


Abstract
We have studied the interactions of five indolocarbazoles withN-[methyl-3H]scopolamine (NMS) and unlabeled acetylcholine at M1-M4 muscarinic receptors, using equilibrium and nonequilibrium radioligand binding studies. The results are consistent with an allosteric model in which the primary and allosteric ligands bind simultaneously to the receptor and modify each other's affinities. The compounds were generally most active at M1 receptors. [3H]NMS binding was enhanced by staurosporine, KT5720, and KT5823 at M1 and M2 receptors, and by K-252a at M1 receptors. Gö 7874 reduced [3H]NMS affinity by up to threefold for all subtypes. A range of cooperative effects with acetylcholine was seen, and, at the M1 receptor, KT5720 had a log affinity of 6.4 and enhanced acetylcholine affinity by 40%. The compounds inhibited the dissociation of [3H]NMS to different extents across the receptor subtypes, with the largest effects at M1 receptors. In equilibrium binding studies the inhibitory potency of gallamine at M1 receptors was not affected by KT5720, indicating that these agents bind to two distinct allosteric sites and have neutral cooperativity with each other. In contrast, gallamine and staurosporine had a negatively cooperative or competitive interaction at M1 receptors. Similarly, the potency and relative effectiveness of KT5720 for inhibiting [3H]NMS dissociation from M1 receptors were not affected by gallamine or brucine, but were affected in a complex manner by staurosporine. These results demonstrate that there are at least two distinct allosteric sites on the M1 receptor, both of which can support positive cooperativity with acetylcholine.
The five subtypes of muscarinic acetylcholine (ACh) receptors are members of the superfamily of G-protein-coupled receptors. In addition to the “primary” sites on the receptor to which agonists and competitive antagonists bind, muscarinic receptors also contain one or more ‘allosteric‘ sites that mediate the effects of various agents on the binding of ligands at the primary site (Ellis, 1997; Holzgrabe and Mohr, 1998; Christopoulos et al., 1998). The effects of most allosteric agents, such as gallamine, strychnine, brucine, alcuronium, tubocurarine, WDuo3, and obidoxime, are consistent with the ternary complex allosteric model, in which the primary and allosteric ligands bind simultaneously to the receptor and modify each other’s affinities (Ehlert, 1988; Lazareno and Birdsall, 1995), and these agents all appear to act at the same “common allosteric site” (Ellis and Seidenberg, 1992; Waelbroeck, 1994; Tränkle and Mohr, 1997; Lazareno, unpublished observations with brucine). The limited evidence for the existence of a second allosteric site comes from studies of very low affinity interactions of obidoxime with Duo3 and WDuo3 at theN-[methyl-3H]scopolamine ([3H]NMS)-occupied m2 receptor (Tränkle and Mohr, 1997) and from the steepness of the concentration-effect curves of tacrine and Duo3 for inhibiting [3H]NMS dissociation (Potter et al., 1989; Tränkle et al., 1996).
The clearest indication that an agent is acting allosterically is its ability to inhibit the dissociation of [3H]NMS, but the potential therapeutic effect of an allosteric drug will be determined by the effect of the agent on the binding of the endogenous ligand ACh. We have recently reported that brucine and some N-substituted analogs act allosterically to enhance the affinity of ACh at one or more muscarinic receptor subtypes (Birdsall et al., 1997, Lazareno et al., 1998), and these observations have been confirmed by others (Jakubik et al., 1997; Murkitt and Wood, 1999). In the course of our screening program to detect allosteric enhancers of ACh affinity we found that staurosporine had allosteric effects, and we describe here the interactions of staurosporine and eight other indolocarbazoles with ACh and the antagonist radioligand [3H]NMS at M1-M4 muscarinic receptors. The results demonstrate the existence of a second allosteric site on these receptors, which also supports positive cooperativity with ACh.
Experimental Procedures
Materials.
[3H]NMS (81–86 Ci/mmol) was from Amersham International, UK, and [35S]GTPγS (1000–1400 Ci/mmol) was from New England Nuclear (Boston, MA). Brucine sulfate, gallamine triiodide, and ACh chloride were from Sigma Chemical Co. (Dorset, UK). Staurosporine was from Sigma and from Alexis Corporation (Nottingham, UK). Gö 7874, Gö 6976, and K-252c were from Calbiochem (Nottingham, UK). K-252a and K-252b were from Alexis and from TCS Biologicals (Buckingham, UK). KT5823 and KT5720 were from TCS, Calbiochem, and Alexis. KT5926 was from TCS and Calbiochem.
Cell Culture and Membrane Preparation.
Chinese hamster ovary (CHO) cells stably expressing cDNA encoding human muscarinic M1-M4 receptors (Buckley et al., 1989) were generously provided by Dr. N. J. Buckley (University of Leeds). These were grown in α-minimum essential medium (GIBCO) containing 10% (v/v) newborn calf serum, 50 U/ml penicillin, 50 μg/ml streptomycin, and 2 mM glutamine, at 37°C under 5% CO2. Cells were grown to confluence and harvested by scraping in a hypotonic medium (20 mM HEPES + 10 mM EDTA, pH 7.4). Membranes were prepared at 0°C by homogenization with a Polytron followed by centrifugation (40,000g, 15 min), were washed once in 20 mM Hepes + 0.1 mM EDTA (pH 7.4), and were stored at −70°C in the same buffer at protein concentrations of 2–5 mg/ml. Protein concentrations were measured with the BioRad reagent, using bovine serum albumin as the standard. The yields of receptor varied from batch to batch but were approximately 10, 1, 2, and 2 pmol/mg of total membrane protein for the M1, M2, M3, and M4subtypes, respectively.
Radioligand Binding Assays.
Unless otherwise stated, frozen membranes were thawed, resuspended in incubation buffer containing 20 mM HEPES + 100 mM NaCl + 10 mM MgCl2 (pH 7.4), and incubated with radioligand and unlabeled drugs for 2 h at 30°C in a volume of 1 ml. Membranes were collected by filtration over glass fiber filters (Whatman GF/B) presoaked in 0.1% polyethylenimine, using a Brandel cell harvester (Semat, Herts, UK); extracted overnight in scintillation fluid (ReadySafe, Beckman); and counted for radioactivity in Beckman LS6000 scintillation counters. Membrane protein concentrations (5–50 μg/ml) were adjusted so that not more than ∼15% of added radioligand was bound. Nonspecific binding was measured in the presence of 10−6 M (±)-quinuclidinyl benzilate (QNB) (an antagonist with picomolar potency) and accounted for 1–5% of total binding. GTP was present at a concentration of 2 × 10−4M in assays containing unlabeled ACh. Data points were usually measured in duplicate. CHO cell membranes do not possess cholinesterase activity (Gnagey and Ellis, 1996; Lazareno and Birdsall, 1993), so ACh could be used in the absence of a cholinesterase inhibitor. The indolocarbazoles and brucine were dissolved in dimethyl sulfoxide, which, at the highest final concentration of 2%, had no effect on binding.
Experimental Designs and Data Analysis.
General data preprocessing, as well as the affinity ratio calculations and routine plots of the semiquantitative equilibrium assay, were performed using Minitab (Minitab, Coventry, UK). The other assays were evaluated by nonlinear regression analysis, using the fitting procedure in SigmaPlot (SPSS, Erkrath, Germany). This procedure is relatively powerful in that it allows the use of two or more independent variables, e.g., concentrations of two drugs.
Equilibrium Binding Assays for Estimation of the Affinity of an Allosteric Agent for the Receptor and the Magnitude of its Cooperativity with [3H]NMS and ACh.
The design and analyses have been described in detail (Lazareno and Birdsall, 1995;Lazareno et al., 1998). Briefly, specific binding of a low concentration of [3H]NMS (one to two times theK
d) was measured in the presence of a number of concentrations of test agent, all in the absence and presence of one or more concentrations of ACh. Specific binding of a high concentration of [3H]NMS (5–10 times Kd) was also measured. Nonlinear regression analysis was used to fit the data to the equation
Above a certain concentration, some allosteric agents, especially those that exhibit neutral or positive cooperativity with [3H]NMS, may slow the kinetics of [3H]NMS binding so much that the binding does not reach equilibrium. In most cases sufficient incubation time was used to allow [3H]NMS binding in the presence of the agent to reach equilibrium. In a few cases, however, the highest concentration of agent would be predicted to slow [3H]NMS kinetics sufficiently to prevent binding equilibrium from being reached, and in these cases the data were better fitted to the equation
If only a single concentration of ACh was used, the data were visualized with affinity ratio plots, where the affinity ratio is the apparent affinity of the primary ligand ([3H]NMS or ACh) in the presence of a particular concentration of test agent divided by the apparent affinity of the primary ligand in the absence of test agent. Theoretically, the EC50 or IC50 of the affinity ratio plot corresponds to the K d of the test agent at the free receptor, and the asymptotic level corresponds to the cooperativity constant for the test agent and primary ligand (Lazareno and Birdsall, 1995). Affinity ratios were calculated from the specific binding data as follows (Lazareno and Birdsall, 1999).
The affinity ratio of [3H]NMS in the presence of a single concentration of test agent is
With assays containing a number of ACh concentrations, affinity ratio plots were calculated using the parameter estimates from the fit of the data set to eq. 1 or 2 as appropriate (Lazareno and Birdsall, 1995).
The affinity ratios of [3H]NMS and ACh,r
L and r
A, respectively, are
Off-Rate Assay to Estimate the Affinity of an Allosteric Agent for the [3H]NMS-Occupied Receptor.
A high concentration of membranes (2–4 mg protein/ml) was incubated with a high concentration of [3H]NMS (5 nM) for about 15 min. Then 10-μl aliquots were distributed to tubes that were empty or contained 1 ml of 10−6 M QNB alone and in the presence of a number of concentrations of allosteric agent (typicallyn = 4). Nonspecific binding was measured in separately prepared tubes containing 10 μl of membrane and 2 μl of [3H]NMS + QNB. Some time later, after about 2.5 dissociation half−lives (see Table 2), the samples were filtered. The data were transformed to rate constants, koff, using the formula
Percentage inhibition of [3H]NMS dissociation from muscarinic receptors by indolocarbazoles
GTPγS Binding Assay.
Membranes expressing M1receptors (5–20 μg/ml) were incubated with [35S]GTPγS (0.1 nM), GDP (10−7 M), and ligands in incubation buffer in a volume of l ml for 30–60 min at 30°C. Bound label was collected by filtration over glass fiber filters prewetted with water.
Results
The structures of the compounds examined are shown in Fig. 1. Figure 2shows the effects of staurosporine on equilibrium [3H]NMS binding at M1 receptors in the absence and presence of a fixed concentration of ACh. [3H]NMS binding was increased by staurosporine concentrations up to 10 μM and was reduced at 30 μM. The increase in [3H]NMS binding reflects a decrease in the K d of [3H]NMS rather than an increase in B max (data not shown). The decrease in binding with 30 μM staurosporine is caused by the slowing of [3H]NMS kinetics by high concentrations of staurosporine (see below) and the consequent lack of equilibration of [3H]NMS binding (Lazareno and Birdsall, 1995). The effect of staurosporine on ACh binding is not clear from inspection of Fig. 2, but nonlinear regression analysis of the data, which also takes into account the effects of high concentrations of staurosporine on the kinetics of [3H]NMS, provided a good fit to the data (lines in Fig. 2) and revealed a four fold negative cooperativity between ACh and staurosporine. The independent effects of staurosporine on [3H]NMS and ACh binding across the four receptor subtypes are easier to visualize when the binding data are transformed into affinity ratios (Lazareno and Birdsall, 1995; Lazareno et al., 1998) (Fig. 3). In theory, the EC50 or IC50 of the affinity ratio plot corresponds to the K d of the test agent for the free receptor, and the asymptotic value corresponds to the cooperativity with the primary ligand. Staurosporine showed positive cooperativity with [3H]NMS at M1 and M2 receptors, neutral cooperativity with [3H]NMS at M4 receptors, and was inactive or neutrally cooperative at M3 receptors. It had negative cooperativity with ACh at M1, M2, and M4 subtypes and was neutral with ACh or inactive at M3 receptors. Staurosporine had K dvalues for unoccupied receptors in the μM range (Fig. 3 and Table 1). In two functional assays with M1 receptors measuring the stimulation by ACh of [35S]GTPγS binding, 10 μM staurosporine reduced basal activity and the E max by 17% ± 7% and 25% ± 4%, respectively, and caused a 2.9 ± 0.9-fold decrease in the potency of ACh (data not shown), which is consistent with the 3.6-fold change predicted from the [3H]NMS binding studies.

Compound structures.

Effect of staurosporine on the binding of [3H]NMS (210 pM) at M1, receptors in the absence and presence of 2.2 μM ACh, all in the presence of 0.2 mM GTP. The points are individual observations. The lines show the fit eq.2 (see Experimental Procedures), which yielded a log affinity of 5.95 ± 0.06, a slope factor of 1.01 ± 0.05, cooperativity with [3H]NMS of 1.51 ± 0.06, and cooperativity with ACh of 0.27 ± 0.03. The affinity ratio plots of these data are included in Fig. 3.

Affinity ratio plots of five indolocarbazoles at M1-M4 receptors, where the affinity ratio is the ratio of the apparent affinities of the primary ligand ([3H]NMS or ACh) in the presence and absence of a single concentration of test agent. The points were derived from duplicate observations of [3H]NMS binding in the absence and presence of ACh, as described in Experimental Procedures. The parameter estimates pK (log affinity of the test agent for the free receptor), αNMS (cooperativity with [3H]NMS), and βACh (cooperativity with ACh) were derived from nonlinear regression analysis of the untransformed data, using eq. 1 or 2 as appropriate (see Experimental Procedures). The parameter estimates from a number of similar assays are summarized in Table 1.
Equilibrium binding parameters of indolocarbazoles with [3]HNMS and ACh at muscarinic receptors
Staurosporine also inhibited [3H]NMS dissociation (Fig. 4). All of the curves had slope factors of 1. Staurosporine was most potent and effective at M1receptors, causing apparently complete inhibition of [3H]NMS dissociation with an IC50 of 1 μM (Table 2). It was three- to fourfold weaker at the other receptor subtypes and caused submaximum inhibition of [3H]NMS dissociation, with the smallest effect, 67% inhibition, seen at M3 receptors. The IC50 values for the inhibition of [3H]NMS dissociation correspond in theory to the Kd values of staurosporine for the [3H]NMS-liganded receptors, and the values at M1 and M2 receptors are consistent with the values predicted from the equilibrium binding studies according to the allosteric model (Table 2). There was a twofold disparity between predicted and observed values at M4 receptors, probably because of inaccuracies in measuring the small degree of negative cooperativity with [3H]NMS. In equilibrium binding studies at M3 receptors staurosporine had little or no effect on the binding of either [3H]NMS or ACh; the clear inhibition of [3H]NMS dissociation caused by staurosporine over the same concentration range suggests that staurosporine was neutrally cooperative with [3H]NMS and ACh at M3 receptors, rather than inactive.

Effect of five compounds on the dissociation rate constant (k off) of [3H]NMS at M1-M4 receptors, expressed as a percentage inhibition of the control k off. The points are the mean and range/2 of duplicate observations. The lines show the fit to a logistic function, as described in Experimental Procedures. The parameter estimates from a number of similar assays are summarized in Table 2.
Gö 7874, a ring-opened analog of staurosporine still bearing a positive charge, showed weak negative cooperativity with [3H]NMS and stronger negative cooperativity with ACh at M1, M2 and M4 receptors, and the reversed pattern at M3 receptors (Fig. 3). It was necessary to introduce a slope factor >1 into the binding equation for Gö 7874 to fit the data adequately to the allosteric model (Table 1). Gö 7874 caused apparently complete inhibition of [3H]NMS dissociation at M1, M2, and M4receptors and submaximum inhibition at M3 receptors. The slopes of the curves at M1, M2, and M4 receptors were also greater than 1 (Fig. 4 and Table 2). The ternary complex allosteric model does not predict slope factors different from 1, so it cannot provide a complete mechanistic explanation of these data. Nevertheless, the affinity values of Gö 7874 for the [3H]NMS-occupied receptor predicted by the model from the equilibrium binding studies are in excellent agreement with the observed values at M1 and M4 receptors (Table 2) and show only a three-fold discrepancy at M2 and M3 receptors, possibly caused by a combination of inaccuracies in the measurement of the small cooperative effects that occurred in equilibrium studies at the M2 and M3 subtypes (Table 1) and the small inhibitory effect on [3H]NMS dissociation from M3 receptors.
KT5823, a ring-contracted analog of staurosporine in which the methylamino group is replaced by a methyl ester, caused a large increase in [3H]NMS binding at M1 and M2 receptors and showed neutral or small positive cooperativity with ACh at these receptors. KT5823 was inactive or neutrally cooperative with 3H-NMS and ACh at M3and M4 receptors (Fig. 3). The positive cooperativity with NMS at M1 receptors was confirmed in functional studies in which 1 μM KT5823 increased the potency of ACh 1.9 ± 0.9-fold at M1 receptors for stimulating [35S]GTPγS binding and caused a 3.3 ± 1.7-fold increase in the affinity of unlabelled NMS (n = 2, data not shown). KT5823 inhibited [3H]NMS dissociation completely at M1receptors, 80% at M2 receptors, and 30–40% at M3 and M4 receptors (Fig. 4). The affinity of KT5823 for the [3H]NMS-occupied receptor estimated from equilibrium studies at M1 and M2 receptors was very similar to the values measured directly. The inhibition of [3H]NMS dissociation seen at M3 and M4 receptors may indicate that KT5823 is neutrally cooperative with [3H]NMS and ACh at these receptors, rather than inactive.
KT5720, a hexyl ester analog of KT5823, was positively cooperative with both [3H]NMS and ACh at M1 receptors (Fig. 2and Table 1). The small (40%) increase in ACh affinity was confirmed in more detailed assays (Fig. 5). KT5720 had little or no effect at M3 receptors and showed neutral cooperativity with [3H]NMS and negative cooperativity with ACh at M4 receptors. The effects of KT5720 at M2 receptors are unclear; earlier batches had small inhibitory effects with [3H]NMS and ACh (Fig. 3), whereas a later batch had small positive effects with [3H]NMS (data not shown). No batch-dependent effects were noted at the other subtypes. KT5720 caused incomplete inhibition of [3H]NMS dissociation at M1, M3, and M4receptors, with little or no effect at M2 receptors (again, earlier batches showed no effect at M2 receptors, whereas a later batch showed a small inhibition (Fig. 4)). The largest effect was seen with M1 receptors, and, at this subtype alone, low concentrations of KT5720 caused a small but consistent increase in [3H]NMS dissociation. This phenomenon was observed in 10 of 11 single-time point assays, with the dissociation rate constant (k off) of [3H]NMS increased by 11 ± 1% (n = 10) in the presence of the most effective concentration between 10 and 300 nM KT5720, and in two full time-course studies in which k off in the presence of 0.1 μM KT5720 was increased by 16.3 ± 0.5 % (data not shown). The affinity of KT5720 for the [3H]NMS-occupied receptor estimated from equilibrium studies at M1 and M4receptors was similar to the values measured directly (Table 2).

Effect of various concentrations of KT5720 on the inhibition of [3H]NMS (50 pM) binding at M1receptors by ACh in a volume of 3 ml. The points are the mean and range/2 of duplicate observations. The lines show the fit to eq. 1 with the slope factor for KT5720 binding set to 1. The parameter estimates were log affinity of KT5720, 6.6 ± 0.1; cooperativity with [3H]NMS, 1.9 ± 0.1; cooperativity with ACh, 1.6 ± 0.2. The inset shows affinity ratio plots derived from these parameters (see Experimental Procedures). The -log IC50 values of ACh in the presence of increasing concentrations of KT5720, from independent logistic fits of the curves, were 5.28, 5.33, 5.40, and 5.42.
K-252a, in which the methoxy group of KT5823 is replaced by a hydroxyl group, showed positive cooperativity with [3H]NMS at M1 receptors and neutral or small negative cooperativity with ACh (Fig. 3 and Table 1). Little or no effect was seen in equilibrium binding studies with the other subtypes. K-252a inhibited [3H]NMS dissociation at M1receptors, apparently by 100%. Slope factors greater than 1 were required to fit the data adequately. Only small, though consistent, effects on the [3H]NMS off-rate were seen at the other subtypes (Fig. 4 and Table 2).
K-252b, K-252c, KT-5926, and Gö 6976 at concentrations up to 10 μM had little or no effect on equilibrium binding of [3H]NMS and ACh and on [3H]NMS dissociation (data not shown) and were not studied further.
We have attempted to determine whether some of the allosteric effects described above occurred through an interaction at the same site on the receptor at which other known allosteric agents act. Figure 6A shows the interaction between KT5720 and gallamine on equilibrium [3H]NMS binding at M1 receptors. Gallamine had its expected inhibitory effect on [3H]NMS binding, and KT5720 showed the expected positive cooperativity with [3H]NMS. If gallamine and KT5720 were acting at the same site, then gallamine should have become less potent in the presence of KT5720 and the nonlinear regression analysis would have indicated strong negative cooperativity between the two agents. In fact, the analysis revealed neutral cooperativity, i.e., in equilibrium binding studies gallamine and KT5720 interact allosterically at M1 receptors through distinct and apparently noninteracting sites. In similar experiments with staurosporine and gallamine at M1 receptors, however, there was a negatively cooperative or competitive interaction between the compounds (Fig. 6B).

Inhibition by gallamine of [3H]NMS binding at M1 receptors in the presence of various concentrations of (A) KT5720 and (B) staurosporine. The points are individual observations. The lines show the fit of the data to eq. 9(see ), where the cooperativity estimates of gallamine with KT5720 and staurosporine were not significantly different from 1 and 0, respectively, and were set to those values. The slope factors for gallamine, KT5720, and staurosporine were not different from 1 and were set at that value. From three such assays, similarly constrained, KT5720 had a log affinity of 6.22 ± 0.17 and a cooperativity with [3H]NMS of 2.39 ± 0.08. From three such assays, similarly constrained, staurosporine had a log affinity of 5.75 ± 0.11 and a cooperativity with [3H]NMS of 1.62 ± 0.13. From these six assays gallamine had a log affinity of 5.05 ± 0.05 and a cooperativity with [3H]NMS of 0.11 ± 0.01. The insets show the effect of the test agent on the -log IC50 of gallamine, obtained from nonlinear regression analysis of the individual curves.
To study the site(s) on the M1 receptor at which KT5720 acts to affect [3H]NMS dissociation, the concentration-related effect of KT5720 was measured alone and in the presence of two or three concentrations each of gallamine, brucine, and staurosporine. Very similar results were obtained in two independent assays; the combined data are shown in Fig.7. The data in each condition are shown in two forms: as a percentage inhibition of the overall control (i.e., true)k off measured in the absence of any test agent and, for each curve, as a fraction of its own controlk off measured in the presence of the test agent and the absence of KT5720. This latter fractional effect measure has useful properties: if the interaction between KT5720 and the test agent is competitive, then in the presence of the test agent the EC50 will increase and the asymptotic fractional effect will also change; if the interaction is noncompetitive and noninteracting (i.e., with neutral cooperativity), and if maximum concentrations of test agent completely inhibit [3H]NMS dissociation, then in the presence of the test agent both the EC50 and asymptotic levels are unchanged (see the).
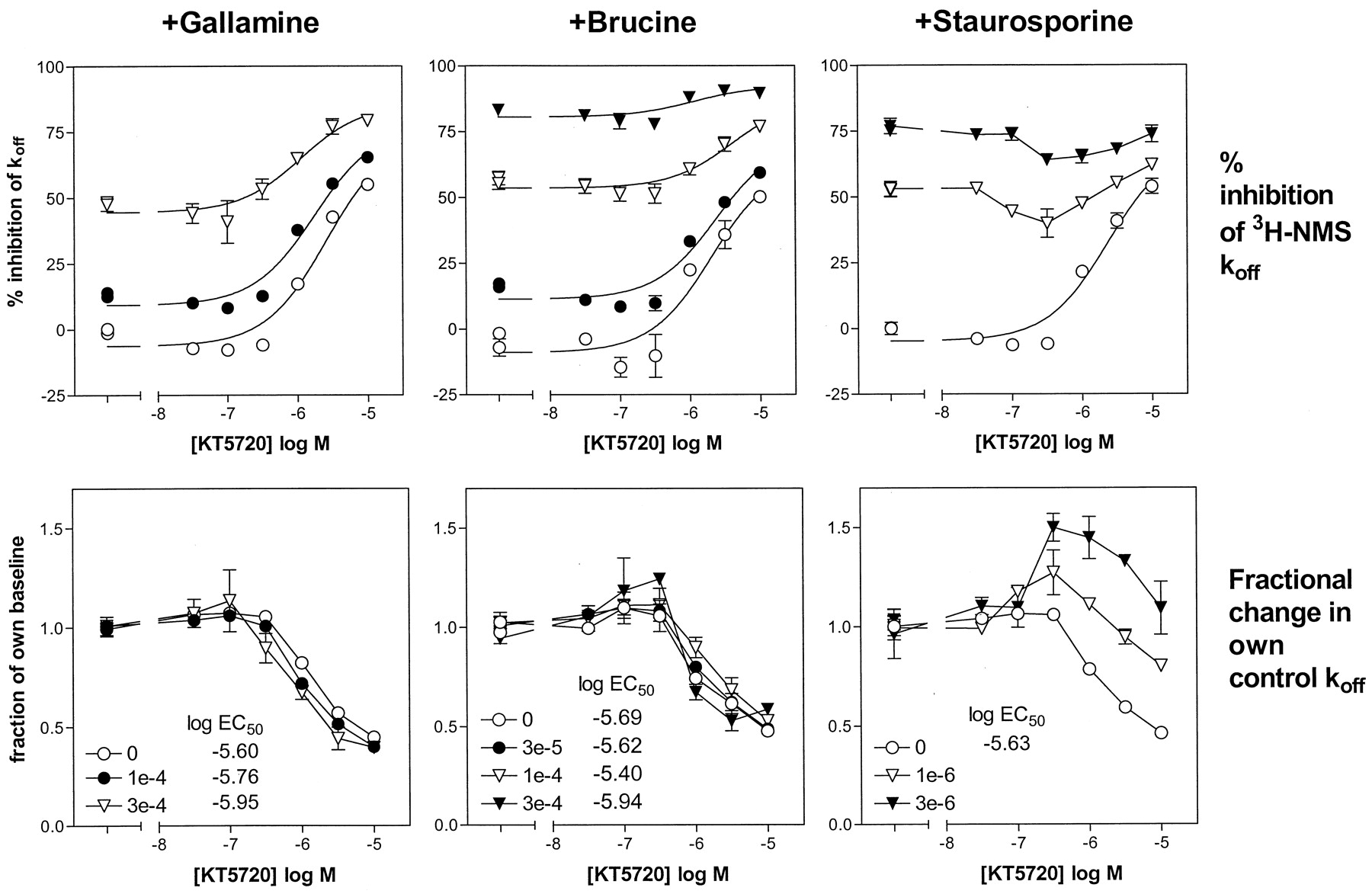
Effect of KT5720 on [3H]NMS dissociation from M1 receptors, alone and in the presence of other allosteric agents, measured at a single time point, as described in Experimental Procedures. The points show the mean and S.E.M. of quadruplicate observations obtained in two assays, except for 10−4 M gallamine, and 3 × 10−5 M and 3 × 10−4 M brucine, which show the mean and range/2 of duplicate observations. The lines in the top panel show the fits of the individual curves to a hyperbolic function, except for those in the presence of staurosporine. The estimates of the log EC50 of KT5720 derive from those fits. The top panel shows the data as percentage inhibition of the controlk off of [3H]NMS. The lower panel shows shows Ef, the [3H]NMSk off values in the presence of KT5720, and a certain concentration of test agent (gallamine, brucine, or staurosporine) as a fraction of the k off values in the presence of that concentration of test agent alone (see).

The lines in the top row of Fig. 7 (except in the presence of staurosporine) are hyperbolic fits to the data. The effect of low concentrations of KT5720 of increasing [3H]NMS dissociation was apparent in all of the curves. When the data are expressed as a fractional effect of own control, the curves for KT5720 in the presence of various concentrations of gallamine or brucine overlap, i.e., they have the same EC50 and asymptotic level. There was a small concentration-related increase in potency in the presence of gallamine, but this is probably experimental noise, because a positively cooperative interaction would result in decreases in the asymptotic level of the fraction of own control plots. These data therefore demonstrate that KT5720 acts at a site different from those at which gallamine and brucine act to inhibit [3H]NMS dissociation from M1 receptors.
A quite different pattern of results was seen with staurosporine. The stimulating effect of low concentrations of KT5720 became more apparent, and the curves tend to converge at high concentrations of KT5720 more than in the presence of gallamine or brucine. It was not possible to measure EC50 values accurately, but inspection of the fractional effect plot suggests that staurosporine reduced the potency of KT5720. These results may indicate that staurosporine and KT5720 compete for the site that mediates inhibition of [3H]NMS dissociation. They strongly suggest that staurosporine can act at site(s) different from those of gallamine or brucine.
Discussion
Five of the nine indolocarbazoles that we have studied act allosterically at muscarinic receptors. Of these, four have similar structures and a number of similarities in their allosteric effects, whereas the fifth, Gö 7874, lacks the tetrahydrofuran/pyran ring system, which may account for its somewhat different effects.
In equilibrium binding studies the four active staurosporine-like compounds (staurosporine, KT5823, KT5720, and K-252a) showed only positive or neutral cooperativity with [3H]NMS, or were apparently inactive, whereas positive, neutral, and negative cooperativity was observed with ACh. The four compounds showed their highest affinity and largest positive effects with [3H]NMS, at the M1 receptor, whereas they were inactive (or neutrally cooperative with [3H]NMS and ACh) at M3 receptors. These compounds bound with slope factors of 1, except for KT5823 at M1 receptors, and this exception may be partly accounted for by artefacts arising from the strong (7–10-fold) positive cooperativity with [3H]NMS seen with this compound. Gö 7874, the other positively charged ligand in addition to staurosporine, also showed selectivity for the M1 receptor, but, in contrast to the other four compounds, it showed negative cooperativity with [3H]NMS and both neutral and negative cooperativity with ACh, and it bound with slope factors greater than 1.
The four staurosporine-like compounds also showed selectivity for the [3H]NMS-occupied M1 receptor, but this was manifested more clearly in the magnitude of inhibition of [3H]NMS dissociation than in the affinity. Again, these compounds bound to the [3H]NMS-occupied receptor with slopes of 1, except for K-252a at M1 receptors. Gö 7874 inhibited [3H]NMS dissociation completely from M1, M2, and M4 receptors with slope factors significantly greater than 1.
There seems to be a relationship between the activity of the compounds in equilibrium binding assays and the maximum degree of inhibition of [3H]NMS dissociation: an ad hoc correlation for the current data is that compounds showing less than 50% inhibition of [3H]NMS dissociation at a particular subtype appear inactive in equilibrium studies, whereas those slowing [3H]NMS dissociation by >50% show activity in equilibrium studies. This rule works in 17 of 20 cases, the exceptions being staurosporine at M3, Gö 7874 at M3, and KT5720 at M4 receptors. The positive relationship between allosteric activity at equilibrium and the degree of inhibition of [3H]NMS dissociation may reflect the degree to which binding of the allosteric agent perturbs the primary ligand recognition site on the receptor. Those cases where the test agent inhibits [3H]NMS dissociation but appears to be inactive at equilibrium may actually reflect a lack of cooperative effect, i.e., neutral cooperativity, rather than a lack of binding of the test agent at equilibrium.
According to the allosteric model, the affinity of a test agent for the [3H]NMS-occupied receptor may be estimated in two independent ways: from direct measurement of effects on [3H]NMS dissociation, and from the product of affinity for the free receptor and cooperativity with [3H]NMS, measured at equilibrium. In this study there are 11 instances where these measures have been determined with sufficient precision to allow comparison. There was good agreement between the measures: three comparisons differed by about threefold, one by about twofold, and the rest (seven) by 60% or less, and there was no obvious bias, because in five cases the equilibrium estimate was larger than the directly measured value and in seven cases it was smaller. These results suggest that the data can be accounted for by the allosteric model, even though the steep slopes seen with Gö 7874 and K-252a are not predicted by the model.
The simple model also cannot account for the effects of KT5720 on [3H]NMS dissociation at M1 receptors, with an initial speeding of dissociation by about 15% at submicromolar concentrations, followed by submaximum inhibition of dissociation at higher concentrations. In the presence of staurosporine the speeding effect became more prominent, whereas the potency of KT5720 for slowing [3H]NMS dissociation appeared to be reduced, suggesting that KT5720 may be exerting its effects at two distinct sites, only one of which can also be occupied by staurosporine. In contrast, the presence of gallamine or brucine had no effect on the potency of KT5270 or its fractional asymptotic effect, suggesting that, unlike staurosporine, gallamine and brucine act at a site different from the site(s) at which KT5720 modulates [3H]NMS dissociation, and that there is no interaction (i.e., neutral cooperativity) between the binding of KT5270 and that of brucine or gallamine.
A similar conclusion can be drawn from equilibrium binding studies at M1 receptors, in which KT5720 showed no interaction with gallamine. In contrast, similar equilibrium binding studies at M1 receptors with staurosporine and gallamine revealed a negatively cooperative or competitive interaction. If the interaction between gallamine and staurosporine is truly competitive, this would imply that bound staurosporine occludes both the gallamine and the KT5720 site. A more parsimonious explanation is that staurosporine binds to the KT5720 site and has negative cooperativity with gallamine. The different interactions with gallamine shown by staurosporine (negative) and KT5720 (neutral) may be related to the fact that staurosporine, like gallamine, is a positively charged molecule, whereas KT5720 is neutral.
These results demonstrate that KT5720, and possibly other indolocarbazoles, bind to an allosteric site on muscarinic receptors that is distinct from the common allosteric site to which gallamine and most other allosteric agents bind. Previously reported allosteric agents have a positively charged nitrogen that is thought to be important for their action. Staurosporine and Gö 7874 are also positively charged, but the other active indolocarbazoles are neutral, which suggests that there is no necessity for a positively charged nitrogen at this new allosteric site. The observed affinities and cooperativities are sensitive to small changes in the chemical structure of the analogs. For example, increasing the alkyl chain length of the ester function of K-252a or methylation of its hydroxyl group increases affinity by 3–15-fold, whereas removal of the methyl group on the ester of K-252a or the alkoxy substitution of the indolocarbazole ring generate apparently inactive compounds.
The agents studied here are known to be potent inhibitors of various protein kinases (Tamaoki et al., 1986; Kase et al., 1987;Kleinschroth et al., 1995), and in most cases the agents have much higher affinity for these targets than for muscarinic receptors, but it is worth noting that KT5720 has only about a sixfold higher potency for its preferred target, protein kinase A (PKA), than for the M1 receptor (log affinity of 7.2 at PKA versus 6.4 at M1 receptors; Kase et al., 1987).
One of our aims has been the development of drugs that enhance the affinity of ACh at M1 receptors while having no effect on ACh binding and function at the other subtypes. The detection of the allosteric properties of KT5720 may be a step toward that goal. KT5720 was the most potent compound at M1 receptors with a log affinity for the free receptor of 6.4, and it showed a small (40%) but consistent positive cooperative effect with ACh. In addition, it may have had little or no effect on ACh affinity at the other subtypes, so KT5720 is close to displaying an absolute subtype selectivity for the M1 receptor, i.e., a positive or negative interaction with ACh at one receptor subtype and neutral cooperativity at the others, so that whatever concentration of agent is administered, only the one receptor subtype is affected functionally (Lazareno and Birdsall, 1995).
In conclusion, we have detected quite potent allosteric interactions of staurosporine and some other indolocarbazole analogs at muscarinic receptors that, at least in the case of KT5720, occur at a site distinct from the common allosteric site. The active indolocarbazoles cause different maximum effects on [3H]NMS dissociation, and the size of the maximum effect on [3H]NMS dissociation is a good predictor of the activity detected in equilibrium studies, suggesting a common mechanism for the two effects. In general, the results from equilibrium and dissociation assays were mutually consistent with the ternary allosteric complex model as the underlying mechanism of the observed effects, but the steep binding slopes seen with two of the compounds and the complex effects of KT5720 on [3H]NMS dissociation indicate that the current model cannot fully account for all of the data. Finally, KT5720 is the most potent agent described so far, showing positive cooperativity with ACh at M1 receptors.
Binding of Radioligand L in the Presence of Allosteric Agents X and Y, Which Act at Different Allosteric Sites
Equilibrium Binding
K L, K X, andK Y are affinities of L, X, and Y, respectively. α is the cooperative constant between L and X at R; β is the cooperative constant between L and Y at R, γ is the cooperative constant between X and Y at R, and δ is the cooperative constant between X and Y at LR. If γ and δ = 0, then the interaction behaves as though X and Y are competitive. If γ and δ = 1, then the binding of X and Y is noninteracting.
Total receptor concentration is R = R + LR + RX + RY + LRX + LRY + RXY + LRXY. Bound radioligand as a fraction of total receptor is
Kinetic Effects on Equilibrium Binding
To estimate the apparent association rate constant in the presence of X and Y, k
obsXY, we start with the simple definition for radioligand alone:
Kinetic Effects of Two Allosteric Agents on Radioligand k off
Here we consider the binding of two agents to the radioligand-occupied receptor. We assume that the allosteric agents have rapid kinetics compared to the radioligandk off and that there may be cooperative effects between the agents.
Assume that each agent, X and Y, can bind to a different site on the radioligand-occupied receptor, with affinities ofK
Xo and K
Yo, respectively (= α · K
X and β · K
Y, respectively), and with cooperativity δ. If δ = 0, then the interaction behaves as though X and Y are competitive with each other. If δ = 1, then the binding of X and Y is noninteracting. k
off is the dissociation rate constant of L from LR; ρ is the ratio of the dissociation rate constants of L from the occupied and free receptor, i.e., k
off*ρX is the dissociation rate constant of L from LRX; k
off*ρY is the dissociation rate constant of L from LRY; k
off*ρXY is the dissociation rate constant of L from LRXY.B
LXY is the bound radioligand in the presence of X and Y at any dissociation time, assuming rapid kinetics. Equation E11Now we consider the effect of a fixed concentration of Y on the parameters of concentration-effect curves of X and the observed radioligand dissociation rate constant.
Equation E11Now we consider the effect of a fixed concentration of Y on the parameters of concentration-effect curves of X and the observed radioligand dissociation rate constant.
The Effect of Y on the EC50 of X.
From eq. 11,
The EC50 of X in the presence of Y is therefore
The Effect of Y on the Efmax of X.
Define EfXY, the k
offobs with varying concentrations of X in the presence of a fixed concentration of Y as a fraction of the k
offobs with the same concentration of Y alone: Equation E20The change in Efmax of X as a function of Y may be expressed as the ratio
Equation E20The change in Efmax of X as a function of Y may be expressed as the ratio
In summary, for concentration-effect curves of X onE fXY (the k offobs in the presence of X and Y expressed as a fraction of thek offobs in the presence of Y alone), if the radioligand cannot dissociate from Y-occupied receptors (ρY = 0 and ρXY = 0), then Y has the same fractional effect on EC50XY and EfmaxXY, with a limiting value of 1/δ, where δ is the cooperativity between X and Y. If δ = 0, then the interaction between X and Y is competitive, and there is no limiting effect of Y. If δ = 1, then the curves of EfXYversus [X] in the presence of any concentration of Y are superimposable.
Footnotes
- Received January 27, 2000.
- Accepted March 22, 2000.
-
Send reprint requests to: Dr. S. Lazareno, MRC Technology, 1-3 Burtonhole Lane, Mill Hill, London NW7 1AD, UK. E-mail:slazare{at}nimr.mrc.ac.uk.
-
This work was funded by Sankyo Co. Ltd, Tokyo, Japan, and the Medical Research Council, UK.
Abbreviations
- Ach
- acetylcholine
- NMS
- N-[methyl-3 H]scopolamine, CHO cells, Chinese hamster ovary cells
- QNB
- (±)-quinuclidinyl benzilate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}