


Abstract
We examined the subtype-selective binding site of the β-adrenergic receptors (βARs). The β1/β2-chimeric receptors showed the importance of the second and seventh transmembrane domains (TM2 and TM7) of the β2AR for the binding of the β2-selective agonists such as formoterol and procaterol. Alanine-substituted mutants of TM7 of the β2AR showed that Tyr308, located at the top of TM7, mainly contributed to β2 selectivity. However, Tyr308 interacted with formoterol and procaterol in two different ways. The results of Ala- and Phe-substituted mutants indicated that the phenyl group of Tyr308 interacted with the phenyl group in the N-substituent of formoterol (hydrophobic interaction), and the hydroxyl group of Tyr308 interacted with the protonated amine of procaterol (hydrophilic interaction). In contrast to β2AR, TM2 is a major determinant that β1-selective agonists such as denopamine and T-0509 bound the β1AR with high affinity. Three amino acids (Leu110, Thr117, and Val120) in TM2 of the β1AR were identified as major determinants for β1-selective binding of these agonists. Three-dimensional models built on the basis of the predicted structure of rhodopsin showed that Tyr308 of the β2AR covered the binding pocket formed by TM2 and TM7 from the upper side, and Thr117 of the β1AR located in the middle of the binding pocket to provide a hydrogen bonding for the β1-selective agonists. These data indicate that TM2 and TM7 of the βAR formed the binding pocket that binds the βAR subtype-selective agonists with high affinity.
The β-adrenergic receptors (βARs) are members of the seven transmembrane G protein-coupled receptor family and are activated by catecholamine and related molecules. The ligand-binding site of the βAR has been extensively characterized by the use of a variety of techniques (Wong et al., 1988; Dohlman et al., 1988; Savarese and Fraser, 1992; Strader et al., 1994; Hockerman et al., 1996). Initial deletion mutagenesis of the hamster β2AR showed that the hydrophilic loop regions connecting TMs of the receptor are not important for agonist or antagonist binding (Dixon et al., 1987). Point mutations of the hamster β2AR have revealed a key amino acid residue in TM3 (Asp113) that is essential for high-affinity binding of both agonists and antagonists, as well as key residues in TM5 (Ser204 and Ser207) that are assumed to interact with two hydroxyl groups of the catechol ring and be critical for agonist activation of the receptor (Strader et al., 1988, 1989). These data suggested that the ligand-binding domain of the βAR resided within the hydrophobic TMs. Recently, Wieland et al. (1996) reported that Asn293 of the β2AR in TM6 interacts with the β-hydroxyl group of βAR ligands and is responsible for stereoselectivity.
Binding domains of βAR subtype-selective antagonists and an agonist, norepinephrine, were also studied by several groups. Frielle et al. (1988) reported that TM6 and TM7 of βAR appear to play an important role in determining binding of the β1- and β2-selective antagonists such as betaxolol and ICI118551. They also reported that the selectivity of norepinephrine, which shows about 10 times higher affinity for the β1AR than for the β2AR, is largely determined by TM4 of the β1AR. Dixon et al. (1989) showed that TM4 is responsible for β1-selective binding of norepinephrine, using chimeras of the hamster β2AR and the human β1AR. Marullo et al. (1990) examined the ligand-binding regions for β1- or β2-subtype-selective antagonists by analyzing chimeric receptors that replaced several TMs of the β1AR or the β2AR with corresponding TMs of the β2AR or the β1AR. They concluded that no single TM could be responsible for the selectivity of βAR antagonists. Because they exchanged two or more TMs at same time, their methods might not estimate the contribution of a particular single TM to the subtype selective binding. Furthermore, they did not analyze the binding site(s) of highly selective βAR agonists.
Thus, the domains and amino acids responsible for the high-affinity binding of β1- and β2-selective agonists have not been examined so far. We determined recently that the β2-selective agonist binding domain was mainly located in TM7 by using β1/β2-chimeric receptors and β2-selective agonists such as TA-2005 and salmeterol (Isogaya et al., 1998; Kikkawa et al., 1998). We also determined that Tyr308 in TM7 was a main amino acid that determined the high-affinity binding of β2-selective agonists by analyzing alanine-substituted mutants. In the present study, we extended the previous finding to other β2-selective agonists such as formoterol and procaterol. We found that Tyr308 bound β2-selective agonists via hydrophobic or hydrophilic interactions, which were dependent on the structures of ligands. We also determined the amino acids most important for the β1AR to bind the subtype selective agonists with high affinity. We built three-dimensional models of βAR-subtype-selective agonist complexes based on the predicted structure of rhodopsin and the results of mutagenesis experiments.
Experimental Procedures
Materials.
The plasmid constructs pBC-β1 and -β2 encoding for the human β1- and β2ARs were kindly provided by Dr. R. J. Lefkowitz (Duke University, Durham, NC). IPS-339 {(t-butyl-amino-3-ol-2-propyl) oximino-9-fluorene-p-hydroxy-benzoate}, salbutamol, salmeterol, procaterol, formoterol, T-0509 {(−)-(R)-1-(3,4-dihydroxyphenyl)-2-[(3,4-dimethoxyphenethyl)amino] ethanol}, xamoterol, prenalterol, T-1583 {α-(3,4,5-trimethoxyphenethylaminomethyl)-[3,4-dihydroxybenzyl-alcohol] hydrochloride}, denopamine, and dobutamine were synthesized at the Lead Optimization Research Laboratory, Tanabe Seiyaku (Saitama, Japan). The structure of these β1- or β2AR-selective agonists is shown in Fig. 1. (−)Norepinephrine-bitartrate, (±)propranolol, and DEAE-dextran were obtained from Sigma Chemical Co. (St. Louis, MO). Dulbecco’s Modified Eagle’s medium and gentamicin were from Life Technologies, Inc., (Rockville, MD). Taq and Pfu DNA polymerases were obtained from Takara (Siga, Japan) or Stratagene (La Jolla, CA), respectively. GTP was purchased from Seikagaku (Tokyo, Japan).125I-labelled cyanopindolol (125I-CYP) was obtained from Amersham Pharmacia Biotech (Arlington Heights, IL) or New England Nuclear (Boston, MA). Fetal bovine serum was from JRH Biosciences (Lenexa, KS).
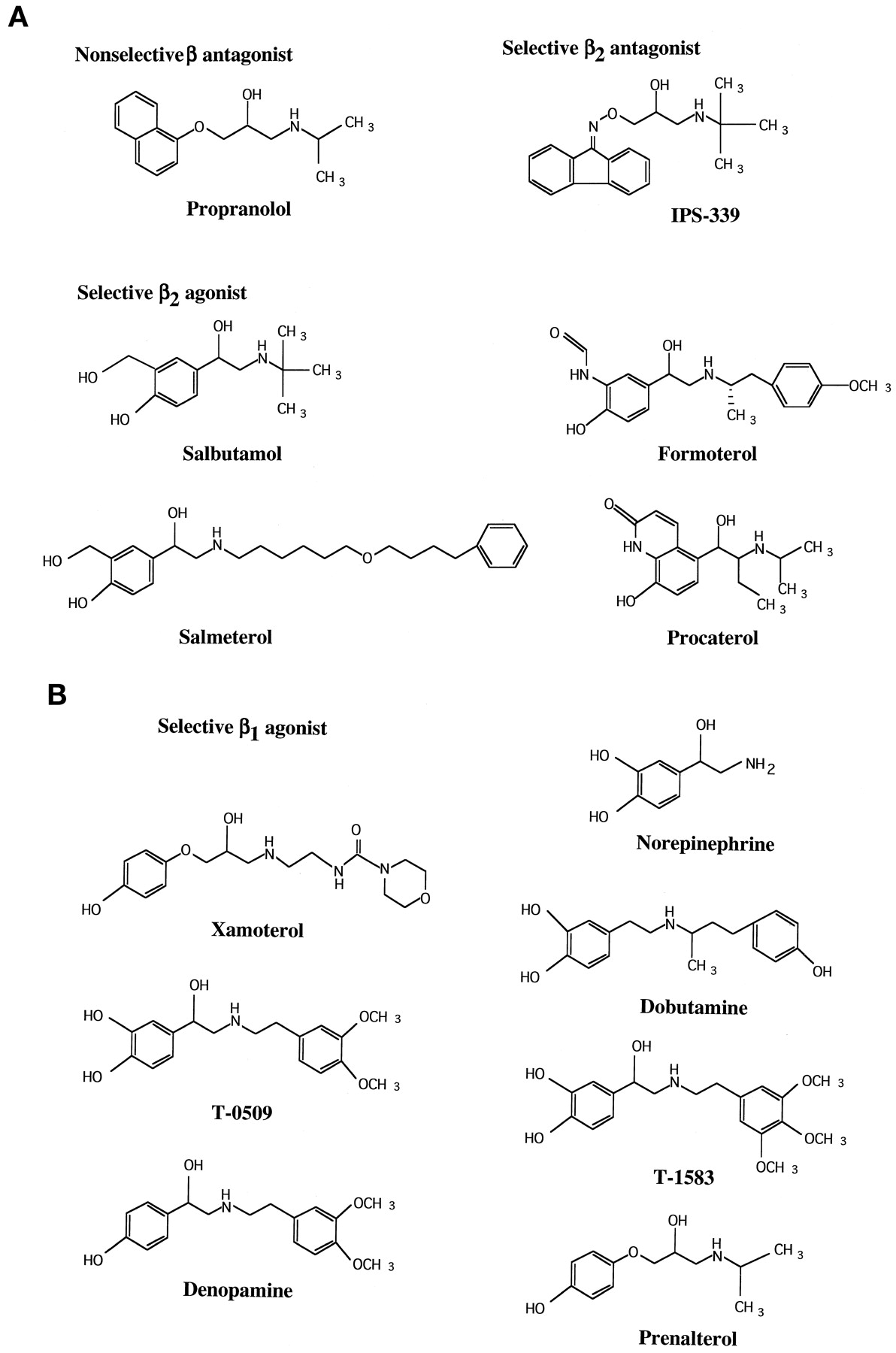
A, the structures of β2AR-selective or -nonselective agonists and antagonists. B, the structures of the β1AR-selective agonists.
Construction of Chimeric β1/β2ARs and Alanine-Substituted βAR Mutants.
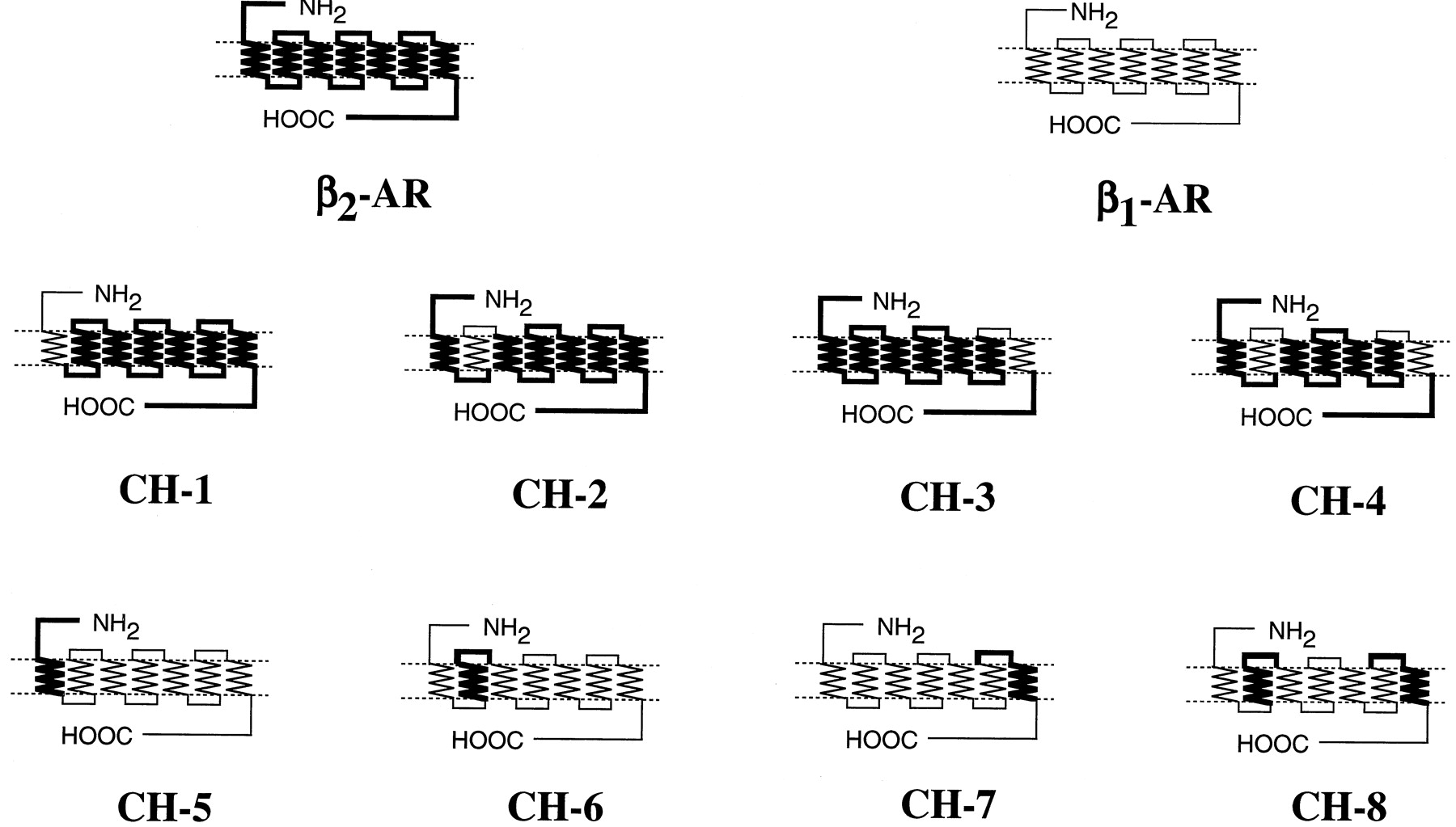
Chimeric β1/β2-receptors were constructed by polymerase chain reaction techniques withTaq or Pfu DNA polymerase as described (Higuchi, 1989). The sequences of the amplified regions were confirmed by the dideoxy chain termination method (Sanger et al., 1977). The amplified region was combined with the rest of the β1- or β2AR sequences to obtain full-length β1/β2 chimeras, and chimeric cDNAs were finally inserted into the EcoRI andBamHI or EcoRI and SalI sites of mammalian expression vector pCMV5. The positions of the junction for individual β1/β2-chimeric receptors are as follows (numbers refer to amino acid positions in the human β1- and β2AR sequences): CH-1, β11–84/β2 60–413; CH-2, β2 1–71/β197–131/β2 107–413; CH-3, β2 1–295/β1347–381/β2 331–413; CH-4, β2 1–71/β197–131/β2 107–295/β1347–381/β2 331–413; CH-5, β2 1–59/β1 85–477; CH-6, β1 1–96/β272–106/β1 132–477; CH-7, β1 1–346/β2296–330/β1 382–477; CH-8, β1 1–96/β272–106/β1 132–346/β2296–330/β1 382–477 (Fig. 2). Alanine-substituted mutants of the β1- and β2ARs and phenylalanine-substituted mutant of the β2AR were constructed by PCR using the Quick Change site-directed mutagenesis method as described by Isogaya et al. (1998). After the mutations were confirmed, the fragments containing the substitutions were ligated with other portions of the receptors. The expression vector pCMV5 was used for the alanine-substituted mutants except for the M98A-β1AR. The expression vector pEF/myc/cyto was used for the mutant due to low expression with pCMV5.
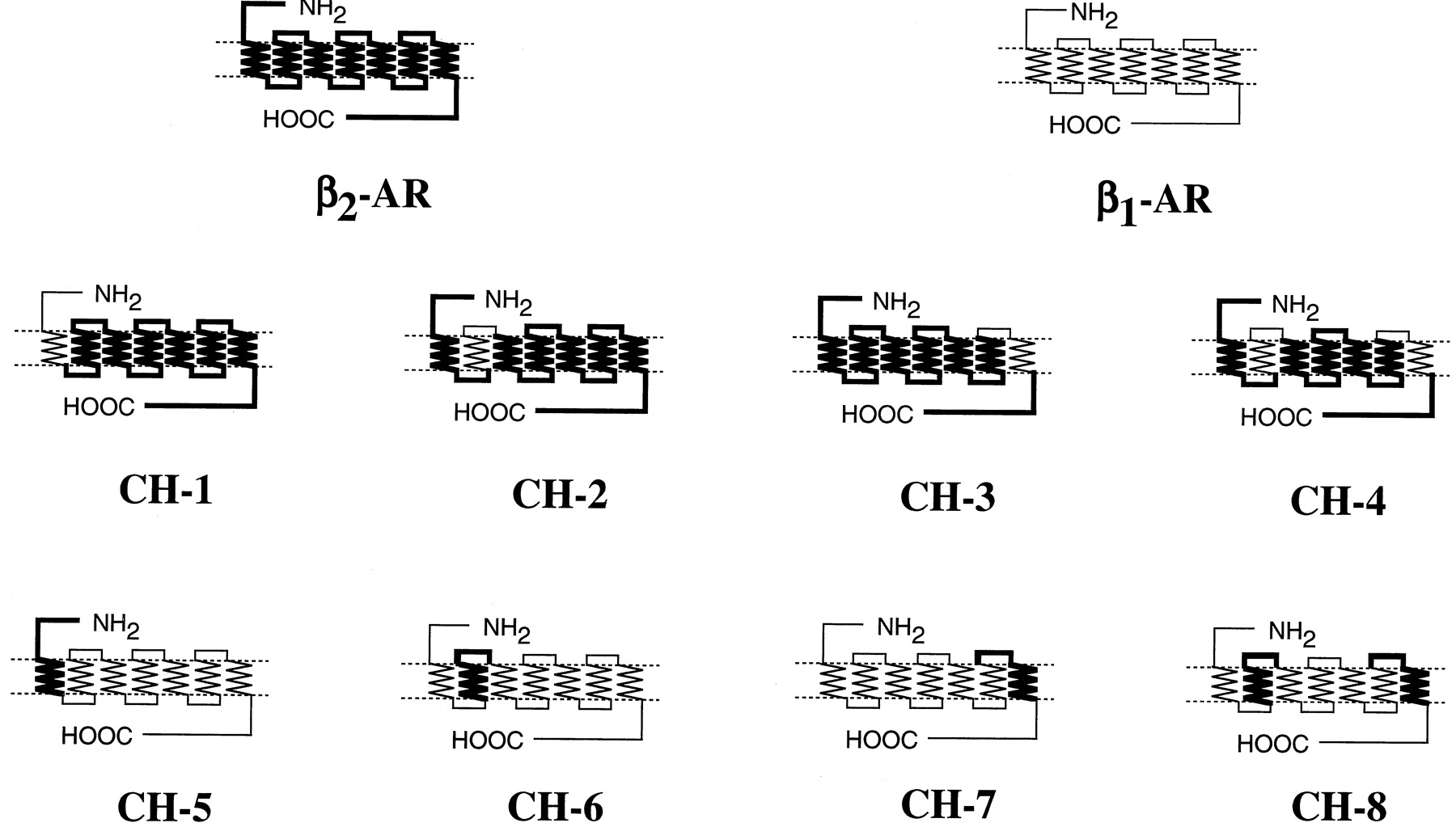
The structures of β1/β2chimeric (CH) receptors. The peptide sequences of the β1AR are shown by thin lines and those of the β2AR are indicated by thick lines. The positions of the junctions are described under Experimental Procedures.
Transient Expression of Wild Type (WT) or β1/β2-Chimeric Receptors in COS-7 Cells.
The cDNAs encoding for the human β2AR in pBC12BI, the human β1AR, or the β1/β2-chimeric receptors in pCMV5 were transfected into COS-7 cells by the DEAE-dextran method (Cullen, 1987). Before the day of transfection, COS-7 cells were seeded at 1.0 to 1.5 × 106 cells/100-mm dish. The amount of the WT or β1/β2-chimeric receptor cDNAs was 5 μg/100-mm dish. All cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and gentamicin (10 μg/ml). Two to three days after the transfection, the cells were harvested for preparation of the crude membrane fraction.
Membrane Preparation.
The COS-7 cells were rinsed with 10 ml of ice-cold PBS and mechanically detached in 1 ml of an ice-cold lysis buffer containing 10 mM Tris-HCl (pH 7.4), 5 mM EDTA, 5 mM EGTA, 10 μg/ml benzamidine, 10 μg/ml soybean trypsin inhibitor (TypeII-S), and 5 μg/ml leupeptin. The lysate was centrifuged at 45,000g for 10 min at 4°C. The pellet was resuspended in 1 ml of a lysis buffer with a Potter type homogenizer and stored at −80°C until use. Protein concentration was determined by the method of Lowry et al. (1951).
Radioligand Binding Assay.
Radioligand binding studies were carried out in a buffer containing 75 mM Tris-HCl (pH 7.4), 12.5 mM MgCl2, and 2 mM EDTA in the presence of 100 μM GTP at 37°C for 60 min using 0.2 to 10 μg of membrane protein. Competition binding assays were performed using the indicated concentration of 125I-CYP and various concentrations (0–10 mM) of unlabeled ligands in the presence of 100 μM GTP. The binding reaction was terminated by the rapid filtration over Whatman GF/C filters and washed three times with the solution containing 25 mM Tris-HCl (pH 7.4) and 1 mM MgCl2. Nonspecific binding was determined in the presence of 5 μM (±)propranolol. The radioactivity remaining on the filter was counted by a gamma counter.
Data Analysis.
All data shown are mean values ± S.E. for n determinations. Equilibrium dissociation constants were determined from saturation isotherms. Radioligand binding data obtained from competition curves were analyzed by a nonlinear regression analysis to determine EC50 values andK i values using PRISM software (GraphPad Software Inc., San Diego, CA). Statistical significance was assessed with one-way ANOVA for the multiple comparisons using JMP software (SAS Institute, Cary, NC). ANOVA post hoc comparisons were made with the Dunnett’s test.
Computer Modeling of β-Selective Agonists-β1- or β2AR Complexes.
The initial coordinates of the backbone and side chain atoms were modeled by assigning the amino acids of the β1- and β2ARs to the model of rhodopsin built byBaldwin et al. (1997), using the Biopolymer module of SYBYL software package (TRIPOS Assoc., St. Louis, MO). The side chain conformations were optimized by the dead-end algorithm with the “large-size” rotamer library (Desmet et al., 1992; Tanimura et al., 1994). The selective agonists (procaterol, formoterol, or denopamine) were docked to the β2- or β1ARs manually by satisfying the following established interactions: Asp113 of the β2AR (Asp134 of the β1AR) with the protonated amine, Ser204(Ser229) and Ser207(Ser232) with catechol or equivalent entities, Asn293 (Asn344) with the hydroxyl group at the β position, and Phe290(Phe341) with the phenyl ring or equivalent groups. After docking procedures, the entire structures were energy minimized with positional restraints on the Cα atoms in the transmembrane helices by MAXIMIN2 of SYBYL software (TRIPOS Assoc.).
Results
Affinities of Propranolol for β1/β2AR Chimeras.
We constructed a series of β1/β2-chimeric receptors. Because a nonselective agonist isoproterenol binds to the βAR at at least three sites (i.e., Asp113, Ser204, and Ser207) and because β-selective agonists often have substituents at an amino group, we focused on TM1, TM2, and TM7 of the βAR to allow the βAR to bind the subtype-selective agonists with high affinity. We used salbutamol, formoterol, and procaterol for β2-selective agonists (see Fig.1A for structures) and T-0509, T-1583, xamoterol, denopamine, dobutamine, and norepinephrine for β1-selective agonists (Fig. 1B). One of TM1, TM2, or TM7, or both TM2 and TM7 of the β2AR were replaced by the homologous regions of the β1AR. These chimeric receptors were termed CH-1, CH-2, CH-3, and CH-4. On the other hand, one of TM1, TM2, or TM7 or both TM2 and TM7 of β1AR were replaced by the homologous regions of the β2AR. They were termed CH-5, CH-6, CH-7, and CH-8 (Fig.2). Table 1shows that the affinities of propranolol for CH-1 to CH-4 were essentially the same as those of the WT β2AR. Although the affinities of propranolol for the three chimeras (CH-5, CH-7, and CH-8) were significantly changed by the introduction of TMs of the β2AR into the β1AR, the changes in the affinities were relatively small compared with those of selective agonists (Table 1). Furthermore, the increases of the affinities for the chimeras did not accompany the decreases of the affinities for the reciprocal chimeras. This suggested that the increases of the affinities for propranolol were not specific for a particular TM.
Effects of replacement of transmembrane regions with corresponding portions of the β1AR on ligand-binding characteristics of the β2AR
Affinities of Formoterol and Procaterol for β1/β2AR Chimeras.
Formoterol and procaterol are highly β2 selective. The ratios of K i values [K i(β1) toK i(β2)] of formoterol or procaterol were 88 or 114, respectively (Table 1). The replacement of TM2 of the β2AR with that of the β1AR (CH-2) decreased the affinities of the two agonists about 7- to 17-fold (Table 1). The contribution of TM1 and TM7 to β2 selectivity of the two agonists was low compared with the contribution of TM2. The affinities of the two agonists were further decreased by the replacement of TM7 together with TM2 of the β2AR (CH-4), and the affinities for the reciprocal mutant of CH-4 (CH-8) were increased nearly to the same values as for the WT β2AR (Table 1). This indicated that both TM2 and TM7 determined the high-affinity binding of formoterol and procaterol.
Affinities of Salbutamol for β1/β2AR Chimeras.
Salbutamol was less potent and selective than formoterol and procaterol [ratio ofK i(β1) toK i(β2) is about 10] (Table 1). The affinity of salbutamol for the β2AR was 2 to 3 orders of magnitude lower than those of the other β2-selective agonists. The affinities of salbutamol were significantly decreased by the replacement of TM2 of the β2AR with that of the β1AR and were increased by the introduction of TM1 or TM2 of the β2AR into the β1AR (Table 1). Although the affinity of salbutamol was decreased in CH-1, the change in the affinity was relatively small compared with that of CH-2 (less than 3-fold versus more than 6-fold). These results suggested that the contribution of TM1 to β2-selective binding of salbutamol was small. The replacement of TM7 of the β2AR with that of the β1AR did not change the affinity of salbutamol for the resulting chimera (CH-3). When both TM2 and TM7 of the β2AR (or the β1AR) were replaced with those of the β1AR (or the β2AR), the chimeras (CH-4 or CH-8) showed the decreased (or increased) affinities for salbutamol (Table 1). These results suggested that the β2 selectivity of salbutamol was mainly determined by both TM2 and TM7.
Affinities of the β2-Selective Antagonist IPS-339 for β1/β2AR Chimeras.
One of the β2AR-selective antagonists, IPS-339, showed about 40 times higher affinity for the β2AR than for the β1AR (Table 1). When TM1, TM2, or TM7 of the β2AR were replaced by the corresponding regions of the β1AR (CH-1 to CH-4), the affinities of IPS-339 for these chimeras were decreased by 3.5- to 8.2-fold (Table 1). On the other hand, the transfer of TM1 or TM2 from the β2AR to the β1AR (CH-5 and CH-6) did not increase the affinities of IPS-339 for these chimeras (Table 1). The slightly increased affinity of IPS-339 caused by transferring TM7 from the β2AR to the β1AR suggested that the major determinant of β2AR selectivity of IPS-339 was TM7, in spite of structural differences between IPS-339 and the β2AR-selective agonists.
Affinities of Synthetic β1-Selective Agonists for β1/β2AR Chimeras.
We examined the βAR selectivity of T-0509, denopamine, xamoterol, dobutamine, T-1583, and prenalterol. These are known as β1-selective agonists when administered to whole animals or to isolated tissues. Dobutamine, T-1583, and prenalterol showed little β1 selectivity in the binding experiments. The ratios of K ivalues [K i(β1) toK i(β2)] of these agonists were less than 3-fold (Table 2). We therefore did not study these agonists in detail. Among these agonists, T-0509, xamoterol, and denopamine showed significantly higher affinities for the β1AR than for the β2AR. The binding experiments using the recombinant βARs expressed in COS-7 cells showed that the selectivity of these agonists was relatively low, compared with the selectivity of β2-selective agonists such as procaterol and formoterol (Table 2). Replacement of TM2 of the β1AR with the homologous region of the β2AR (CH-6) decreased the affinities of these three agonists, and transfer of TM2 of the β1AR to the β2AR (CH-2) increased the affinities of these agonists to nearly same values as for the WT β1AR (Table 2). The effect of replacement of TM2 together with TM7 on xamoterol binding was essentially the same as the effect of replacement of TM2 alone. Although the affinities of T-0509 and denopamine for CH-6 (the β1AR with TM2 of the β2AR) were decreased, those for the CH-8 (the β1AR with TM2 and TM7 of the β2AR) were increased, for an unknown reason (Table 2). These data suggested that TM2 of the β1AR determines the β1selectivity, even though it is not a sole determinant of the β1-selective binding site.
Effects of replacement of transmembrane regions with corresponding portions of the β2AR on ligand-binding characteristics of the β1AR
Affinities of the Endogenous β1-Selective Agonist Norepinephrine for β1/β2AR Chimeras.
Norepinephrine is the endogenous β1-selective agonist. We confirmed the β1 selectivity of norepinephrine (ratio of Ki(β2) to Ki(β1) is about 9.0) (Table 3). We also examined the affinities of norepinephrine for the various β1/β2AR chimeras. The replacement of TM7 but not TM2 of the β1AR with the homologous region of the β2AR decreased the affinity of norepinephrine. These results indicated that TM7 contributed to β1-selective binding of norepinephrine, which constrasted with synthetic β1-selective agonists. The importance of TM7 for β1-selective binding was further supported by the finding that the introduction of TM7 of norepinephrine of the β1AR into the β2AR (CH-3) increased the affinity of norepinephrine. These data suggested the contribution of different TMs to subtype-selective binding of structurally different β1-selective agonists.
Effects of replacement of transmembrane domains of β1AR with corresponding regions of the β2AR on binding characteristics of norepinephrine
Effects of Substitution of Amino Acids with Alanine in TM7 of the β2AR on Binding of Procaterol and Formoterol.
We have recently reported that TM7 of the β2AR played an important role in determining the high-affinity binding of β2-selective agonists such as TA-2005 and salmeterol, and Tyr308 in TM7 was the most important amino acid for the high-affinity binding (Isogaya et al., 1998; Kikkawa et al., 1998). To examine the role of TM7 for binding of procaterol and formoterol in detail, we expressed alanine-substituted mutants of the β2AR, in which the amino acids in TM7 of the β2AR different from those of the β1AR were individually changed to alanine. Because the effect of exchange of TMs between the β1AR and β2AR on the binding characteristics of salbutamol is relatively small compared with formoterol and procaterol, we did not examine the binding characteristics of salbutamol for the alanine-substituted mutants. TheK d values of these muatants for125I-CYP is essentially the same as that of the WT β2AR, indicating that the substitution did not cause nonspecific alterations of the binding sites (Table4). Among mutants, the Y308A-β2AR is the only one that showed significantly decreased affinities for formoterol and procaterol. A similar conclusion indicating the importance of Tyr308 was obtained from the previous reports (Isogaya et al., 1998; Kikkawa et al., 1998), using TA-2005 and salmeterol as β2-selective agonists. Procaterol also showed decreased affinity for the L324A-β2ARs. The amino acid of the β1AR at the homologous position of Tyr308 is Phe instead of Ala. We made a mutant in which Tyr308 is replaced with Phe, and we examined the binding characteristics of these agonists. Although the affinity of formoterol was not significantly decreased by the replacement, the affinity of procaterol was decreased by the replacement. The K i value of procaterol for the Y308F-β2AR is essentially the same as that of the Y308A-β2AR. The differential susceptibility of formoterol and procaterol to hydroxyl group at Tyr308 indicated that the high-affinity binding of procaterol but not formoterol required the hydroxyl group of Tyr308. We also found that the affinity of salmeterol did not decrease in the Y308F-β2AR, indicating the importance of hydrophobic interaction for β2-selective, high-affinity binding of salmeterol (Table 4). The affinity of the nonselective β-agonist isoproterenol for the Y308F-β2AR showed essentially the same value as that for the WT β2AR. The differential susceptibility of isoproterenol and β2-selective agonists to the removal of the hydroxyl group from Tyr308 indicated that the effect of the removal of the hydroxyl group from Tyr308 is specific for β2-selective agonists.
Effects of replacement of amino acids in TM7 of the β2AR with alanine or phenylalanine on ligand-binding characteristics of the β2AR
Effects of Substitution of Amino Acids with Alanine in TM2 of β2AR on Binding of Procaterol and Formoterol.
To examine the contribution of individual amino acids in TM2 to the binding of β2-selective agonists, the amino acids in TM2 of the β2AR that were different from those of the β1AR were changed to alanine. It is assumed that alanine makes a hole at the position of the replacement without altering the conformation of the amino acid with the cognate ligand (Clackson and Wells, 1995; Holst et al., 1998). TheK d values of 125I-CYP for these mutants were almost same as that of the WT β2AR, indicating that TM2 did not contribute to the binding of 125I-CYP. It is reasonable to assume that TM2 retained the same conformation as in the WT-β2AR (Table5). Among eight mutants, only H93A-β2AR showed significantly decreased affinity for procaterol. The other mutants did not show significantly decreased affinities for procaterol, formoterol, or salmeterol. Although replacement of TM2 of the β2AR with that of β1AR decreased the affinities 32-fold for salmeterol and 7-fold for formoterol (Table 1, and see Table 1 inIsogaya et al., 1998, for salmeterol), we could not identify the specific amino acid(s) that contributed to the high-affinity binding for these agonists. These results suggested that TM2 contributes to selective binding as a whole entity and that a specific amino acid is not important for the high-affinity binding of β2-selective agonists.
Effects of replacement of amino acids in TM2 of the β2AR with alanine on ligand-binding characteristics of the β2AR
Effects of Substitution of Amino Acids with Alanine in TM2 of β1AR on Binding of T-0509, Xamoterol, and Denopamine.
The contribution of each amino acid in TM2 of the β1AR to β1-selective agonists was examined by expressing and characterizing the alanine-substituted β1AR mutants. The affinities of T-0509 and denopamine were significantly decreased in the mutants that substituted alanine at Leu110, Thr117, and Val120 (Table6). The structure of denopamine is the same as that of T-0509 except that denopamine lacks a hydroxyl group at the meta position in the catechol ring (Fig. 1B). It is reasonable to assume, therefore, that the mutation of same amino acids decreased the affinities for both agonists. The decreases of the affinities of T-0509 and denopamine were significant but slightly smaller (2- to 6-fold) than the decreases in affinities of these substances for the chimeric receptors (∼10-fold). The affinities of xamoterol were not significantly decreased by any substitution of amino acids in TM2.
Effects of replacement of amino acids in TM2 of the β1AR with alanine on ligand-binding characteristics of the β1AR
Computer Modeling of β-selective Agonist-βAR Complexes.
We built three-dimensional models of β-selective agonist-βAR complexes to visualize the binding pocket. The structural models of the β1 and β2ARs were built on the basis of the predicted structure of rhodopsin simulated by Baldwin et al. (1997) (Fig.3). General features of the present models are as follows. First, the amino acids of TM2 and TM7 form a binding pocket that can interact with N-substituents of the selective agonists. Second, Tyr308 in TM7 locates at the top of the binding pocket and covers the binding pocket from the upper side. Third, the binding pocket mainly consists of hydrophobic residues. Procaterol, formoterol, and denopamine were well fitted to the binding pocket of the model. The phenyl group of Tyr308 of the β2AR covers the β2-selective agonists (procaterol and formoterol) from the top of the pocket and acts like a “barrier” to prevent the ligand from moving freely into extracellular space (Fig. 3, A and B). The N-substituent of formoterol goes a little farther into the binding pocket formed by TM2 and TM7 than does theN-substituent of procaterol (Fig. 3A). Then Tyr308 interacts with the phenyl ring of theN-substituent of formoterol, mainly through hydrophobic interaction. This is consistent with the result that the change of Tyr308 to Phe did not significantly decrease the affinity of formoterol. When Tyr308 is mutated to alanine, alanine cannot inhibit movement of theN-substituents of the agonists to extracellular space. This mutation resulted in the receptors that showed decreased affinities for the β2-selective agonists.

Three-dimensional models of β2- and β1-selective agonist-βAR complexes. Three-dimensional models were built by the predicted structure of rhodopsin by simulating with SYBYL. The possible interaction sites between the βAR and selective agonists are shown. A, side view of formoterol-β2AR complex. B, side view of procaterol-β2AR complex. C, side view of denopamine-β1AR complex. The interaction sites of formoterol and procaterol with the β2AR are as follows: Ser204 and Ser207 in TM5 with catechol or an equivalent group of the ligands, Phe290 in TM6 with a phenyl or carbostiryl group of the ligands, Asn293 in TM6 with a hydroxyl group at the β position, Asp113 in TM3 with the protonated amine. Putative interaction sites of denopamine with the β1AR are as follows: Ser232 in TM5 with a hydroxyl group at the para position, Phe341 in TM6 with a phenyl group, Asn344 in TM6 with a hydroxyl group at β position, Asp138 in TM3 with the protonated amine. Phe359 in TM7 of the β1AR is the homologous amino acid of Tyr308of the β2AR. Other amino acids that were assigned to interact with the ligands are mentioned in the text.
The N-substituent (isopropyl group) of procaterol locates near Tyr308 due to the presence of an ethyl group at the α position, and the protonated amine of procaterol is positioned to interact with the hydroxyl group of Tyr308 (Fig. 3B). The replacement of Tyr308 with Phe decreased the affinity of procaterol, possibly due to disruption of the interaction between the hydroxyl group of Tyr308 and the protonated amine. Although the replacement of His93 caused a decreased affinity for procaterol, it is unlikely that His93 interacts directly with procaterol on the basis of the model (Fig. 3B).
The amino acids of TM2 and TM7 of the β1AR form a binding pocket as they do in β2AR (Fig. 3C). The results of Ala substitution in β1AR showed that Leu110, Thr117, and Val120 were important amino acids for β1-selective agonist binding (Table 6). Of these three amino acids, only Thr117 seemed to be positioned to interact directly with the methoxy group of theN-substituents of denopamine and T-0509. The results also suggested that Leu110 and Val120 contribute indirectly to the β1-selective binding through hydrophobic interaction, because Leu110 and Val120 cannot reach the methoxy group (Fig. 3C).
Space-filling models of TM2 and TM7 showed the differences between the binding pockets (distribution and orientation of hydrophobic and polar amino acids) of the β1- and β2ARs (Fig. 4). In the β1AR, polar amino acids of TM2 and TM7 faced each other and hydrophobic aromatic amino acids, which are assumed to interact with the N-substituents of the β-selective agonists located near extracellular space. The β1AR model suggested that the selective ligands may not deeply enter the binding cleft consisting of hydrophobic amino acids in TM2 and TM7, because the side chains of polar amino acids may interfere with the access of the N-substituents of the ligands to the binding pocket. There are not as many polar amino acids in TM2 and TM7 of the β2AR as there are in the β1AR. Hydrophobic interaction between theN-substituents of ligands and TMs of the β2AR may be more stable than the interaction between the β1-selective ligands and the amino acids in TM2 and TM7 of the β1AR.

Space-filling models of TM2 and TM7 of the β1- and β2ARs. The left panel is a model of TM2 and TM7 of the β1AR, and the right panel is a model of TM2 and TM7 of the β2AR. Hydrophobic (aromatic) amino acids consisting of Phe, Trp, and Tyr are shown in green. Hydrophobic (aliphatic) amino acids consisting of Ala, Ole, Leu, Met, Val, and Pro are shown in yellow. Polar or charged amino acids consisting of Asn, Asp, Cys, Gln, Glu, Arg, Lys, Ser, His, and Thr are shown in cyan. The back bone is shown in white.
Discussion
We have demonstrated that the affinities of the synthetic β1-selective agonists such as T-0509, xamoterol, and denopamine were increased or decreased by transferring TM2 of the β1AR to the β2AR or TM2 of the β2AR to the β1AR. This indicates that TM2 of the β1AR is a major determinant of the high-affinity binding of the β1-selective agonists. In contrast with the β1AR, the replacement of TM2 of the β2AR with the homologous region of the β1AR decreased the affinities of β2-selective agonists, and introduction of TM2 or TM7 into the β1AR partially restored the high-affinity binding. Furthermore, the affinities for the β1AR with both TM2 and TM7 of the β2AR became close to the values for the WT β2AR. These data on loss-of-function and gain-of-function mutants suggest that TM2 of the β1AR is a major determinant for the β1-selective agonist to bind to the receptor with high affinity, and that both TM2 and TM7 of the β2AR determine the high-affinity binding of the β2-selective agonists.
There are several reports that the specific amino acids in TM2 and TM7 are close together and are functionally interacting (Zhou et al., 1994;Sealfon et al., 1995; Perlman et al., 1997). In addition to these reports, Ballesteros et al. (1998) recently reported that Arg, which is located at the bottom of TM3 and is well conserved among G protein-coupled receptors, interacts with Asn in TM2 and Asp in TM7 in the gonadotropin-releasing hormone receptor. It is possible that amino acids in TM2 and TM7 form the binding pocket in a cooperative manner and provide the site for high-affinity binding of the β-selective agonists.
The structures of T-0509, T-1583, and denopamine are similar to each other (Fig. 1B). Among these agonists, T-1583 did not show β1AR selectivity. This suggests that the positions of methoxy groups on the phenyl ring extending from the protonated amine are important for the β1-selective binding, and three methoxy groups are not accommodated by the binding pocket formed by TM2 and TM7. The selectivity of denopamine was lower than that of T-0509, suggesting that the hydroxyl group of the phenyl ring at the metaposition also contributes in part to β1selectivity, possibly through interaction with Ser229 in TM5 of the β1AR or the homologous amino acid Ser204 in TM5 of the β2AR. Xamoterol has a long side chain extending from the protonated amine and shows relatively high affinity for the β1AR, compared with other β1-selective agonists. It indicates that a long side chain (N-substituent) may be necessary to reach TM2, which plays an important role in the β1selectivity.
Norepinephrine is the endogenous catecholamine that shows β1AR selectivity. Two groups of researchers have reported (Frielle et al., 1988; Dixon et al., 1989) that TM4 of the β1AR is the region that is responsible for the β1-selective binding of norepinephrine. In contrast with the previous works, the present study showed that TM7 was a primary region that determined the β1-selective binding of norepinephrine. However, the authors of the previous studies evaluated the β1 selectivity based on the ratios ofK i values of norepinephrine and epinephrine. When they evaluated the β1selectivity with the K i values of norepinephrine, their results are consistent with our findings. The replacement of TM1 to TM6 of the β1AR with those of the β2AR did not confer the high-affinity binding of norepinephrine on the chimera (Frielle et al., 1988).
Figure 5 illustrates the amino acids in TM2 and TM7 that are different between the β1AR and the β2AR. Because agonist binding domains are assumed to be located within TMs, and 42% of the amino acids in TM7 of the β1AR and the β2AR are different, compared with 29% in TM2, TM7 may be a more favorable target for the subtype-selective agonist.

The aligned amino acid sequences of TM2 and TM7 of the human β1- and β2ARs. The numbers refer to the first methionine of the β1- or β2AR as first amino acid. The same amino acids between the β1- and β2ARs are shown in the squares.
Space-filling models suggested that the polar amino acids of the binding pockets consisting of TM2 and TM7 of the β1- and β2ARs are differentially located and orientated. Although the selectivities of β1AR-selective agonists are at most 10-fold, the affinities of β2-selective agonists are high for the β2AR and low for the β1AR. This difference may be explained by the differential location of polar amino acids in TM2 and TM7. The β1AR contains Phe359 at a position homologous to Tyr308 of the β2AR, an amino acid that is critical for the high-affinity binding of the β2-selective ligands. However, because there are polar amino acids around Phe359, the N-substituents of the β1-selective agonists cannot be accommodated by polar amino acids in TM2 and TM7. This also suggests that the design of β1-selective agonists may be more complex than that of β2-selective agonists because ligands should contain both hydrophobic and hydrophilic parts in an appropriate position and orientation to interact with polar and hydrophobic amino acids in TM2 and TM7. T-0509 and denopamine, but not T-1583, showed the β1 selectivity. The only difference between the three agonists is that T-1583 contains three methoxy groups in itsN-substituent, compared with the other two agonists, which have two methoxy groups. The interaction between the β1-selective agonists and TM2 and TM7 may be interfered with by repulsion between polar amino acids in TM2 and TM7 of the β1AR and the third methoxy group of T-1583.
A three-dimensional model of β-selective agonist-βAR complexes revealed a unique binding pocket formed by TM2 and TM7, which can explain the binding characteristics of β-selective agonists for the mutated β1- or β2ARs. We previously reported that Tyr308 in TM7 of the β2AR played a major role in the binding of β2-selective agonists such as TA-2005 and salmeterol with high affinity (Isogaya et al., 1998; Kikkawa et al., 1998). We extended the previous observation to other β2-selective agonists such as procaterol and formoterol and proposed that Tyr308, which is located at the top of TM7, plays two roles in the binding of selective agonists, as determined by mutagenesis and three-dimensional modeling. The first role of Tyr308 is to provide high-affinity binding via hydrophobic or hydrophilic interactions with the β2-selective agonists. The second role of Tyr308 is to prevent N-substituents of the selective agonists from freely moving into extracellular space. The affinities of salmeterol and formoterol were decreased in Y308A-β2AR but not Y308F-β2AR. However, the affinities of procaterol were decreased in both Y308A- and Y308F-β2ARs. This discrepancy could be explained by the different types of interactions between the β2-selective agonists and Tyr308, that is hydrophobic and hydrophilic interactions. Alanine substitution cannot complement the interaction and block free movement of the N-substituent of the ligand from the binding pocket. The chimeric receptor, in which TM7 of the β1AR is introduced into the β2AR, did not show significantly decreased affinities for procaterol and formoterol. However, the Y308A-β2AR mutant, in which Tyr308 in TM7 is replaced with alanine, did show decreased affinities for both agonists. This apparent discrepancy can be explained by the fact that the amino acid of the β1AR that is homologous to Tyr308 of the β2AR is Phe.
It is interesting to try to understand how the amino acids contributing to high-affinity binding participate in the activation steps, because TM7 changes the conformation and contributes to activation of the receptors upon agonist binding (Wess et al., 1993; Abdulaev and Ridge, 1998). It is possible that TM2 and/or TM7 involve not only the high-affinity binding of the selective agonists but also the activation step.
In conclusion, binding domains of βAR subtype-selective agonists appeared to be localized in TM2 and TM7. We showed that TM2 was especially important for β1 selectivity, that both TM2 and TM7 were important for β2selectivity, and that interaction of the binding pocket formed by TM2 and TM7 of the β1- or β2ARs with N-substituents of the subtype-selective agonists is essential for high-affinity binding. We identified several amino acids that are important for the β1 or β2 selectivities. However, our data do not exclude the possibility that other amino acids in TM2 and TM7 participate in subtype-selective binding for the agonists that have different structures from those of the agonists examined in this report.
Acknowledgments
We thank Dr. R. J. Lefkowitz for the pBC-β1 and pBC-β2plasmids.
Footnotes
- Received May 27, 1999.
- Accepted July 22, 1999.
-
Send reprint requests to: Hitoshi Kurose, Ph.D., Laboratory of Pharmacology and Toxicology, Graduate School of Pharmaceutical Sciences, University of Tokyo, 7–3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail:kurose{at}mol.f.u-tokyo.ac.jp
-
↵1 Current address: Toray Industries, Inc., Basic Research Laboratories, 1111 Tebiro, Kamakura, Kanagawa 248-8555, Japan.
-
↵2 Current address: Lead Optimization Research Laboratory, Tanabe Seiyaku Co., Ltd., 2-2-50 Kawagishi, Toda-shi, Saitama 335-8505, Japan.
-
This work was supported in part by grants from the Ministry of Education, Science, Sports, and Culture of Japan (to T.N.) and the Mochida Memorial Foundation for Medical and Pharmaceutical Research (to H. Kurose).
Abbreviations
- βAR
- β-adrenergic receptor
- TM
- transmembrane domain
- CYP
- cyanopindolol
- CH
- chimera
- WT
- wild type
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}