


Abstract
Rifampicin, an antibiotic widely used in tuberculosis therapy, is known to exert psychotropic side effects in some patients. Recently, rifampicin has been reported to activate the glucocorticoid receptor (GR) in human hepatocytes. Because there is evidence that increased levels of glucocorticoids may induce cognitive impairment, sometimes culminating in depression, the side effects of rifampicin may result from GR activation in central nerve cells. Therefore, we used reporter gene assays to determine whether rifampicin displays glucocorticoid-like effects in human neuroblastoma SK-N-MC cells or mouse hippocampal HT22 cells. Rifampicin was unable to elicit any detectable transactivation of GR in both cell types, whereas cortisol or dexamethasone led to a potent transcriptional response. Rifampicin was also inactive in the same HepG2 cell line that was originally used to demonstrate the effect of rifampicin on GR. Moreover, rifampicin was unable to compete with dexamethasone for binding to GR. Finally, by blocking the multidrug resistance P-glycoprotein transporter (a xenobiotic extrusion pump) with verapamil or cyclosporin A, we excluded the possibility that the lack of effect by rifampicin was due to its export from the cell. Our results establish that rifampicin does not activate GR, and rule out the hypothesis that the psychotropic side effects of rifampicin treatment are a consequence of GR activation.
The glucocorticoid receptor (GR) belongs to the superfamily of ligand-modulated transcription factors (Mangelsdorf et al., 1995) that can regulate gene transcription by activation as well as by repression (Beato et al., 1995). GR is activated on binding of glucocorticoids (GCs) in various tissues, e.g., lung, liver, bone marrow, immune system, and brain, and thus plays a central role in many physiological and developmental processes (Evans-Storms and Cidlowski, 1995; Schmid et al., 1995; De Kloet et al., 1998).
Because GR shows anti-inflammatory and immunosuppressive activities, numerous synthetic GCs have been developed over the past decades. Some of these are used in the treatment of chronic inflammatory diseases such as asthma, rheumatoid arthritis, inflammatory bowel disease, and autoimmune diseases (Kimberly, 1994). Recently, the antibiotic rifampicin has been presented as a novel and unexpected ligand for GR that can induce its transcriptional regulation capabilities (Blanchard, 1998; Calleja et al., 1998b). This finding is surprising to the clinician because one would expect immunosuppressive side effects if rifampicin did indeed exert GC-like properties at GR. However, despite its widespread use among patients suffering from tuberculosis (Raviglione et al., 1995), these side effects have not been reported for rifampicin. This raises some questions about the interaction of rifampicin with GR. In fact, with human alveolar A549 cells, Jaffuel et al. (1999) recently found that rifampicin did not induce a GR-dependent promoter, did not repress an activator protein-1-dependent promoter, and did not enhance nuclear translocation of GR. Moreover, rifampicin did not elicit a GR response in mouse AtT20 cells and in COS7 cells (Ray et al., 1998). It has been argued that the discrepancy with the report by Calleja et al. (1998b) might be due to differences in cell type, species, or promoter specificity (Ray et al., 1998). Alternatively, the intracellular concentration of rifampicin may have been reduced by induction of the P-glycoprotein transporter (Calleja et al., 1998a), which is the product of the multidrug resistance gene and functions as an extrusion pump for xenobiotics (Uhr et al., 1999). Species specificity is unlikely to explain the discrepancy, however, because both the alveolar A549 cells used by Jaffuel et al. (1999) and the HepG2 cells used by Calleja et al. (1998b) are of human origin.
Despite these seemingly conflicting results, the finding that rifampicin activates GR is intriguing to the psychiatrist because rifampicin does have known psychotropic side effects. Several observations suggest a link between GR activation and such side effects. There is evidence that increased levels of GCs induce cognitive impairment and, frequently, a variety of depressive syndromes (Brown and Suppes, 1998). Similar clinical conditions are observed with Cushing's disease (Sonino and Fava, 1998), which is characterized by hypercortisolism. Moreover, it has been suggested that lowering the level of cortisol by treating depressive patients with drugs that block cortisol biosynthesis, e.g., metyrapone or ketoconazole, may have beneficial effects predominantly among hypercortisolemic depressives (Murphy, 1997). In fact, encouraging results emerged from a double blind study with ketoconazole (Wolkowitz et al., 1999).
Given all these findings, we hypothesized that the discrepancy in the above-mentioned literature may be explained by tissue-specific effects. GC-like activity of rifampicin in some cells, e.g., neuronal cells, but not in others, could explain why rifampicin has psychotropic side effects, but not the other adverse effects one would expect from a general GR agonist, e.g., immunosuppression, osteoporosis, diabetes, arterial hypertension.
Thus, our objectives in this study were 2-fold. First, we wished to test the hypothesis that the central side effects of rifampicin, which often lead to severe psychiatric syndromes, are due to its GC-like activity. To this end, we used two neuronal cell lines to test binding to and transactivation of GR by rifampicin under different experimental conditions. Second, we aimed to clarify whether rifampicin indeed activates GR because the answer to this question has far-reaching implications both for steroid receptor research and for its clinical applications. Therefore, we addressed the remaining arguments (cell type, antibiotic export from the cell, or promoter specificity) that could potentially explain the discrepancy described above, i.e., we included HepG2 cells in our experiments, tested inhibitors of the P-glycoprotein transporter, and used two different types of promoters.
Materials and Methods
Cell Culture.
Mouse clonal hippocampal HT22 cells [a subclone of the HT4 hippocampal cell line (Morimoto and Koshland, 1990)] and human neuroblastoma SK-N-MC cells (American Type Culture Collection no. HTB-10) were kindly provided by the laboratory of C. Behl (Max Planck Institute of Psychiatry, Munich, Germany). The cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (FCS), 36 mg/l sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin sulfate, 0.25 μg/ml amphotericin (all purchased from Life Technologies, Rockville, MD) and 4.5 g/l glucose at 37°C and 10% CO2. Mouse liver HepG2 cells were kindly provided by F. Kiefer (GSF, Neuherberg, Munich, Germany) and cultivated under the same conditions except that glucose was at 1.0 g/l.
Cell Transfection and Induction.
Cells (106) were seeded into plastic dishes (60-mm diameter) 2 days before transfection in medium containing 10% charcoal-stripped, steroid-free FCS. Dextran T-70 (Pharmacia, Uppsala, Sweden) was used for charcoal-stripping of FCS (Damm, 1994). Putative residual cortisol concentration was determined by HPLC and found to be below the detection limit, i.e., below 0.036 nM in the medium containing the steroid-free FCS (data not shown). In addition, the luciferase activity of HT22 cells incubated without or with the GR antagonist RU486 (30 μM) showed no difference in the reporter assay described below, whereas RU486 efficiently suppressed cortisol induction of the glucocorticoid responsive element (GRE)-dependent reporter gene (data not shown). Thus, the activity of putative residual steroids is negligible.
When cells reached ∼50 to 70% confluency, medium was renewed, and calcium phosphate-precipitated plasmid DNA was distributed over the cells. The precipitate was prepared 30 min before adding to the cells and consisted of 1.6 μg of steroid-responsive luciferase reporter plasmid MTV-Luc (Hollenberg and Evans, 1988) or tk-(GRE)2-Luc (Rupprecht et al., 1993), 2 μg of simian virus 40 promoter-driven β-galactosidase expression vector pCH110 (Pharmacia LKB, Freiburg, Germany), and, where indicated, 0.4 μg of pRK7GR that expresses human GR (Hollenberg et al., 1985) from the cytomegalovirus-promoter of the vector pRK7 (Spengler et al., 1993) in 200 μl of Hanks' balanced salt buffer [137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4(H2O], 6 mM d-glucose, 20 mM HEPES, pH 7.1), to which 10 μl of 2.5 M CaCl2 slowly was added. Cells were cultured for an additional 5 to 6 h, the medium was removed, 2 ml of PBS [0.2 g/l KCl, 0.2 g/l KH2PO4, 8 g/l NaCl, 2.16 g/l Na2HPO4(H2O)] supplemented with 15% glycerol was added for 2 min, and cells were washed twice with PBS. Cells were cultured for 16 h in fresh medium (containing 10% steroid-free FCS), supplemented with dexamethasone, cortisol, rifampicin, verapamil, or cyclosporine A (all purchased from Sigma Chemical Co., Deisenhofen, Germany) in the combinations and concentrations indicated in the text. Dexamethasone, cortisol, and cyclosporine A were dissolved in ethanol, and rifampicin and verapamil were dissolved in dimethyl sulfoxide (DMSO). Additional solvent was added to each cell dish so that the total concentration of solvent was the same throughout each experimental setup. The activity of rifampicin was verified by its dose-dependent inhibition of bacterial growth (data not shown).
Luciferase and β-Galactosidase Assay.
Medium was removed and 1 ml of lysis buffer (0.1 M KHPO4, pH 7.8, 1 mM dithiothreitol) was added to each plate (60-mm diameter). Cells were scraped from the plate, transferred to a 1.5-ml reaction tube, and subjected to three freeze (3 min at −80°C) and thaw (3 min at 37°C) cycles. After centrifugation (Heraeus Biofuge, 13,000 rpm, room temperature, 4 min), 50 μl of each supernatant (corresponding to ∼1–2 × 105 cells) was transferred to a 96-well plate. Then 150 μl of 33 mM KHPO4, pH 7.8, 1.7 mM ATP, 3.3 mM MgCl2, 13 mM Luciferin (Roche Biochemicals, Mannheim, Germany) was added to each sample by the injector of an automatic luminometer (Luminat LB 96; Wallac GmbH, Freiburg, Germany) and light emission was measured for 10 s. The absolute numbers of the luminometer measurements varied depending on the cell number and transfection efficiency and typically were between 200 and 500 cps for noninduced SK-N-MC cells, between 250 and 600 cps for noninduced HepG2 cells, between 400,000 and 1,200,000 cps for HepG2 or SK-N-MC cells induced with 300 nM cortisol, between 350 and 1200 cps for noninduced HT22 cells, and between 4,000 and 20,000 cps for HT22 cells induced with 300 nM cortisol. To correct for variations in transfection efficiencies, values of the luciferase assay were normalized with β-galactosidase activities that were measured as follows: 50 μl of cell extract was added to 100 μl of galactosidase buffer (60 mM Na2HPO4, 40 mM NaH2PO4, 10 mM KCl, 1 mM MgCl2, 50 mM β-mercaptoethanol) on a 96-well plate. Then 20 μl of 2 mg/ml O-nitrophenyl β-d-galactopyranoside was added and the reaction was incubated at 37°C. After 10 to 30 min, absorption was measured at 405 nm in a multiphotometer (Dynatech MR5000).
Receptor-Binding Assay.
HepG2 cells were transfected with the GR-expressing plasmid pRK7GR as described above. After 16 h, cells were pelleted, resuspended in 1 ml of binding buffer [5 mM Tris-HCl, pH 7.4; 5% glycerol; 1 mM EDTA; 10 mM Na2MoO6(2H2O); 2 mM β-mercaptoethanol], homogenized, and centrifuged (35,000 rpm at 2°C for 60 min in a Ti 70.1 rotor). Then 100 μl of the cytosol (corresponding to 40–60 μg of protein) was incubated (18 h, 2°C) in a total volume of 150 μl of binding buffer with radiolabeled dexamethasone (specific activity was 80 Ci/mmol; Amersham, Braunschweig, Germany) alone or in combination with either unlabeled dexamethasone, RU486, or rifampicin as competitors at the concentrations indicated in the text. With 100 μl of the incubation reaction, bound and free steroids were separated by gel filtration by using Sephadex LH-20 (Pharmacia), radioactivity was measured with 600 μl of the total eluate of 1000 μl in a liquid scintillation counter (Beckman LH 6500) and protein concentration was determined by the Lowry method (Lowry et al., 1951) to normalize the data. The amount of picomoles bound radiolabeled dexamethasone per milligram of protein was between 0.3 and 0.6.
Results
Rifampicin Does Not Activate GR in a Mouse Hippocampal Cell Line.
To test the hypothesis that GR activation by rifampicin accounts for its psychotropic effects, we initially used the mouse cell line HT22 to characterize the interaction of rifampicin with GR. This cell line is derived from the hippocampus (Morimoto and Koshland, 1990), a brain area richly endowed with corticosteroid receptors. Thus, the hippocampus most likely would be a primary target of a GC-like activity of rifampicin. We tested activation of the steroid-dependent mouse mammary tumor virus (MMTV) promoter, which contains several GREs, by increasing amounts of either cortisol or rifampicin in transient transfection assays (Fig. 1A). This promoter clearly was activated on incubation with cortisol (or dexamethasone; data not shown). However, rifampicin was unable to elicit any response from this promoter at any concentration used, even at those clearly exceeding the reported K dof rifampicin binding to GR of 9.9 nM. This difference with the data reported by Calleja et al. (1998b), who used HepG2 cells, could be explained by selective binding of rifampicin to human, but not murine, GR and/or by a lower level of GR in HT22 cells compared with overexpressed GR in HepG2 cells (Calleja et al., 1998b). Therefore, we assayed rifampicin in HT22 cells while overexpressing human GR. Again, rifampicin did not stimulate transcription from the MMTV promoter, whereas cortisol increased transcription up to ∼50-fold (Fig. 1B). In these assays, we also used a luciferase reporter plasmid containing an MMTV promoter with a deletion of the GREs to verify that the transactivation observed with cortisol was, in fact, GR-dependent (data not shown).
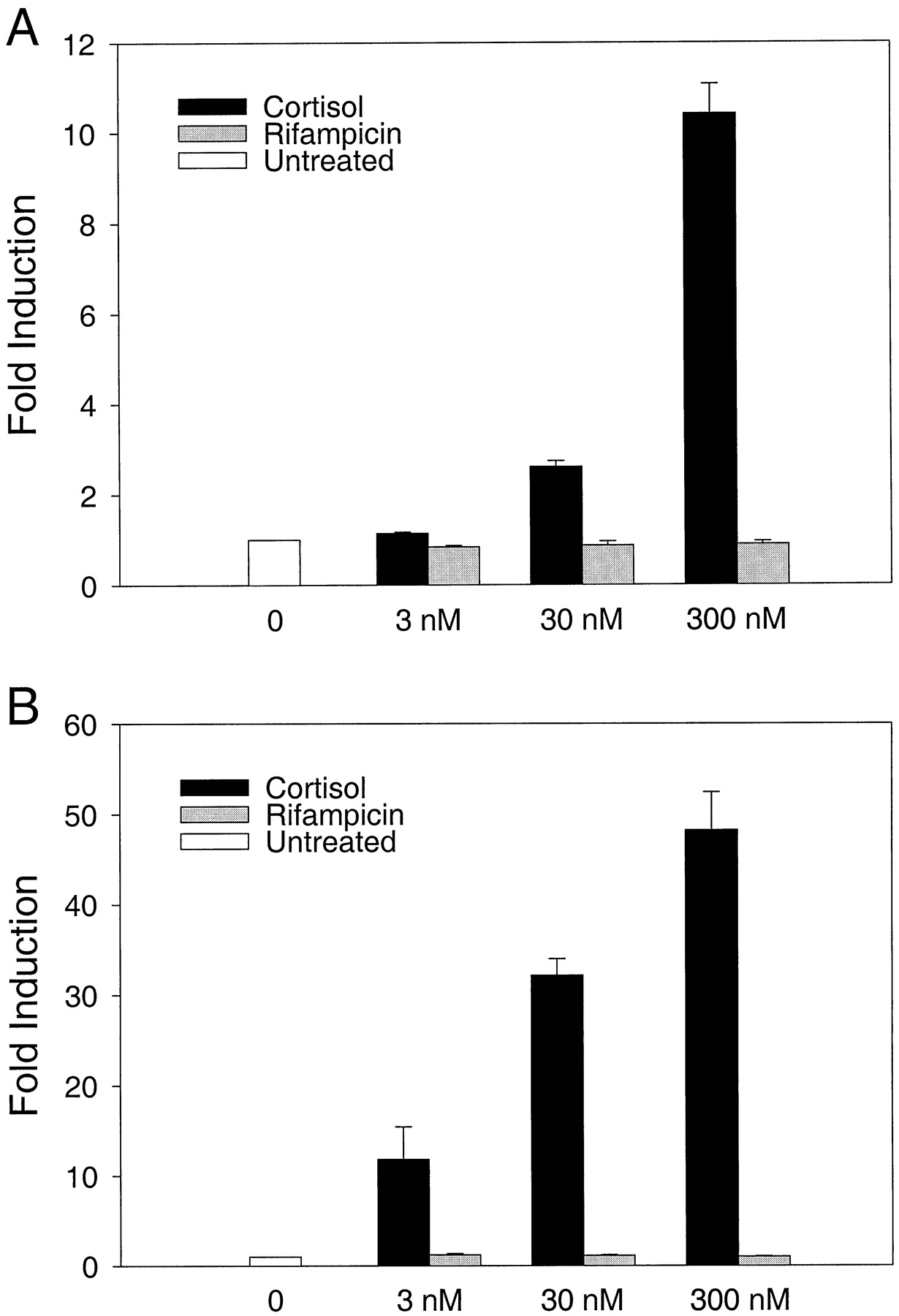
Inactivity of rifampicin in HT22 cells. A, murine hippocampal HT22 cells were transfected with a GR-dependent reporter plasmid (MTV-Luc) and a β-galactosidase expressing control plasmid (pCH110). After transfection, cells were cultivated either with DMSO and ethanol alone (white column), or with increasing amounts of either cortisol (black columns) or rifampicin (gray columns). Luciferase activities were corrected by the data of the β-galactosidase assay and are presented as fold induction with untreated cells as reference. B, a human GR-expressing plasmid (pRK7GR) has been cotransfected, all other conditions were the same. Results represent means ± S.E. of three independent experiments.
Rifampicin Does Not Activate the Glucocorticoid Receptor in a Human Neuronal Cell Line.
Cell type specificity has been reported for GC-like activity of rifampicin (Calleja et al., 1998a). Therefore, it was possible that rifampicin is not an activator of GR in hippocampal cells, but may be in other cells of the brain. We used human neuroblastoma SK-N-MC cells to investigate the effect of rifampicin on GR activity, again with an MMTV promoter containing reporter plasmid in transient transfection assays. The results demonstrate that rifampicin did not induce GR-dependent transcription at any concentration tested (3, 30, and 300 nM; Fig. 2A), whereas cortisol increased transcription up to ∼2000-fold (as did dexamethasone; data not shown).

Inactivity of rifampicin in SK-N-MC (A) and HepG2 (B) cells. Cells were transfected with a GR-dependent reporter plasmid (MTV-Luc), a human GR-expressing plasmid (pRK7GR), and a β-galactosidase-expressing control plasmid (pCH110). After transfection, cells were cultivated either with DMSO and ethanol alone (white column), or with increasing amounts of either cortisol (black columns) or rifampicin (gray columns). Luciferase activities were corrected by the data of the β-galactosidase assay and are presented as fold induction with untreated cells as reference. Results represent means ± S.E. of three independent experiments.
Rifampicin Does Not Activate GR in HepG2 Cells.
The GC-like behavior of rifampicin originally was described for the human hepatocyte cell line HepG2. Thus, it was possible that receptor binding and activation by rifampicin does not occur in neuronal cells, but may occur in other tissues. We applied the reporter assay described above for HT22 and SK-N-MC cells to HepG2 cells. As before, we found no activity for rifampicin (Fig. 2B), whereas cortisol showed strong induction of the GR-dependent MMTV promoter of up to ∼2000-fold (similar to dexamethasone; data not shown). In conclusion, we found no induction of the transcriptional activity of GR by rifampicin, irrespective of the cell line used.
Rifampicin Is Not a Ligand for GR.
Although rifampicin did not activate GR in three different cell lines, the possibility remained that rifampicin is a ligand for GR as described recently (Calleja et al., 1998b). Therefore, we overexpressed GR in SK-N-MC and HepG2 cells for 20 h after transfection with the plasmid pRK7GR, which expresses GR from the cytomegalovirus promoter. We tested the ability of rifampicin to compete with simultaneously added, radiolabeled dexamethasone for binding to GR in cytosolic extracts. As shown in Fig.3, rifampicin was unable to compete for binding to GR, even when it was present in 10,000-fold excess over dexamethasone. In contrast, unlabeled dexamethasone or RU486 (mifepristone), a GR antagonist (Baulieu, 1990), did efficiently reduce binding of radiolabeled dexamethasone to GR. We conclude that rifampicin is unable to bind to the hormone binding site of GR.

Rifampicin is unable to compete with dexamethasone for binding to GR. GR was overexpressed in SK-N-MC (A) and HepG2 (B) cells, cytosolic extracts were prepared and incubated with either 10 nM [3H]dexamethasone alone (black column) or with 10 nM [3H]dexamethasone and various nonradioactive competitors [1000-fold molar excess of dexamethasone (dex), white column; 100-fold molar excess of RU486, bright gray column; 100-fold molar excess of rifampicin (RIF), gray column; 10,000-fold molar excess of rifampicin, dark gray column]. GR-bound [3H]dexamethasone was determined and normalized to the protein content. Results represent means ± S.E. of four independent experiments.
Rifampicin Has No Effect on Cortisol- or Dexamethasone-Induced Activation of GR.
We also addressed the question of whether rifampicin, although not a ligand for GR, may act as a cofactor that increases the GC-induced response of GR because an additive effect was observed when rifampicin was used in equimolar concentrations with dexamethasone (Calleja et al., 1998b). As shown for HT22 cells (Fig.4), rifampicin did not change cortisol-induced activation of the MMTV promoter at any concentration tested (3, 30, and 300 nM, for both cortisol and rifampicin). The same results were obtained with dexamethasone (three independent experiments; data not shown). Rifampicin also did not change cortisol- or dexamethasone-induced transcription in SK-N-MC and HepG2 cells (three independent experiments for both cell lines and for cortisol and dexamethasone, respectively; data not shown). Rifampicin also had no influence on GR-dependent transcription when it was used in 10-fold excess over dexamethasone in either SK-N-MC or HepG2 cells (three independent experiments with each cell line; data not shown).

Rifampicin does not function as a coactivator for GR. HT22 cells were transfected with a GR-dependent reporter plasmid (MTV-Luc) and a β-galactosidase-expressing control plasmid (pCH110). After transfection, cells were cultivated either with DMSO and ethanol alone (white column), or with increasing amounts of either cortisol alone (black columns) or cortisol and rifampicin (gray columns). Luciferase activities were corrected by the data of the β-galactosidase assay and are presented as fold induction with untreated cells as reference. Results represent means ± S.E. of three independent experiments.
Inhibitors of P-Glycoprotein Transporter Do Not Unmask GC-Like Activity of Rifampicin.
It has been argued that rifampicin is inactive in some cell lines due to its export by the P-glycoprotein transporter (Calleja et al., 1998a). P-glycoprotein may be intrinsically active or induced by the drug rifampicin. We tested the influence of the two most widely used inhibitors of P-glycoprotein, verapamil and cyclosporin A (Volm, 1998), on MMTV transcription in the presence of either GC or rifampicin. In HT22 cells, verapamil enhanced cortisol-induced MMTV transcription up to 1.8-fold (Fig.5A). Similar effects of verapamil were observed for dexamethasone-induced transcription (three independent experiments; data not shown). However, even in the presence of verapamil, rifampicin still was unable to elicit any GC-like response (Fig. 5A). Similarly, cyclosporin A (1 μM) enhanced cortisol-induced transcription in HT22 cells, but did not unmask a GC-like activity of rifampicin (three independent experiments; data not shown). In HepG2 cells, there was virtually no effect of verapamil on cortisol-induced transcription (Fig. 5B), and rifampicin still remained inactive in the presence of verapamil, both when tested as an activator alone (Fig.5B), or when tested as a coactivator in combination with cortisol (three independent experiments; data not shown). The same results were obtained when rifampicin was tested as a coactivator for dexamethasone, or when cyclosporin A was used to inhibit P-glycoprotein (three independent experiments for each combination; data not shown). Finally, all experiments performed with HepG2 cells also were performed with SK-N-MC cells with almost indistinguishable results (data not shown). Thus, we conclude that the putative GC-like activity of rifampicin is not masked as a result of export from the cell by P-glycoprotein.

Inhibitors of the P-glycoprotein do not unmask GC-like activity of rifampicin. HT22-cells were transfected with a GR-dependent reporter plasmid (MTV-Luc), and a β-galactosidase-expressing control plasmid (pCH110). After transfection, cells were cultivated either with DMSO and ethanol alone (white column), or with increasing amounts of either cortisol alone (black columns) or verapamil (Vera) alone (bright gray column), or cortisol plus verapamil (dark gray columns), or rifampicin (RIF) alone (white hatched columns), or rifampicin plus verapamil (gray hatched columns). Verapamil was 20 μM in all experiments. Luciferase activities were corrected by the data of the β-galactosidase assay and are presented as fold induction with untreated cells as reference. B, analogous experiments were performed with HepG2 cells that were cotransfected with the GR-expressing plasmid pRK7GR in addition to MTV-Luc and pCH110. Results represent means ± S.E. of three independent experiments.
Inactivity of Rifampicin in GR-Dependent Transcription Is Not Due to Promoter Specificity.
It has been speculated that promoter specificity is a factor in the GC-like behavior of rifampicin (Calleja et al., 1998a; Ray et al., 1998). Therefore, we used a second reporter plasmid, tk-(GRE)2-Luc, that contains two palindromic GREs (Rupprecht et al., 1993) in our transient transfection assays. As demonstrated in Fig. 6, cortisol efficiently induced this promoter in SK-N-MC cells, but rifampicin failed to do so. Verapamil had no influence on these results (three independent experiments; data not shown). The same experiments were performed with HepG2 cells with similar results (data not shown). In conclusion, we found no evidence that rifampicin behaves like a GC under any circumstance.

The inactivity of rifampicin is not specific to the MMTV promoter. SK-N-MC cells were transfected with a GR-dependent reporter plasmid (tk-[GRE]2-Luc), a human GR-expressing plasmid (pRK7GR), and a β-galactosidase-expressing control plasmid (pCH110). After transfection, cells were cultivated either with DMSO and ethanol alone (white column), or with increasing amounts of either cortisol (black columns) or rifampicin (gray columns). Luciferase activities were corrected by the data of the β-galactosidase assay and are presented as fold induction with untreated cells as reference. Results represent means ± S.E. of three independent experiments.
Discussion
The psychotropic side-effects of rifampicin treatment posed a conundrum that seemed closer to solution with the discovery of the GC-like activities of rifampicin (Calleja et al., 1998b). However, we report in this study that rifampicin is unable to induce transcriptional activation from a GR-dependent promoter in three cell lines and does not bind to the hormone-binding site of GR. Recently, data have been presented showing that rifampicin does not activate GR in A549 human alveolar cells (Jaffuel et al., 1999), in murine AtT20, and in COS7 cells (Ray et al., 1998). It has been argued that the GC-like activity of rifampicin may be species, cell type, or promoter specific, or that rifampicin may be exported from the cells by the P-glycoprotein transporter (Calleja et al., 1998a), thereby lowering the intracellular concentration and masking the activity of rifampicin in the cell. We consider it very unlikely that anyone of these arguments can explain the discrepancy between our findings and the data reported by Calleja et al. (1998b), for the following reasons. We used cells derived from mouse and human sources, and from brain and liver tissues, including the same HepG2 cell line that was used in the original report of the GC-like activities of rifampicin (Calleja et al., 1998b). In addition, we transfected two different GRE-responsive promoters, and neither showed a response to rifampicin. Finally, we used two well documented inhibitors of the P-glycoprotein transporter, verapamil and cyclosporin A (Volm, 1998), and found that rifampicin remains inactive, even in the presence of these inhibitors. Thus, the discrepancy cannot be explained by an insufficient level of rifampicin, even more so when considering that the concentration range of up to 300 nM that we applied by far exceeded the reportedK d of 9.9 nM. Collectively, there is now firm evidence from this study and two other reports (Ray et al., 1998;Jaffuel et al., 1999) that rifampicin does not act as a GC.
Therefore, other properties of rifampicin must account for its clinical side effects. For example, the report of a patient who was under rifampicin therapy and required higher than expected doses of the antidepressant nortriptyline to obtain therapeutic drug levels (Bebchuk and Stewart, 1991) can be explained by the ability of rifampicin to induce cytochrome P450 3A genes and the P-glycoprotein transporter (Schuetz et al., 1996; Salphati and Benet, 1998; Wacher et al., 1998; Uhr et al., 1999), thereby either oxidizing nortriptyline or excluding it from target cells. This also is consistent with the fact that normal doses of nortriptyline were sufficient when rifampicin treatment was stopped (Bebchuk and Stewart, 1991).
Thus, although rifampicin does not behave like a GC, it nevertheless has an effect on steroid metabolism by induction of cytochrome P450 3A genes. Oxidation of steroids by cytochrome P450 most likely also accounts for the insufficient suppression of cortisol by dexamethasone in patients treated with rifampicin (Keven et al., 1998). According to the findings reported herein, the induction of cytochrome P450 3A genes by rifampicin and the subsequent oxidation of steroids and other compounds (Edwards et al., 1974; Elansary and Earis, 1983; McAllister et al., 1983) cannot be explained by activation of GR. Instead, these effects probably are mediated by recently described nuclear receptors termed pregnane X receptor (Kliewer et al., 1998, 1999), or hPAR (Bertilsson et al., 1998)). Whether activation of these receptors is involved in generating the psychotropic side effects of rifampicin remains to be elucidated.
Acknowledgments
We thank F. Kiefer for providing HepG2 cells, C. Behl for providing HT22 (initially a kind gift from P. Maher, The Scripps Research Institute, La Jolla, CA) and SK-N-MC cells and the plasmid MTV-Luc, and D. Spengler for providing the plasmids tk-(GRE)2-Luc and pRK7GR. We also are indebted to A. Gesing and H. Reul for their help in setting up the receptor-binding assay, to M. Uhr for HPLC analysis, and to D. Spengler for critical reading of the manuscript.
Footnotes
- Received September 30, 1999.
- Accepted December 20, 1999.
-
Send reprint requests to: Dr. Theo Rein, Max Planck Institute of Psychiatry, Kraepelinstrasse 10, D-80804 Munich, Germany. E-mail: theorein{at}mpipsykl.mpg.de
Abbreviations
- GR
- glucocorticoid receptor
- GC
- glucocorticoid
- FCS
- fetal calf serum
- GRE
- glucocorticoid responsive element
- DMSO
- dimethyl sulfoxide
- MMTV
- mouse mammary tumor virus
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}