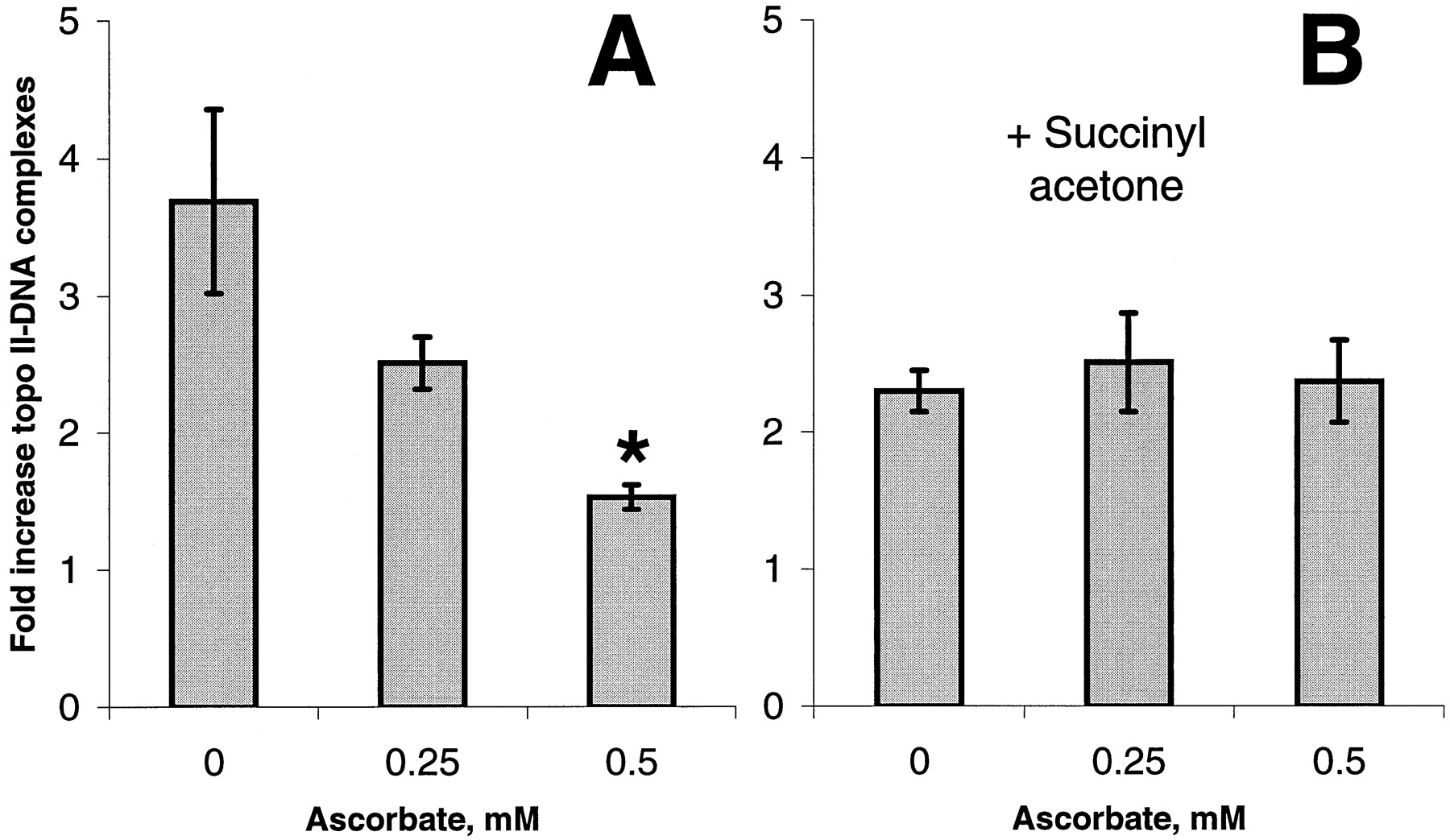
Our previous studies demonstrated that free radical activation of etoposide to its phenoxyl radical enhanced its ability to induce topo II–DNA complexes (Yalowich et al., 1996). We suggested that oxidation of etoposide to its phenoxyl radical by endogenous myeloperoxidase might contribute to topo II poisoning in HL60 cells. Consequently, ascorbate should diminish etoposide activity through its ability to prevent the myeloperoxidase-catalyzed formation of etoposide phenoxyl radicals. To test this hypothesis, we first studied the effects of ascorbate on etoposide-induced topo II-DNA covalent complex formation in HL60 cells. We found that a 2-h pretreatment of HL60 cells with ascorbate (0–0.5 mM) produced a concentration-dependent decrease in etoposide (20 μM)-induced topo II–DNA complexes (Fig. 10A). When HL60 cells were treated for 48 h with 0.5 mM succinyl acetone, a heme synthesis inhibitor (Nonaka et al. 1992), myeloperoxidase activity was reduced to 5% of control levels. Under these conditions etoposide-induced topo II–DNA complexes were reduced (Fig. 10B) compared with myeloperoxidase-replete HL60 cells (Fig. 10A). Etoposide-induced topo II–DNA complex formation in succinyl acetone-treated HL60 cells represents the activity of unmetabolized (by myeloperoxidase) VP-16. Most importantly, these myeloperoxidase-depleted cells were insensitive to ascorbate (Fig.10B). Similarly, ascorbate did not affect etoposide-induced topo II-DNA complex formation in human erythroleukemia K562 cells that lack myeloperoxidase (Lozzio and Lozzio, 1979) (results not shown). These results indicate that the effects of ascorbate to protect against formation of etoposide-induced topo II-DNA complexes in HL60 cells (Fig. 10A) are associated with reduction of etoposide phenoxyl radicals generated by myeloperoxidase.

Effect of ascorbate on etoposide-induced topo II–DNA covalent complexes in HL60 cells. Cells were treated for 48 h in the absence (A) or presence (B) of 0.5 mM succinyl acetone (a heme synthesis inhibitor) to deplete cells of myeloperoxidase activity before further experimentation. Cells were then treated for 2 h with ascorbate (0–0.5 mM) in DMEM media containing 1% FBS. Cells were washed and resuspended in serum-free buffer, incubated 1 h with 20 μM etoposide, and assessed for topo II-DNA complexes as described in Materials and Methods. Results are expressed as fold increase over controls not containing etoposide. Bars, A, mean ± S.E.M. from three experiments, ∗, significantly different (p = .033) compared with etoposide incubated in the absence of ascorbate; B, mean ± range from two experiments.
Discussion
This study demonstrates for the first time that etoposide is a substrate for both purified myeloperoxidase and endogenous myeloperoxidase activity in human leukemia HL60 cells. Etoposide undergoes one-electron oxidation to form the etoposide phenoxyl radical that can be redox-cycled by intracellular thiols such as GSH and, to a much less extent, by protein sulfhydryls.
The idea that free radicals (e.g., etoposide semiquinone radicals) may be responsible to some degree for the cytotoxic effects of etoposide is not new (Van Maanen et al., 1988; Usui and Sinha, 1990). What is new is the idea that etoposide phenoxyl radicals are formed by a myeloperoxidase-catalyzed reaction, before any accumulation of quinones or other secondary oxidation and/or decomposition products, which have been suggested to be mainly responsible for the cyto- and genotoxic effects of etoposide (Mans et al., 1992). Etoposide phenoxyl radicals can oxidize thiols, triggering formation of thiyl radicals and subsequently oxygen radicals thus causing redox-cycling and damage in myeloperoxidase-rich cells (e.g., in HL60 cells and possibly in bone marrow myelogenous progenitor cells; Fig.11).

Potential cyto- and genotoxic pathways triggered by myeloperoxidase-catalyzed generation of etoposide phenoxyl radicals and their blockade by antioxidants.
Our results lead to a model in which the genotoxic effects of etoposide are mediated by a redox-cycling mechanism with potentially three amplifying cascades (Fig. 11):
1) Etoposide is oxidized in a reaction catalyzed by myeloperoxidase to its phenoxyl radical, etoposide-O⋅.
2) Etoposide-O⋅ is reduced back to etoposide via oxidation of intracellular thiols (RSH in Fig. 11), i.e., glutathione and sulfhydryl groups of proteins (potentially including topo II). Reduced etoposide (etoposide-OH) is thus repeatedly available as a substrate for myeloperoxidase, at the expense of intracellular thiols, which undergo one electron oxidation to reactive thiyl radicals (RS⋅).
Our attempts to directly detect etoposide-driven formation of thiyl radicals by EPR spin trapping were not successful. No EPR-detectable DMPO-GS adducts were found when either purified myeloperoxidase or HL60 cell homogenates were incubated under conditions favorable for production of thiyl radicals (i.e., in the presence of etoposide, H2O2, and GSH). Production of DMPO-GS nitrone adducts was, however, revealed by a more sensitive HPLC procedure developed for detection of EPR-silent DMPO-GS nitrones (Stoyanovsky et al., 1996). Accumulation of etoposide-dependent thiyl radical-derived DMPO-GS nitrones was documented when etoposide, GSH, H2O2, and DMPO were incubated with either purified myeloperoxidase or HL60 cell homogenates. Thus the results of the study for the first time provide quantitative information on the generation of thiyl radical by myeloperoxidase-catalyzed oxidation of etoposide. The extent of thiyl radical production, however, is probably significantly underestimated here due to the technical limitations posed by the potential for DMPO itself to compete with etoposide as a substrate for peroxidase.
3) Thiyl radicals (RS⋅) can further react to generate disulfide anion-radicals (RS-S-⋅R), which can donate an electron to oxygen. Superoxide anion radical (O2 − ⋅) thus produced can form, in the presence of transition metal complexes, the extremely reactive hydroxyl radical (HO⋅), which damages DNA and other critical biomolecules, ultimately inducing genotoxicity.
The fact that our EPR measurements did not detect etoposide phenoxyl radical-dependent production of thiyl radicals in HL60 cells, which was only detectable by a much more sensitive HPLC procedure, indicates that thiyl radical steady-state concentrations may be relatively low. Even if the effectiveness of direct modification of DNA by free radicals (thiyl radicals, superoxide, and hydroxyl radicals) is limited, the extensive genotoxic effects of etoposide may be due to oxidative modification of topo II and subsequent topo II-associated damage to DNA (Henichart et al., 1997). In line with this, our work revealed that ascorbate, which prevented formation of etoposide phenoxyl radicals, was also effective in diminishing etoposide-induced topo II–DNA complexes. It should be mentioned that AML is characterized by distinct translocations involving the chromosome 11q23 region (Felix et al., 1995; Aplan et al., 1996). An 8.3-kb breakpoint cluster region of the putative oncogene ALL1 (also called MLL; located on chromosome band 11q23) has been associated with myeloid leukemias arising subsequent to treatment with topo II inhibitors such as etoposide, teniposide, doxorubicin (Aplan et al., 1996). These chromosome band 11q23 translocation breakpoints have recently been identified by Felix et al. (1995) to be in close proximity to topo II consensus cleavage sites. Our results imply that myeloid stem cells may be unusually susceptible targets for the leukemogenic effects of etoposide because of high constitutive myeloperoxidase activity and resultant formation of etoposide phenoxyl radicals.
Our experiments clearly indicate that concentrations of etoposide as low as 10 to 20 μM are sufficient to trigger its myeloperoxidase-catalyzed one-electron oxidation to phenoxyl radicals. These concentrations of etoposide are well within the range of its levels in plasma attainable in vivo during most commonly used regimens of chemotherapy (Nguyen et al., 1998). Moreover, concentrations of etoposide used in our study are much below the plasma concentrations of etoposide attainable during a high-dose etoposide chemotherapy regimens, a strategy recently evolved to improve the treatment outcome in patients with relapsed and/or refractory tumors (Rick et al., 1998).
As a consequence of our model, which is based on the central role of etoposide-O⋅ in genotoxicity, chemopreventive nutritional strategies can be targeted at minimizing etoposide conversion to its phenoxyl radical and/or minimizing the damaging effects of the radicals, whereas leaving the direct topo II-mediated antitumor effects of unmetabolized (by myeloperoxidase) etoposide unchanged. Hence, the biochemical effect of etoposide treatment may be shifted toward direct targeting of tumor cells and away from carcinogenesis in nontumor cells containing high levels of enzymes that convert etoposide to its phenoxyl radical. In particular, the etoposide redox-cycling can be interrupted by nutritional antioxidants that can either directly reduce etoposide-O⋅ phenoxyl radicals and preserve endogenous targets of oxidation (e.g., ascorbate, approach 1) or compete with etoposide for myeloperoxidase and produce low reactive phenoxyl radicals that do not react with thiols (e.g., PMC, approach 2).
In the present work, we experimentally tested these approaches by studying the effects of ascorbate (approach 1) and a vitamin E homolog, PMC (approach 2), on the formation of etoposide phenoxyl radicals in HL60 cell homogenates. We found that ascorbate indeed outcompetes intracellular thiols in the reduction of the etoposide phenoxyl radical. This suggests that ascorbate can protect thiols against oxidation by etoposide phenoxyl radicals and prevent myeloperoxidase-dependent genotoxic effects. Most importantly, this hypothesis is directly supported by our results demonstrating that ascorbate can diminish the formation of topo II–DNA complexes in myeloperoxidase-rich HL60 cells treated with therapeutically relevant low concentrations of etoposide. In addition, when myeloperoxidase activity is depleted by incubation of HL60 cells with succinyl acetone, there is no protective effect of ascorbate against etoposide-induced topo II–DNA complex formation. It is important to note that under conditions in which myeloperoxidase activity is inhibited, etoposide-induced topo II–DNA complexes are diminished to a level representing the activity of unmetabolized (by myeloperoxidase) VP-16. Hence, myeloperoxidase-mediated formation of etoposide phenoxyl radicals provides an incremental enhancement in topo II–DNA complex formation. It is this activity that may be responsible for the genotoxic effects of etoposide in myeloid progenitor cells that contain myeloperoxidase. It is this myeloperoxidase-mediated enhancement of etoposide activity that is protected against by treatment with ascorbate.
One limitation of this approach is that ascorbate is rapidly consumed in the course of the reaction and does not ultimately prevent etoposide redox-cycling. Thus, the antioxidant effectiveness of ascorbate will depend on the maintenance of its sufficiently high intracellular concentration through nutritional manipulations and application of additional chemopreventive strategies.
We further found that a vitamin E homolog PMC, whose phenoxyl radicals do not oxidize endogenous thiols (Kagan et al., 1990), outcompetes etoposide as a substrate for myeloperoxidase. This offers another opportunity to reduce myeloperoxidase-catalyzed metabolism of etoposide without triggering thiol-dependent redox-cycling. The fact that PMC could inhibit the formation of etoposide-O⋅ at much lower concentrations relative to etoposide suggests that PMC is indeed a preferred substrate for myeloperoxidase.
Finally, ascorbate can synergistically interact with vitamin E and its homologs and recycle them from their phenoxyl radicals (Kagan et al., 1990). Hence, a combination of ascorbate and a vitamin E homolog may more effectively prevent etoposide phenoxyl radical formation and oxidative modification of thiols in cells not only through etoposide radical reduction but also via recycling the vitamin E homolog.
Redox cascade reactions of etoposide may also play a role in its antitumor effects. Indeed, in earlier studies in which we enhanced etoposide oxidation by use of an exogenous source of free radicals, 1-2,2′-azobis-2,4-dimethylvaleronitryle, we found a small but significant augmentation of topo II inhibition (40%) (Tyurina et al., 1995). Thus, to evaluate the extent nutritional antioxidants may weaken the tumoricidal action of etoposide, it will be important to examine in greater detail the effects of myeloperoxidase-catalyzed redox-cycling of etoposide and nutritional antioxidants on topo II-DNA covalent complex formation.
In conclusion, a critical point of this study is the possibility that there may be a therapeutic window for the use of etoposide in combination with nutritional antioxidants, so that its genotoxic side effects in myeloid cells are minimized and its antitumor effects are maintained. According to our hypothesis, the genotoxic effects of etoposide are due mainly to its myeloperoxidase-catalyzed oxidation to phenoxyl radicals and subsequent redox-cycling that may be prevented or reduced by nutritional antioxidants. Etoposide has a direct inhibitory effect on topo II, a crucial component of its antitumor activity, and this will be unchanged by any manipulations that alter the redox cascade we propose. Studies are now underway to test the validity of our results on normal, human myeloid bone marrow progenitor cells.
Acknowledgments
We are especially grateful to Prof. Billy W. Day for stimulating discussions and his assistance with this and related studies.
Footnotes
- Received November 24, 1998.
- Accepted June 16, 1999.
-
Send reprint requests to: Dr. Valerian E. Kagan, Department of Environmental and Occupational Health, University of Pittsburgh, 260 Kappa Dr., RIDC Park, Pittsburgh, PA 15238. E-mail:Kagan{at}vms.cis.pitt.edu
-
↵1 On leave from the Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Science, St. Petersburg, Russia.
-
Supported by grants from the American Institute for Cancer Research 97-B128, American Cancer Society DHP-125, National Institutes of Health CA 74972, National Institutes of Health ES 09387, the Johns Hopkins Center for Alternatives to Animal Testing 9829 R3, the National Cancer Institute Oncology Research Faculty Development Program (to V.A.T.), and Leukemia Research Foundation fellowship (to Y.Y.T.).
Abbreviations
- PnA
- cis-parinaric acid (Z-9, E-11, Z-15-octadecatetraenoic acid)
- PMC
- 2,2,5,7,8-pentamethyl-6-hydrochromane
- GSH
- glutathione
- DMSO
- dimethyl sulfoxide
- DFO
- deferoxamine mesylate [′-demethylepipodophyllotoxin-9-(4,6-O-ethylidene-β-d-glucopyranosine)]
- PMSF
- phenylmethyl-sulphonyl fluoride
- DMPO
- 5,5-dimethyl-1-pyrrolineN-oxide
- DTPA
- diethylentriaminepentaacetic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}