


Abstract
Many Regulators of G proteinSignaling (RGS) proteins accelerate the intrinsic GTPase activity of Giα and Gqα-subunits [i.e., behave as GTPase-activating proteins (GAPs)] and several act as Gqα-effector antagonists. RGS3, a structurally distinct RGS member with a unique N-terminal domain and a C-terminal RGS domain, and an N-terminally truncated version of RGS3 (RGS3CT) both stimulated the GTPase activity of Giα (except Gzα) and Gqα but not that of Gsα or G12α. RGS3 and RGS3CT had Gqα GAP activity similar to that of RGS4. RGS3 impaired signaling through Gq-linked receptors, although RGS3CT invariably inhibited better than did full-length RGS3. RGS3 potently inhibited GqαQ209L- and G11αQ209l-mediated activation of a cAMP-response element-binding protein reporter gene and GqαQ209L induced inositol phosphate production, suggesting that RGS3 efficiently blocks Gqα from activating its downstream effector phospholipase C-β. Whereas RGS2 and to a lesser extent RGS10 also inhibited signaling by these GTPase-deficient G proteins, other RGS proteins including RGS4 did not. Mutation of residues in RGS3 similar to those required for RGS4 Giα GAP activity, as well as several residues N terminal to its RGS domain impaired RGS3 function. A greater percentage of RGS3CT localized at the cell membrane than the full-length version, potentially explaining why RGS3CT blocked signaling better than did full-length RGS3. Thus, RGS3 can impair Gi- (but not Gz-) and Gq-mediated signaling in hematopoietic and other cell types by acting as a GAP for Giα and Gqα subfamily members and as a potent Gqα subfamily effector antagonist.
A variety of hormones, neurotransmitters, and physical stimuli trigger intracellular responses by binding to seven transmembrane receptors. These receptors link to downstream signaling pathways by activating heterotrimeric G proteins and, as such, are designated G protein-coupled receptors (GPCRs). In their inactive state heterotrimeric G proteins are composed of three subunits: α, β, and γ (see reviews by Bourne et al., 1991; Hepler and Gilman, 1992;Gudermann et al., 1995; Neer, 1995). There are 23 α-subunits divided into four major subfamilies based on primary sequence homology and common downstream effectors termed Gsα, Giα, Gqα, and G12/13α. There are five different β-subunits and 10 different γ-subunits. Upon ligand binding a GPCR stimulates the α-subunit of a heterotrimeric G protein to exchange GDP for GTP. In the GTP-bound form, Gα dissociates from Gβ γ, each of which can activate downstream effectors. Signaling is halted when the GTP-bound Gα-subunits hydrolyze GTP to GDP, which results in reassembly with Gβ γ-subunits to form inactive heterotrimers.
Recent genetic and biochemical experiments have revealed the existence of a novel family of proteins termed Regulators ofG protein Signaling (RGS) that act as GTPase-activating proteins (GAPs) for the Giαand Gqα subfamilies (De Vries et al., 1995;Berman et al., 1996b; Dohlman et al., 1996; Druey et al., 1996; Hunt et al., 1996; Koelle and Horvitz, 1996; Watson et al., 1996). Recently, p115 RhoGEF, which contains a highly diverged RGS domain, was shown to be a G12α GAP (Kozasa et al., 1998); however, no Gsα GAP has been shown to exist. Many RGS proteins bind tightly to the GDP-AlF4 −-activated forms of Giα and Gqα , a conformation that mimics the transition state in the GTPase reaction, and thereby accelerate the intrinsic rate at which the Gα-subunits hydrolyze GTP (Berman et al., 1996a, b; Hunt et al., 1996; Watson et al., 1996; Hepler et al., 1997;Popov et al., 1997). Analysis of crystals of RGS4 complexed with Giα 1-GDP-AlF4 −revealed that the 120-amino acid RGS domain (also referred to as the RGS box) forms a four-helix bundle that directly contacts the three “switch regions” in Giα 1 (Tesmer et al., 1997). These regions undergo the greatest conformational change during GTP hydrolysis, and specific amino acids in RGS4 appear to stabilize them in a transition state facilitating the hydrolysis reaction. The specificity of RGS4 protein for the Giα and Gqα subfamilies likely relies on the structure of the switch regions. Based on the RGS4-Giα 1 and Gsα crystal structures, the failure of RGS4 to bind Gsα is secondary to specific amino acids in Gsα and RGS4 that disrupt the interaction by steric overlap, charge repulsion, and creations of small cavities at the interface (Sunahara et al., 1997; Tesmer et al., 1997). The failure of RGS4 to act as a GAP for G12α is more easily explained because amino acid differences in the G12α switch regions would disrupt the surface and charge complementarity of the interface observed between RGS4 and Giα 1 (Tesmer et al., 1997).
Several studies have indicated that the RGS protein RGS3 impairs Gi- and Gq-mediated signaling. RGS3 inhibited interleukin-8 induced mitogen-activated protein kinase activation (Druey et al., 1996) and inositol triphosphate (IP3) production in response to signaling through the gonadotropin-releasing hormone (GnRH) receptor (Neill et al., 1997), Gi- and Gq-linked signaling pathways, respectively. A truncated form of RGS3 (RGS3CT) impaired Gq- and Gi-mediated signaling as well as Gs-triggered signaling, whereas a full-length version inhibited only Gi-mediated signaling (Chatterjee et al., 1997a). In contrast, expression of a full-length RGS3 in a human mesangial cell line partially blocked an endothelin-1-induced calcium flux, a Gq-mediated response (Dulin et al., 1999). The present study explored the relative effectiveness of RGS3, RGS3CT, and RGS4 in modulating Gq-mediated signaling. We provide information concerning the relative GAP activity of RGS3, RGS3CT, and RGS4 for Gqα, as well as Giα, Gzα, Gsα, and G12α. RGS3 emerges as a potent inhibitor of Gq-mediated signaling by acting not only as a Gq GAP but also as an antagonist of GTP-bound Gq signaling.
Experimental Procedures
Plasmids.
To make His6-RGS3 and His6-RGS3CT (amino acids 314–520) polymerase chain reaction (PCR) fragments generated from RcCMV-RGS3 were inserted into the NdeI/XhoI sites of the bacterial expression vector pET15b (Novagen, Madison, WI) in frame with the hexahistidine tag. To make glutathione S-transferase (GST)-RGS3, a PCR product generated from RcCMV-RGS3 was directionally cloned in the BamHI and EcoRI sites of the bacterial expression vector pGEX2T. To make FLAG-RGS3NT (RGS3 1–313), FLAG-RGS3CT (314–520), and FLAG-RGS3, the appropriate PCR products were subcloned into pFLAGCMV-2. FLAG-RGS3 E419A/N420A (EN mutant), FLAG-RGS3 R499A/F500A (RF mutant), FLAG-RGS3 K350A/K353A/liter356A (KKL mutant), and FLAG-RGS3 E386A/E387A (EE mutant) were created by site-directed mutagenesis of pFLAGCMV-2 RGS3 (Stratagene, La Jolla, CA). Expression vectors for the beta-adrenergic receptor, GqαQ209L and G11αQ209L, were kindly provided by Dr. S. Gutkind (National Institutes of Health, Bethesda, MD). The expression vectors for RGS1, RGS2, RGS3, and RGS4 have been previously described (Druey et al., 1996). Dr. P. Casey (Duke University, Durham, NC) and Dr. J. Gunzburg (Institut Curie, Paris, France) kindly provided the RGS10 and RGS14 expression vectors, respectively. The RGS5 expression vector was created by PCR with known sequence information and subcloned in-frame with a hemagglutinin (HA)-tag into pCRIII. The cAMP-response element binding (CREB)-β-galactosidase reporter plasmid was kindly provided by Dr. R. Cone (Vollum Institute, OR). The pFA2-Elk1, pFR-luc, and pSRE-luciferase plasmids were purchased (Stratagene).
Purification of Recombinant Proteins.
The His-tagged recombinant RGS protein expressions were performed in Escherichia coli BL21(DE3) by induction with 0.5 mM isopropylthio-β-galactoside at 30°C for 2 h. The recombinant proteins were batch purified under nondenaturing conditions with NiNTA beads (Qiagen, Santa Clara, CA) and eluted with an imidazole gradient. The purified protein fractions were dialyzed against the wash buffer and stored at −70°C. To make the GST fusion proteins, the appropriate constructs were transformed into E. coli BL21(DE3) pLysS, and induced with 0.5 mM isopropylthio-β-galactoside for 2 h at 30°C. Recombinant protein purification was carried out in ice-cold phosphate-buffered saline (PBS)/1% Triton X-100 with glutathione-Sepharose beads (Pharmacia, Piscataway, NJ). After purification the GST-fusion protein was stored on the beads at 4°C or eluted and kept at −70°C.
Immunocytochemistry.
HEK 293T cells were grown on a cover slip in a 10-cm plate [Dulbecco's modified Eagle's medium (DMEM), 10% fetal calf serum (FCS)] until they were 50% confluent. Transfection with pFLAGCMV-2 RGS3, pFLAGCMV-2 RGS3CT, or empty vector was performed with calcium phosphate. The medium was changed 8 h after transfection, and cells were harvested 2 days later. The cover slips were washed with PBS, covered with 50% acetone/50% methanol, and kept at 4°C. After 1 h the liquid was removed, and the cover slips were air dried. Blocking of nonspecific binding sites was performed for 2 h at room temperature with PBS containing 10% FCS and 2% bovine serum albumin (BSA). Then the slides were incubated with mouse anti-FLAG monoclonal antibody (1:1000) in 2% BSA in PBS for 2 h at room temperature. After the sample was washed with PBS for 10 min, the slides were incubated for 2 h with fluorescein isothiocyanate-conjugated affinity-purified goat anti-mouse Ig 1:1000 in PBS containing 2% BSA. Then, cover slips were washed four times with PBS, air dried, and mounted on slides.
Measurement of GAP Activity
Measurements ofk cat for hydrolysis of GTP for Gzα and G12α were determined as described (Berman et al., 1996b). Direct measurement of thek cat for GTP hydrolysis by Gqαrequired the use of the mutant GqαR183C, which is based on the analogous mutation in Gsα, R174A (Freissmuth and Gilman, 1989), and Giα, R178C (Kleuss et al., 1994). Although this mutation in Giα markedly reduces itsk cat for GTP hydrolysis, the mutant protein retains its responsiveness to RGS proteins (Chediac and Ross, 1999). The method used for Gzα hydrolysis of GTP is a modification of that previously described (Berman et al., 1996b). In this study similar methods were used for Giα to approximate as closely as possible the conditions for Gqα, Gzα, and G12α. Briefly, G protein α-subunits were loaded with [γ32P]GTP (5–10 μM, Amersham, Cleveland, OH) in the presence of 50 mM HEPES (pH 7.4), 0.1 mg/ml BSA, 1 mM dithiothreitol, and either 5 mM EDTA and 0.05% C12E10 (for Giα) or 10 μM free Mg2+, 30 mM (NH4)2SO4, 4% glycerol, and 5.5 mM 3-[(cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (CHAPS; for Gqα). The loading reactions were performed for 20 min at 30°C for Giα or 2 h at 20°C for Gqα. After incubation, free [γ32P]GTP and [32P]orthophosphate were removed by chromatography on Sephadex 25 containing 50 mM HEPES (pH 7.4), 1 mM CHAPS, 1 mM dithiothreitol, 18 μg/ml BSA, and either 0.1% octylglucoside plus 5 mM EDTA (Giα) or 10 μM free Mg2+(Gqα). Hydrolysis of bound [γ32P]GTP was initiated by addition of 1 mM nonradioactive GTP, 10 mM MgCl2 (for Giα), and RGS protein or buffer. Reaction temperatures for Giα and Gqα were 4 and 20°C, respectively. Aliquots were removed at the indicated times and added to 5% (w/v) Norit (Norit Americas Inc., Atlanta, GA) in 50 mM NaH2P04. After the sample was centrifuged at 1500 rpm for 10 min, aliquots of supernatant containing 32Pi were counted by liquid scintillation spectrometry.
Assessment of Reporter Gene Activity
HEK 293T cells were plated in 10-cm dishes and transfected using calcium phosphate when the cells were 50% confluent. For Gq-mediated signaling, HEK 293T were transfected with constructs that direct the expression of the muscarinic type 1 (M1) receptor (2 μg/plate), FLAG-RGS3 or HA-RGS4, and CREB β-galactosidase reporter plasmid (2 μg/plate) receptor. In some experiments 0.5 μg of a cytomegalovirus-luciferase plasmid (Promega) was used to monitor the transfection efficiency. pcDNA was used to normalize the total amount of DNA used per plate. The medium was replaced 8 h later, and 48 h after transfection the cells were stimulated for 6 h with 1 mM carbachol (Sigma, St. Louis, MO) and then harvested. For Gs-mediated signaling, HEK293T cells were transfected with constructs that direct the expression of the beta-adrenergic receptor (2 μg/plate), CREB-β-galactosidase (1 μg/plate), and FLAG-RGS3 or HA-RGS4. Forty-eight hours after transfection, the cells were stimulated for 6 h with 10 μM isoproterenol (Sigma) and harvested. For GqαQ209L- and G11αQ209l-mediated signaling, HEK 293T cells were transfected with constructs that direct the expression of CREB-β-galactosidase (1 μg/plate), GqαQ209L or G11αQ209L (0.5 μg/plate), and different RGS protein expression vectors. The cells were harvested 24 h after transfection. The pelleted cells from the various signaling assays were lysed in 100 μl of reporter lysis buffer (Promega) for 20 min on ice. After the sample was centrifuged, 10 μl of the supernatant were tested for β-galactosidase activity with galactan chemiluminescent substrate (Tropix, Bedford, MA) or luciferase activity with a luciferase substrate (Promega). Data were normalized by protein concentration (Bradford assay, Bio-Rad, Hercules, CA) or by the activity levels of a control reporter gene. The expression levels of various RGS proteins were confirmed by immunoblotting for the appropriate epitope, HA or FLAG.
Western Blotting
The HS-Sultan, Molt-4, Jurkat, COS-7, PC-12, RAMOS, HeLa, and K562 cell lines were obtained from the American Tissue Culture Collection (Rockville, MD). All the lymphoid cell lines were maintained in RPMI 1640 supplemented with 5 to 10% FCS, and the nonlymphoid cells were maintained in DMEM plus 10% FCS. Cell lysates of various cell lines were obtained by adding 1 × 107 cells to a solution containing 150 mM NaCl, 50 mM Tris (pH 7.5), 5 mM EDTA, and 1% Nonidet P-40, along with a cocktail of protease inhibitors for 20 min on ice. The detergent-insoluble material was removed by microcentrifugation for 10 min at 4°C. In some experiments, cells were lysed in hypotonic buffer (20 mM Tris-HCl, pH 7.5, with protease inhibitors), sonicated, subjected to a low-speed spin to remove the nuclei, and fractionated into a membrane-enriched and -depleted fraction by centrifugation at 52,000 rpm for 30 min. A total of 50 to 100 μg of protein (Bio-Rad assay) from each sample were fractionated by SDS-polyacrylamide gel electrophoresis and transferred to pure nitrocellulose. Membranes were blocked with 3% BSA in TTBS (Tris-HCl, NaCl, Tween 20) for 1 h and then incubated with an appropriate dilution of the primary antibody in 1.5% BSA and 0.05% sodium azide in TTBS overnight. The blots were washed twice with TTBS before the addition of a biotinylated goat-anti rabbit Ig (DAKO, Carpinteria, CA) diluted 1:5000 in TTBS containing 10% FCS. After a 1-h incubation, the blot was washed twice with TTBS and then incubated with streptavidin conjugated to horseradish peroxidase (DAKO). The signal was detected by enhanced chemiluminescence following the recommendations of the manufacturer (Amersham). The antisera against RGS3 were used at a 1:400, and the mouse monoclonal antibodies were reactive with FLAG or HA (Covance, Richmond, CA) at a 1:1000 dilution. The RGS3 antiserum used in this study was prepared against recombinant RGS3 in rabbits and recognized recombinant RGS3, transfected RGS3, and a band of similar mobility in cellular lysates. Another rabbit antiserum raised against a conserved peptide in RGS2 and RGS3 also recognized recombinant RGS3, transfected RGS3, and the same bands as did the first antiserum (data not shown).
Inositol Phosphate Production
COS-7 cells were transfected with LipofectAMINE (1:8) after serum starvation for 24 h. Twenty-four hours after transfection, the culture medium was replaced with inositol-free DMEM containing 5% FCS and 1 mM sodium pyruvate for 2 h. Next, 2 μCi/ml ofmyo-[2-3H]inositol (Amersham) were added, and 15 min later, 10 mM LiCl was added. The cells were incubated for an additional 14 h and washed with phosphate-buffered saline, and then 0.5 ml of 20 mM formic acid was added to each well. After an incubation period of 30 min, the supernatant was collected and a second extraction was performed. Each 1-ml extract was neutralized to pH 7.5 with 7.5 mM HEPES and 150 mM KOH. The supernatants were centrifuged for 2 min at 15,000g and collected, and each was loaded onto to a 0.5-ml Dowex AG-X8 column (Bio-Rad), which had been previously washed with 2 ml of 1 M NaOH and 2 ml 1 M formic acid and five washes of 5 ml of water. After the sample was loaded, the column was washed with 5 ml of water, 5 ml of 5 mM borax, and 60 mM sodium formate. The columns were eluted with 3 ml of 0.9 M ammonium formate and 0.1 M formic acid. To 10 ml of CytoScint, 0.2 ml of each elution was added and the sample was analyzed via scintillation counting.
Results
Expression and Localization of RGS3
We generated an RGS3-specific antiserum to delineate the expression of RGS3 in various cell types. To do so we immunized rabbits with recombinant RGS3. The RGS3 was produced as a GST-fusion protein inE. coli and purified on glutathione-agarose before cleaving it from GST with factor Xa. The antiserum, but not the preimmune sera, recognized recombinant and in vitro translated RGS3, and its detection of RGS3 could be blocked by the immunizing peptide (data not shown). The analysis of cellular lysates prepared from a variety of cell lines revealed an approximately 75-kDa protein (Fig.1A) that comigrates with recombinant RGS3 or epitope-tagged RGS3 expressed in HEK 293T cells (data not shown). The migration of RGS3 did not coincide with its predicted molecular mass (54 kDa), suggesting a post-translational modification or simply aberrant migration. A similar migration of RGS3 has been found with a different antiserum (Dulin et al., 1999). A third antiserum raised against a shared epitope in RGS2 and RGS3 also recognized recombinant RGS3 and detected bands in cellular lysates similar to the other two antisera (S. Sinnarajah, unpublished observation). Three cell lines (K562, COS-7, and PC-12) expressed RGS3 at relatively high levels, whereas Hs-Sultan, RAMOS, HEK 293T, and NG108-15 had moderate levels, and HeLa, Nalm-6, Jurkat, K562, and Molt-4 had either low or undetectable amounts. Some cell lines had a doublet at approximately 75 kDa (Nalm-6, Jurkat, K562, and PC-12). We also examined whether lysophosphatidic acid (LPA) raised the levels of RGS3 expression in HS-Sultan cells, a human B lymphocyte cell line in which RGS1 can be induced by treatment with platelet-activating factor (Druey et al., 1996). Treatment with LPA resulted in a rapid enhancement of RGS3 expression with an increase noted by 1 h after stimulation (Fig.1A). Immunofluorescent staining with an epitope-specific antibody localized RGS3 and RGS3CT largely in the cytosol of transfected COS cells (Fig. 1B), although this approach provided little information concerning the amounts of RGS3 and RGS3CT associated with the cell membrane (see below).

Expression and intracellular localization of RGS3. A, Different cell lysates were analyzed by immunoblotting with an anti-RGS3 antiserum (lanes 1–16). Hs-Sultan cells were stimulated with LPA (10−7 M) for the indicated times. Molecular mass markers and the origin of the cell lysates are indicated. B, Localization of RGS3 and RGS3CT. Cos-7 cells were transfected with constructs directing expression of FLAG-RGS3 or FLAG-RGS3CT, or with a control plasmid. Immunofluorescent staining with a FLAG antibody is shown.
RGS3 and RGS3CT Enhanced the GTPase Activity of Giαand Gqα but Not That of Gsα, G12α, or Gz.
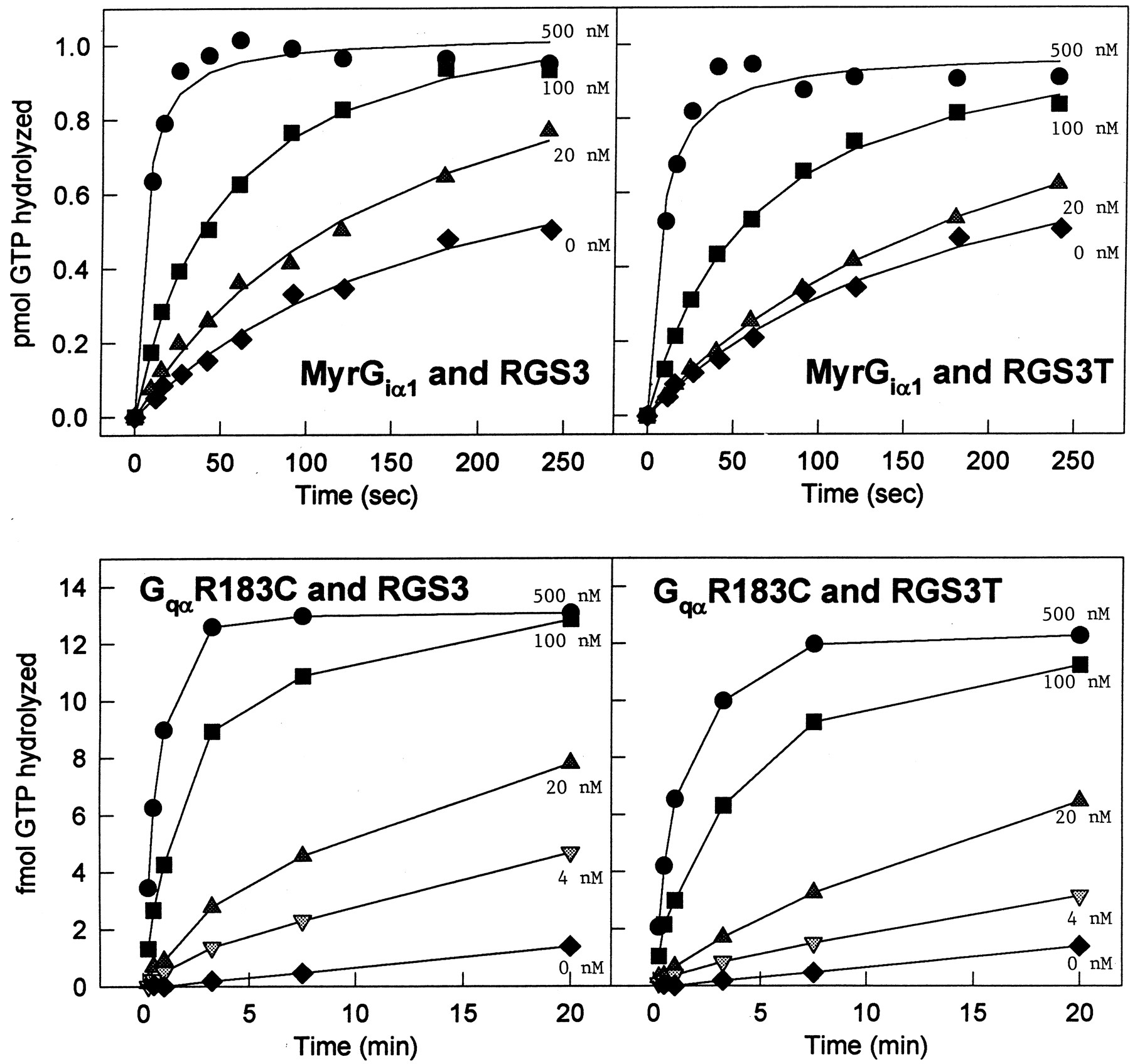
The previously cited differences of RGS3 and RGS3CT on signaling pathways prompted us to compare the GAP activity of RGS3 and RGS3CT for different Gα-subunits. We analyzed the effects of RGS3 and RGS3CT on the GTPase activity of Giα 1 and Gqα during a single catalytic turnover (Fig.2). The GqαGTPase assays required the use of the mutant GqαR183C (Chediac and Ross, 1999), which is based on analogous mutations in Gsα, R174A (Freissmuth and Gilman, 1989), and Giα, R178C (Kleuss et al., 1994). This Giα mutant has a significantly reduced k cat of GTP hydrolysis but retains sensitivity to RGS proteins (Kleuss et al., 1994; Berman et al., 1996a). The recombinant G proteins were loaded with [32P]GTP, and GTP hydrolysis was initiated in the absence or presence of increasing concentrations of RGS proteins. Both RGS3 and RGS3CT stimulated GTP hydrolysis by Giα 1 and Gqα, indicating that the N-terminal domain of RGS3 does not alter the ability of RGS3 to act as a GAP.

RGS3 and RGS3CT enhance the GTPase activity of Giα 1 and Gqα. Different concentrations of H6-RGS3 (left panels) and H6-RGS3CT (right panels) were tested for their ability to accelerate the GTPase activity of MyrGiα 1(top panels) and Gqα R183C (bottom panels). The following concentrations of RGS3 or RGS3CT were added to the MyrGiα 1 GTPase hydrolysis reactions: none (♦), 20 nM (▴), 100 nM (▪), or 500 nM (●). The following concentrations of RGS3 or RGS3CT were added to the GqαR183C GTPase hydrolysis reactions: none (♦), 4 nM (▾), 20 nM (▴), 100 nM (▪), or 500 nM (●).
Next, we compared the effects of RGS3 and RGS3CT with those of RGS4 on the GTPase activity of different Gα-subunits (Fig.3). We found that RGS3 and RGS3CT enhanced the Gqα GTPase activity to a degree similar to that of RGS4, whereas RGS4 was a superior GAP for Giα 1. Strikingly, in the Gzα GAP assay, RGS4 had significant activity, whereas RGS3 had none. Despite the ability of RGS3CT to inhibit Gs-mediated signaling (Chatterjee et al., 1997a), both RGS3 and RGS3CT failed to act as a GAP for Gsα (Fig. 5). Finally, RGS3 and RGS3CT did not alter the GTPase activity of G12α (data not shown).
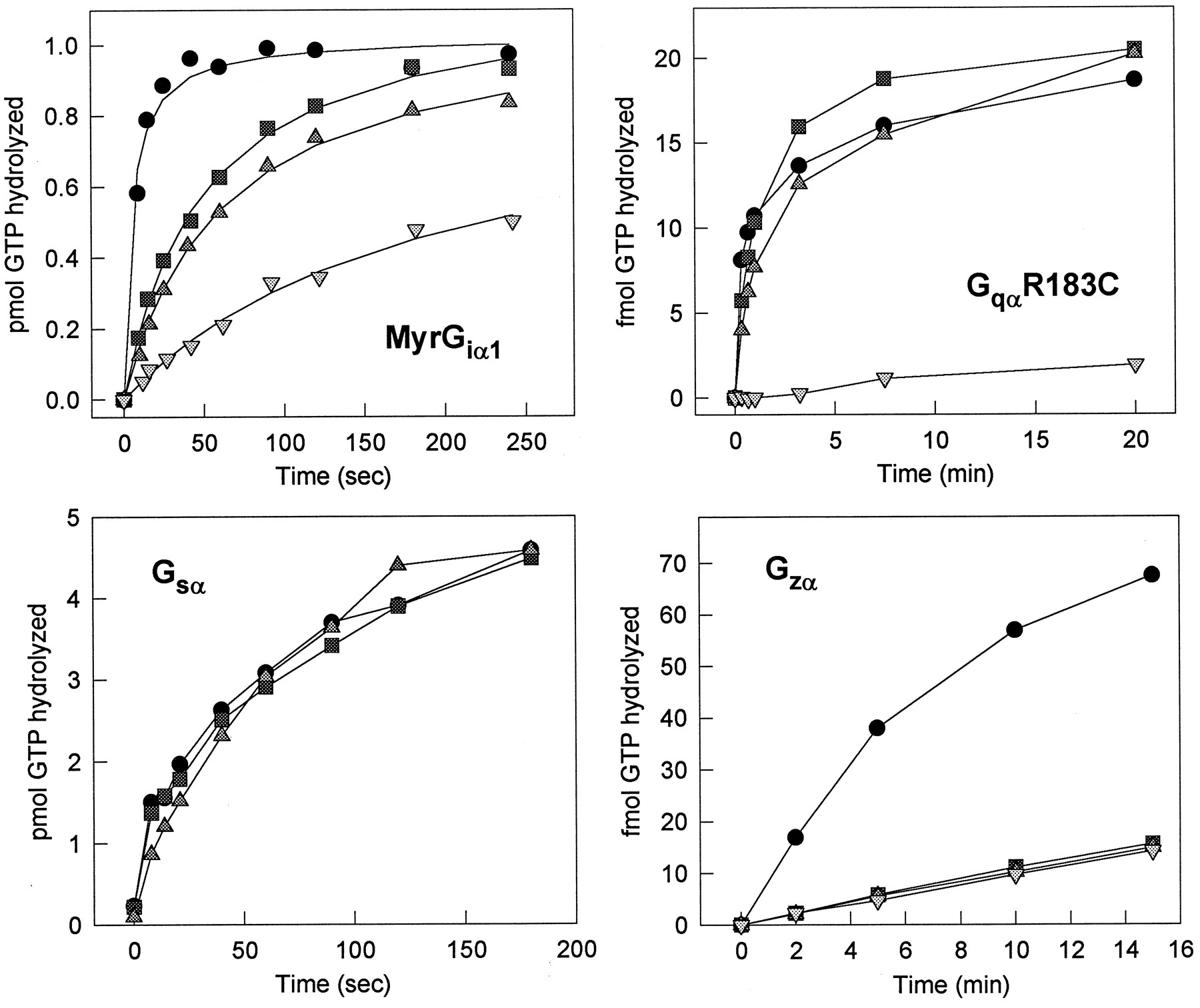
Comparison of RGS3, RGS3CT, and RGS4 on the GTPase activity of Giα 1, Gqα, Gsα, or Gzα. H6-RGS3, H6-RGS3CT, and H6-RGS4 were tested for their enhancement of the GTPase activity of Giα 1[top left: 100 nM RGS4 (●), 100 nM RGS3 (▪), 100 nM RGS3CT (▴), buffer (▾)], GqαR183C [top right: 150 nM RGS4 (●), 150 nM RGS3 (▪), 150 nM RGS3CT (▴), buffer (▾)], Gsα [bottom left: 1 μM RGS3 (●),1 μM RGS3CT (▪), buffer (▴)], or Gzα [bottom right: 150 nM RGS4 (●), 150 nM RGS3 (▪), 150 nM RGS3CT (▴), buffer (▾)].
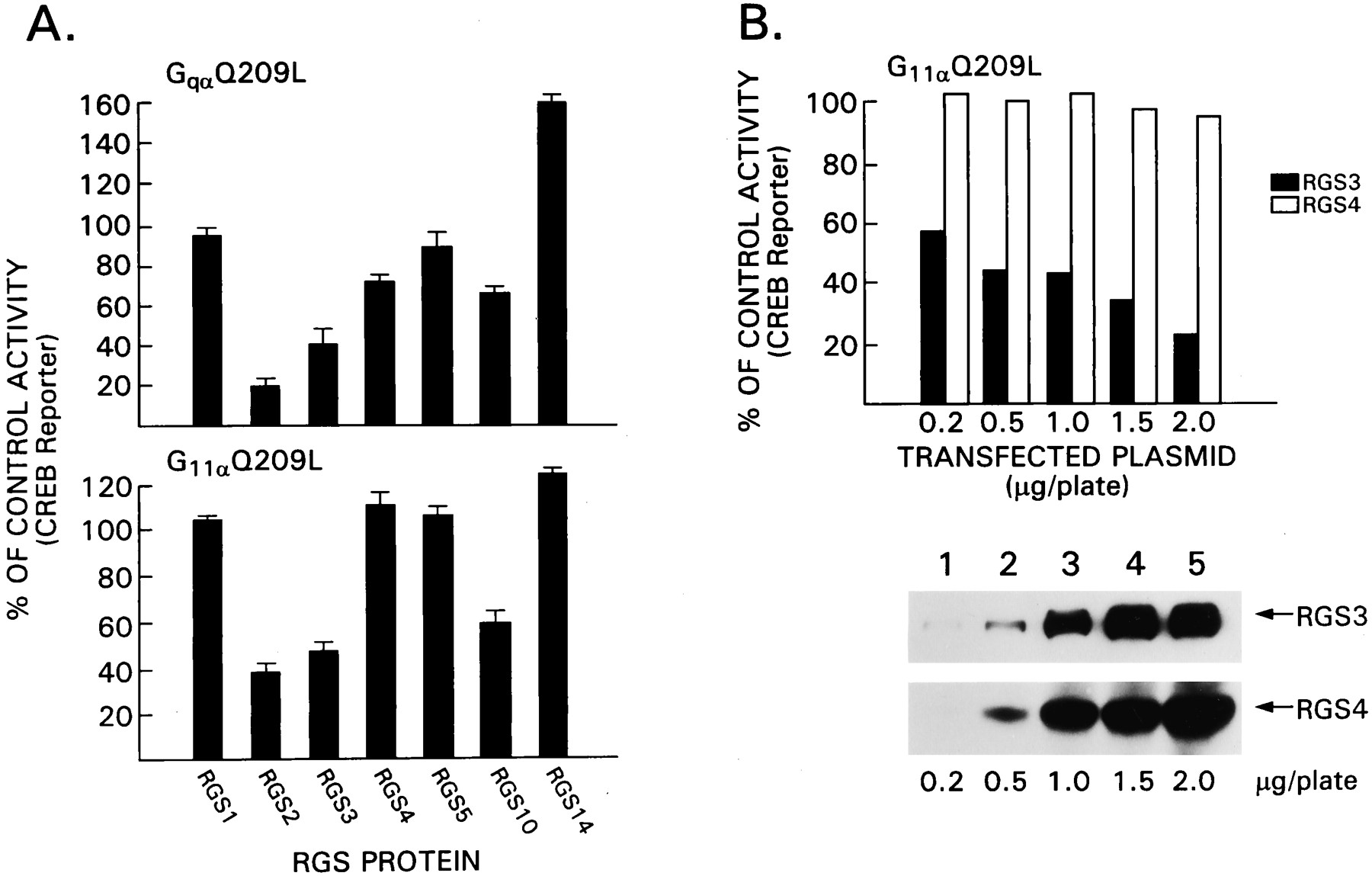
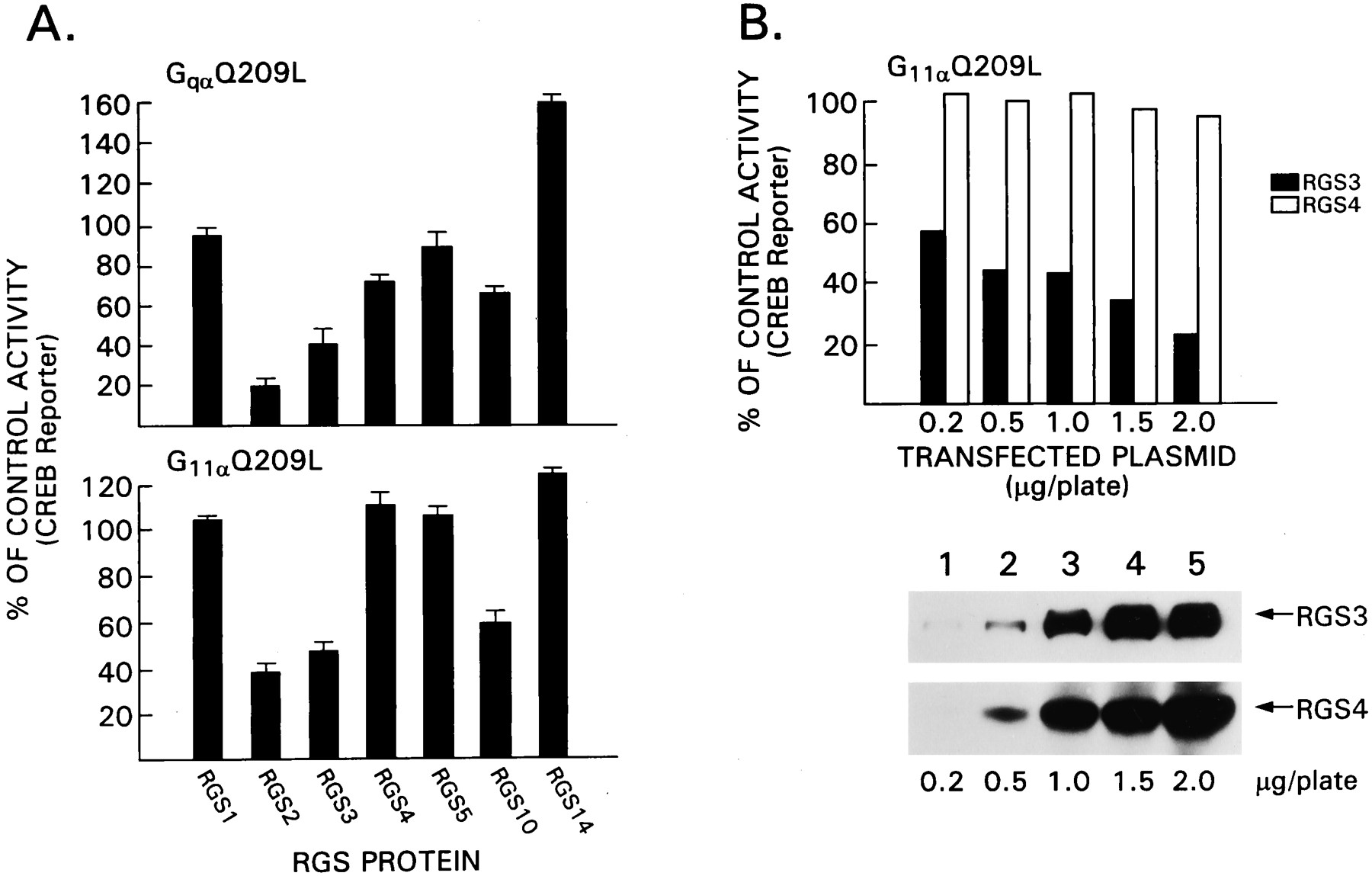
Comparison of the effects of different RGS proteins on Gq signaling. A, GqαQ209L- and G11αQ209l-mediated CREB activation is inhibited by RGS proteins. HEK 293T cells were transfected with constructs directing the expression of GqαQ209L (0.5 μg/plate) or G11αQ209L (0.5 μg/plate) in the presence of CREB-β-galactosidase (0.5 μg/plate) and different RGS protein expression vectors (1 μg/plate). Transfection efficiency was monitored by cotransfection with cytomegalovirus-luciferase (0.1 μg/plate). Luciferase and β-galactosidase activity were measured 24 h after transfection. Data are reported as the percentages of control ± 2 S.D. of three experiments performed in duplicate. The control is cells transfected with the activated G protein in the presence of an irrelevant plasmid. B, A direct comparison of the effects of RGS3 and RGS4 on G11αQ209L signaling. This experiment is similar to that shown in A except increasing concentrations of constructs that direct the expression of FLAG-RGS3 or FLAG-RGS4 were transfected. Data are from one experiment performed in duplicate and representative of two others performed. The data are expressed as percentages of activation compared with control transfections without RGS protein. The immunoblot was performed with a FLAG, and the signal was detected by enhanced chemiluminescence.
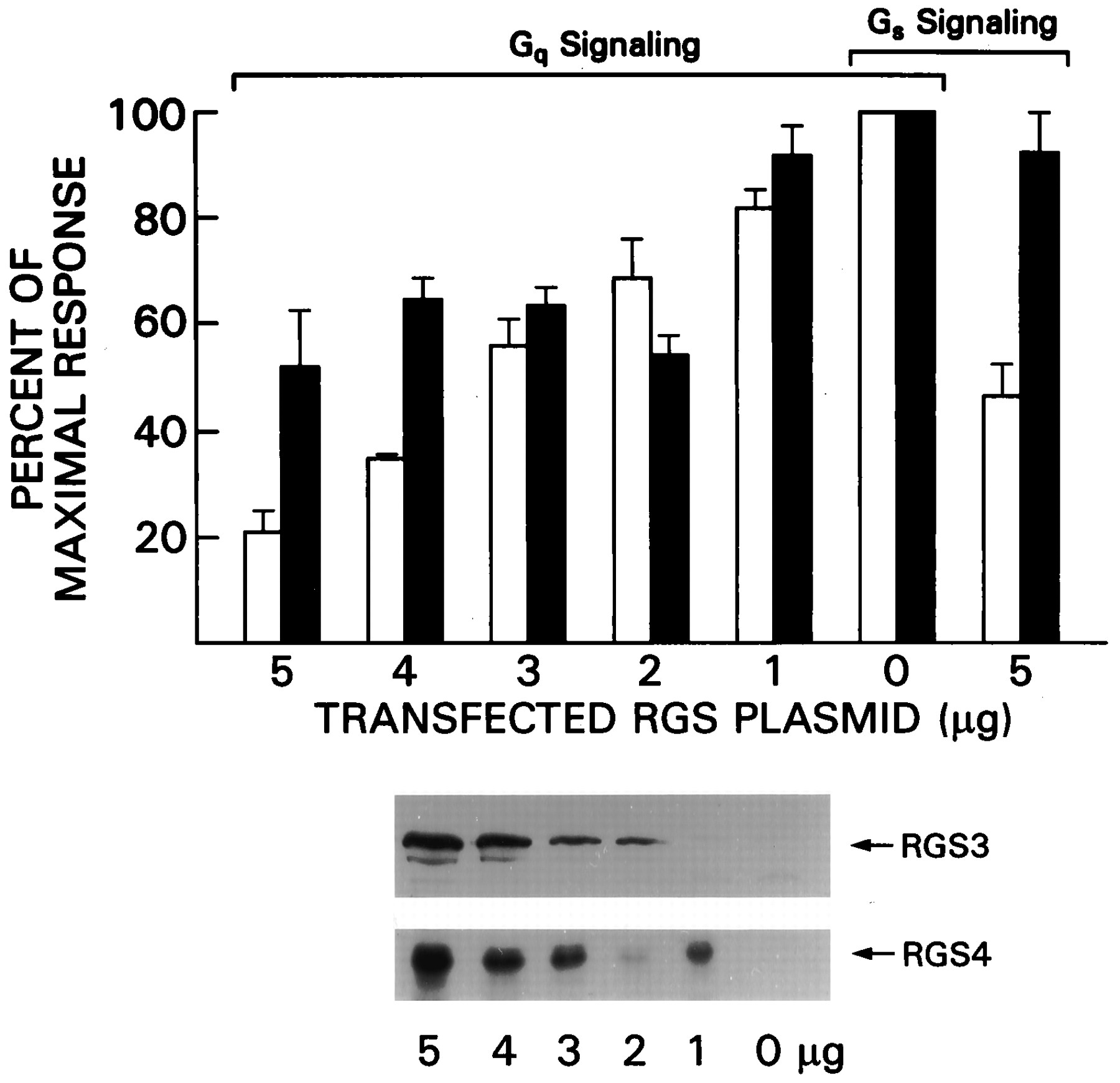
RGS3 Impairs Signal Transduction through the Muscarinic M1 Receptor and Beta-Adrenergic Receptor More Effectively Than Does RGS4
RGS3 inhibited IP3 production in response to signaling through the GnRH receptor (Neill et al., 1997), whereas RGS4 did not despite its ability to inhibit Gq-mediated signaling in other experiments (Hepler et al., 1997; Huang et al., 1997). RGS3CT inhibited platelet-activating factor-induced IP3 production, but RGS3 did not (Chatterjee et al., 1997a). To explore the relative effectiveness of RGS3 and RGS4 in inhibiting signal transduction through another Gq-coupled GPCR, we transiently transfected HEK293T cells with a construct that directs the expression of the M1 receptor in the presence or absence of increasing amounts of expression vectors for RGS3 or RGS4. Signaling through the M1 receptor was monitored with a CREB-driven β-galactosidase reporter plasmid. Activated Gqα is known to stimulate phospholipase C-β to convert phosphatidylinositol bisphosphate into IP3 and diacylglycerol. IP3 stimulates Ca+2 release from intracellular stores activating CaM kinase IV, which in-turn phosphorylates the transcription factor CREB. This results in CREB activation and transcription of the pCREB/β-galactosidase reporter gene (Chen et al., 1995). Carbachol stimulation of the M1-transfected cells resulted in a 10- to 20-fold increase in reporter gene activity. We observed a dose-dependent inhibition of reporter gene activity by RGS3 (Fig.4). RGS4 also inhibited signaling through the M1 receptor; however, the maximal level of inhibition was significantly less than that observed with RGS3. Immunoblotting cell lysates for RGS3 and RGS4 revealed the expected increase in RGS3 and RGS4 expression in the transfected cells (Fig. 4). Similar experiments were performed with RGS3CT, and it inhibited reporter gene activity to an even greater extent than did full-length RGS3 (data not shown). Thus, RGS3 inhibits Gq-mediated signaling through the M1 receptor in HEK293T cells much better than does RGS4.

RGS3 impairs signal transduction through Gq- and Gs-linked receptors. HEK 293T cells were transfected with constructs directing the expression of the M1 receptor (2 μg) or the β-adrenergic receptor (2 μg) and a CREB β-galactodase reporter construct (5 μg) in the presence of varying concentrations of pcDNA-RGS3HA (open bars) or pcDNA-RGS4HA (black bars) expression vector. The transfected cells were exposed to carbachol or isoproterenol for 8 h before analysis of reporter gene activity. Data are shown as percentages of activity obtained in the absence of RGS protein. In all experiments the agonists induced at least a 10-fold increase in reporter gene activity in the absence of RGS proteins. Expression of the transfected RGS plasmid was verified by HA immunoblotting. Results are from 12 independent experiments. Error bars, S.D.
In addition to its inhibition of Gq-mediated signaling, RGS3T reduced the calcitonin gene-related peptide stimulated increase of cAMP levels in BHK cells, a response mediated by Gsα(Chatterjee et al., 1997a). To evaluate whether RGS3 or RGS4 inhibited Gs-mediated signaling in HEK 293T cells, we transfected constructs that direct the expression of the beta-adrenergic receptor and either RGS3 or RGS4. Signaling via the beta-adrenergic receptor was monitored with the same reporter plasmid that was used for Gq-mediated signaling. Gsα-GTP activates adenylyl cyclase, which increases cAMP levels and results in protein kinase A activation. Activated protein kinase A phosphorylates CREB, resulting in CREB activation and enhanced transcription of the pCREB/β-galactosidase reporter gene (Chen et al., 1995). Isoproterenol induced an approximately 10-fold increase in reporter gene activity. To observe a consistent inhibition in reporter gene activation, we needed to transfect 5 μg/plate of RGS3 expression vector, whereas a similar level of RGS4 had only a modest inhibitory effect (Fig. 4). Thus, in HEK 293T cells, high expression levels of RGS3 can inhibit Gs-mediated signaling.
RGS3 Inhibits GqαQ209L and G11αQ209L Signaling
Although RGS3, RGS3CT, and RGS4 had equivalent GAP activity for Gqα, RGS3 better inhibited signaling through the Gq-linked GnRH (Neill et al., 1997) and M1 receptors than did RGS4. Although selective RGS protein-receptor interactions were reported recently (Xu et al., 1999), we suspected that other mechanisms might also be important. RGS2 potently inhibited Gq-signaling irrespective of the receptor used, in a manner superior to that of RGS1, RGS4, and RGS16 (Xu et al., 1999). Perhaps RGS2 and RGS3 inhibit the interaction of GTP-Gqα with its effectors better than do other RGS proteins. To establish an in vivo system to test the effectiveness of RGS proteins to act as effector antagonists for Gq subfamily members, we expressed constitutively active mutants of Gqα and G11α and evaluated their ability to activate three different reporter genes. Varying concentrations of G11αQ209L or GqαQ209L were transfected into HEK 293T cells along with a CREB, a serum response element, or an Elk-1 reporter gene (Table 1). Both G11αQ209L and GqαQ209L potently activated the CREB reporter gene, but not the serum response element and Elk-1 reporter genes. G11αQ209L and GqαQ209L had biphasic dose-response curves for CREB reporter gene activation with an optimal concentration of transfected DNA between 0.1 and 0.5 μg per transfection. When optimized, G11αQ209L activated the CREB reporter 2-fold better than did GqαQ209L.
Fold activation of different reporter genes by G11αQ209L and GqαQ209L
Next, we screened a panel of RGS proteins for their effect on GqαQ209L-mediated activation of the CREB reporter gene. Expression constructs designed to express RGS1, RGS2, RGS3, RGS4, RGS5, RGS10, or RGS14 were transfected into HEK 293T in the presence or absence of GqαQ209L along with the CREB reporter gene (Fig. 5A). Transfection efficiency was monitored with a control plasmid, which expressed luciferase from a cytomegalovirus promoter. All the RGS proteins were epitope tagged and well expressed as assessed by immunoblotting (data not shown). We found that RGS3 inhibited GqαQ209L-induced activation of the CREB reporter gene, whereas RGS4 had only a modest effect. Of the RGS proteins we tested, RGS2 was slightly superior to RGS3, and RGS10 had a modest effect. In contrast, RGS1 and RGS5 had minimal effects, and RGS14 consistently enhanced GqαQ209L-mediated activation of CREB activity. The same panel of RGS proteins was examined with G11αQ209L to activate the CREB reporter gene. Again RGS2 and RGS3 inhibited; however, RGS5 and RGS4 had no effect at the concentration tested. Similar to the analysis of GqαQ209L signaling, RGS10 had a modest inhibitory effec,t and RGS14 augmented the response, although to a lesser degree than previously. Next we directly compared RGS3 and RGS4 (Fig. 5B). The lowest amount of RGS3 expression vector tested (0.2 μg) was superior to the highest concentration of RGS4 tested (2 μg). Thus, despite their similar GAP activity for Gqα, 10-fold less RGS3 inhibits signaling by GqαQ209L and G11αQ209L better than did RGS4.
Analysis of the Effects of RGS3 Mutant Proteins on G11αQ209L Signaling
Constructs that direct the expression of two RGS3 point mutants, EN mutant and RF mutant, were created based on the residues in RGS4 known to be important in its interaction with Giα and necessary for its GAP activity for Giα (Druey and Kehrl, 1997; Tesmer et al., 1997; Srinivasa et al., 1998). Expression vectors for two other RGS3 mutant proteins, KKL mutant and EE mutant were created based on residues noted to be conserved in the N-terminal region of four RGS proteins that are Gqα GAPs (RGS1, RGS2, RGS3, and RGS4). HEK 293T cells were transfected with FLAG-RGS3 or one of the constructs that expresses a FLAG-tagged mutant protein in the presence of G11αQ209L and the CREB reporter gene. Similar levels of expression of the wild-type RGS3 and the RGS3 mutant proteins were observed by immunoblotting with a FLAG monoclonal antibody. We found that the EN and RF mutant proteins had significantly less activity than the wild-type protein, although the mutations did not totally abolish their activity (Fig. 6). In addition both the KKL mutant and the EE RGS3 mutant proteins had less activity than the wild-type protein. The KKL mutation was the more detrimental and indicates that region N terminus to the RGS domain is necessary for optimal inhibition of Gq/11-mediated signaling.

Effects of point mutations in RGS3 on G11αQ209l-mediated CREB reporter gene activation. HEK 293T cells were transfected with constructs directing the expression of G11αQ209L (0.5 μg/plate), CREB-galactosidase (0.5 μg/plate), and FLAG-RGS3 or various FLAG-RGS3 mutant proteins (1 μg/plate). β-Galactosidase levels were measured 24 h later. The data are expressed as percentages of activation in comparison to HEK 293T cells expressing G11αQ209L in the presence of a control plasmid. Each value is the mean ± 2 S.D. of five experiments performed in duplicate. An anti-FLAG immunoblot (bottom) shows that all mutant proteins were expressed.
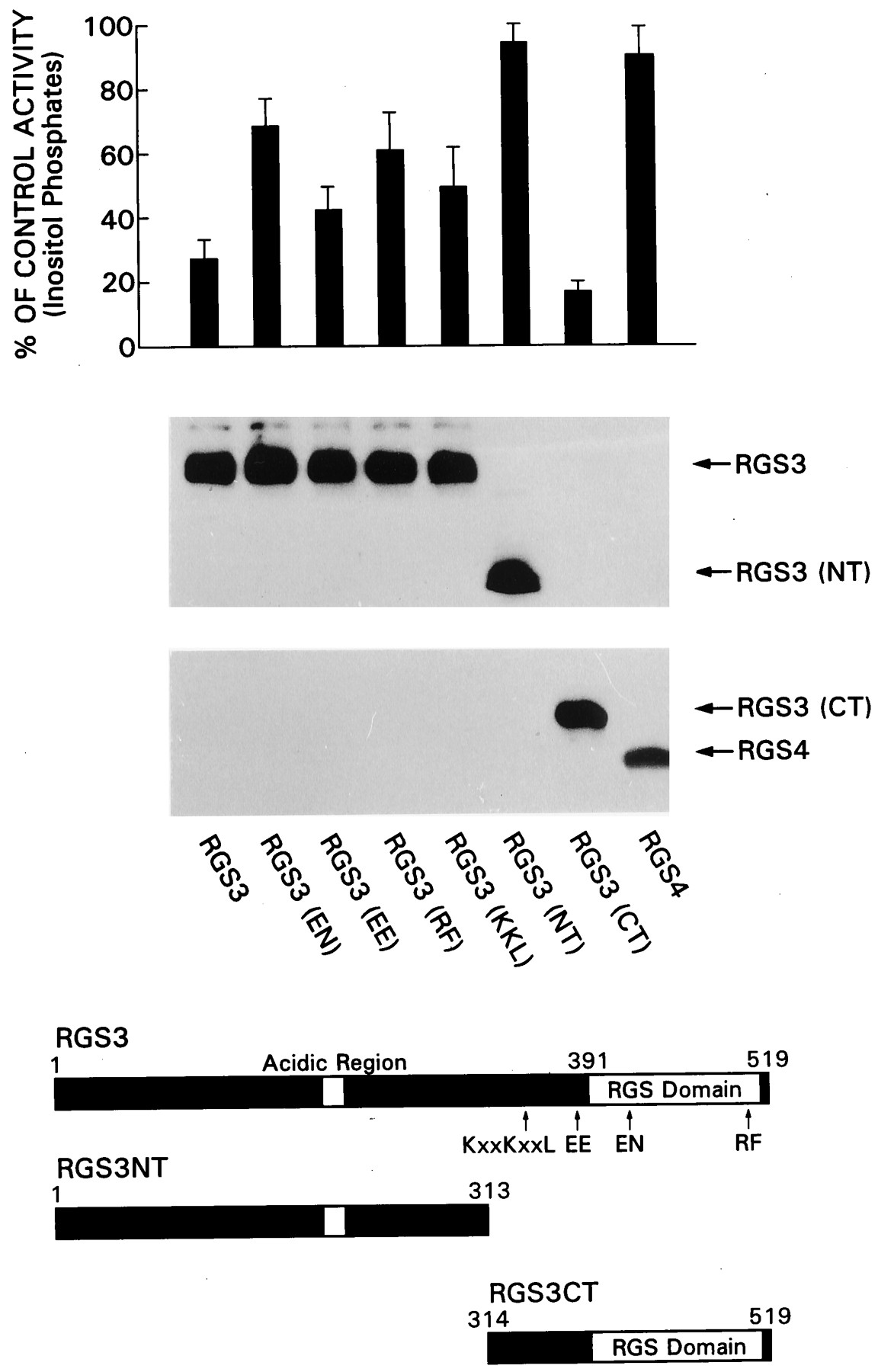
To verify these results in a different cell type and with a different assay system of Gq signaling, we examined the effects of RGS3 and RGS4 on inositol phosphate generation after expression of GqαQ209L in COS-7 cells. The expression of GqαQ209L in COS-7 cells resulted in a 12-fold increase in the generation of inositol phosphates. RGS3 significantly reduced GqαQ209l-induced inositol phosphate accumulation, whereas RGS4 was ineffective (Fig.7). The inhibitory effects of RGS3 were not due its N-terminal 314 amino acids, because the C-terminal 206 amino acids were more effective than the full-length protein, and the N terminus itself had no effect. Again, the RGS3 EN and RF mutants were impaired compared to wild-type RGS3 in their inhibition of GqαQ209L induced inositol phosphate accumulation. Also, similar to the previous results, the RGS3 KKL mutant was significantly compromised, whereas the RGS3 EE mutant was closer to that of wild type.

Top, Effect of point mutations in RGS3 on GqαQ209L-induced inositol phosphate generation. COS-7 cells were transfected with constructs directing the expression of GqαQ209L (0.2 μg/plate) and FLAG-RGS3, FLAG-RGS4, FLAG-RGS3CT, FLAG-RGS3NT or various FLAG-RGS3 mutant proteins (0.8 μg/plate). The accumulations of inositol phosphates were measured during the last 15 h of a 48-h culture period. The data are expressed as percentages of control (no RGS protein), and each value is the mean ± 2 S.D. of five experiments performed in duplicate. Middle, An anti-FLAG immunoblot indicates that all mutant proteins were expressed. Bottom, A schematic of RGS3, RGS3CT, and the various RGS3 mutants are shown.
Finally, to address why RGS3CT outperforms full-length RGS3 in the Gq-mediated-signaling experiments (Chatterjee et al., 1997b; Fig.8A) and yet had equivalent levels of GAP activity for Gqα (Fig. 3), we analyzed whether RGS3 and RGS3CT differed in the subcellular localization before or after expression of GqαQ209L. We transfected Cos-7 cells transfected with epitope-tagged versions of RGS3 or RGS3CT in the presence or absence of GqαQ209L and prepared cell lysates, the membrane-enriched and membrane-depleted fractions of which we fractionated. Immunoblotting for epitope-tagged RGS3 or RGS3CT revealed higher levels of RGS3CT in both the absence of GqαQ209L and following the expression of GqαQ209L (Fig. 8B). Before expression of GqαQ209L, we found 3-fold more RGS3CT than RGS3 in the membrane-enriched fraction, and after expression of the GTPase-deficient G-protein, we detected approximately 75% more RGS3CT than RGS3 in the membrane-enriched fraction. These results indicate that under steady-state conditions the N terminus of RGS3 may negatively effect the localization of the RGS3 to its likely site of action at the plasma membrane, which is in part overcome after cellular activation.

Membrane localization of RGS3 and RGS3CT. A, Dose response to RGS3 and RGS3CT. COS-7 cells were transfected with constructs directing the expression of GqαQ209L (0.2 μg/plate) and various amounts of either FLAG-RGS3 or FLAG-RGS3T. The accumulations of inositol phosphates were measured during the last 15 h of a 48-h culture period. The data are expressed as percentages of control (no RGS protein), and each value is the mean of two experiments. An anti-FLAG immunoblot shows the expression levels of RGS3 and RGS3CT. B, Distribution of RGS3 and RGS3CT at cellular membranes. Cell lysates prepared from COS-7 cells transfected with constructs directing the expression of GqαQ209L and increasing concentrations (0.2, 0.4, and 0.8 μg) of FLAG-RGS3 or FLAG-RGS3CT were fractionated into membrane-enriched (Memb.) and membrane-depleted fractions (Supernat.) and subjected to FLAG immunoblotting. The immunoblot was reprobed with an antiserum directed against Giα proteins to verify equal loading of the cell membranes. This experiment was performed three times with similar results.
Discussion
The observations described above extend our knowledge of RGS3 as a unique member of the RGS family. RGS3 is expressed in multiple cell tissues including in hematopoietic cells. It is a potent inhibitor of Gα-mediated signaling, and it accelerates the GTPase activity of Giα 1and Gqα but not Gzα, G12α, or Gsα. GTPase-deficient forms of Gqα and G11α are potent activators of a CREB reporter gene, and RGS3 inhibits their ability to activate the reporter gene. RGS3 amino acid residues in the putative RGS3/Gqα contact site, as well as N terminal to the RGS domain, are necessary for RGS3 to inhibit the activation of downstream effectors by GTPase-deficient forms of G11α and Gqα . The N terminus of RGS3 may regulate its GAP activity by limiting access of full-length RGS3 to Gα-subunits at the cell membrane.
Analysis of RGS3 mRNA expression has revealed multiple RGS3 mRNA transcripts present at high levels in lung, kidney, and muscle tissue (Druey et al., 1996). One major immunoreactive RGS3 band of 75 kDa was present in the cell lysates (in some instances a doublet was noted). When epitope-tagged versions of RGS3CT and RGS3NT are expressed in COS cells we have observed that RGS3NT is a doublet and RGS3CT is a single band (S. Sinnarajah, unpublished observation). This suggests that the N-terminal portion of RGS3 may be modified to account for the doublet noted by immunoblotting. The original RGS3 cDNA was isolated from a B lymphocyte cDNA library and is predicted to encode for a 54-kDa protein; however, recombinant RGS3 migrates at 75 kDa, as does the epitope-tagged version when expressed in mammalian cells. A smaller RGS3 may be derived from the 1.8-kb RGS3 mRNA transcript observed on Northern blot analysis and can be accounted for by an mRNA that splices from exon 2 to exon 4 deleting exon 3, which encodes the first 326 amino acids of RGS3. Alternatively, truncated forms of RGS3 may arise by another mechanism, perhaps incomplete gene duplication, or by the use of an alternative promoter (Chatterjee et al., 1997b). Our RGS3 antiserum would not recognize such proteins because it fails to detect the N-terminal truncated RGS3. The RGS3 antiserum was also used to examine the effects of stimulation through a GPCR on RGS3 expression in HS-Sultan cells. Similar to RGS1, which can be induced in HS-Sultan cells by treatment with platelet-activating factor (Druey et al., 1996), exposure of HS-Sultan cells to lysophosphatic acid resulted in increased RGS3 expression.
Our results define RGS3 as a particularly potent regulator of Gqα-mediated signaling. Although the GAP assay results suggest that RGS4 and RGS3 have similar levels of activity for Gqα, RGS3 proved more effective than RGS4 in inhibiting signaling through the Gq-linked M1 muscarinic receptor. The efficacy of RGS3 is unlikely related to its N-terminal domain, because RGS3CT is more potent than is RGS3. Also, we can conclude that the N-terminal domain of RGS3 does not markedly influence RGS3 GAP activity because both RGS3 and RGS3CT performed similarly in GAP assays. However, it remains possible that an in vivo post-translational modification of the N terminus of RGS3 could alter its GAP activity. The reported failure of RGS3 to inhibit Gq-mediated signaling in BHK cells suggests that a modification of RGS3 or the presence of an interacting protein may regulate its intracellular localization or GAP activity (Chatterjee et al., 1997a). In fact, our experiments suggest that the N-terminal RGS3 may limit its access to intracellular membranes. Less of the full-length RGS3 localized at cell membranes both before and after expression of GqαQ209L, a stimulus that translocates RGS3 to cellular membranes.
The efficacy of RGS3 in inhibiting Gq-mediated signaling is not likely explained solely on the basis of GPCR-RGS protein interaction (Xu et al., 1999). Both RGS2 and RGS3 are superior to RGS4 in inhibiting Gq-mediated signaling irrespective of the GPCR used. Furthermore, RGS3 and RGS2 markedly inhibited signal transduction initiated by Q209L mutants of Gqα and G11α , whereas RGS4 did not. Because RGS4 does not act as a GAP for GqαQ209L under the same conditions that it does for GqαR183C (P. Chediac, unpublished observation), a likely explanation for the efficacy of RGS3 is that it inhibits GTP-bound Gqα from activating downstream effectors. Previously, RGS2 was shown to be a 10- to 30-fold more potent inhibitor of GTP-γs-bound Gqα-induced activation of phospholipase Cβ than was RGS4 (Heximer et al., 1997). RGS10 also impaired activation of the CREB reporter gene by GTPase-deficient forms of Gq. However RGS1 is like RGS4, a Gqα GAP (Moratz et al., 2000), but incapable of blocking signaling by GTPase-deficient forms of Gq.
What is the structural basis of the success of RGS2 and RGS3 in inhibiting Gq/11αQ209L-mediated signal transduction? Comparison of the RGS domains of RGS2 and RGS3 with those of other RGS proteins that are not good inhibitors does not reveal any compelling differences. The amino acid residues in the three major contact sites defined in the RGS4/Giα 1 crystal structure are very similar between RGS3 and RGS4. Several of these residues are undoubtedly important in the interaction of RGS proteins with Gqα, because mutations of them interferes with the inhibition by RGS3 of G11αQ209L and GqαQ209L signaling. Mutations introduced into the region just N terminal to the RGS domain of RGS3 also impaired RGS3 function. The KKL mutation in RGS3 significantly compromised the inhibitory activity of RGS3, and yet these residues are conserved among many of the RGS proteins that do not behave as Gqα-effector antagonists, suggesting that other critical amino acids remain to be identified. A direct alignment of RGS2 and RGS3CT does reveal a short stretch of conserved amino acids to the C-terminal side of the KxxKxxL sequence (RGS2 residues 55–62 PGKPKTGK and RGS3 residues 366–373 PGAPPAGK) that are not present in other RGS proteins. To approach the importance of this region and the N-terminal portions of RGS2 and RGS3 in general, fusion proteins between the N terminus of RGS3 and the C terminus of RGS4 will be made to test whether we can convert RGS4 into a more potent Gq-effector antagonist.
Because the existence of RGS proteins that act as GAPs for Gsα remains a possibility and because we had observed an inhibition of Gs-mediated signaling, we were interested to examine whether RGS3 had Gsα GAP activity. When tested in a standard GAP assay, RGS3 failed to enhance the GTPase activity of Gsα. One caveat in interpreting the GAP data is that the assays in this study were done in the absence of receptors; therefore, it remains possible that RGS3 is a Gsα GAP in the presence of the appropriate receptor. A precedent for such a possibility is that the Gα specificity of RGS2 was only revealed in the presence of a receptor (Ingi et al., 1999). In preliminary experiments we found that the expression of RGS3 also inhibited the activation of the CREB reporter by a GTPase-deficient form of Gsα (J. Yuen, unpublished observation). Although the physiologic relevance of these observations needs further clarification, this is the third study to show that RGS proteins may modify Gs signaling (Chatterjee et al., 1997a; Tseng and Zhang, 1998).
Recently, we found RGS3 to be a more effective inhibitor than RGS1, RGS2, or RGS4 of interleukin-8 and MCP-1-directed migration of a pre-B lymphocyte cell line (Bowman et al., 1998). Because chemotaxis is dependent on the release of βγ-subunits from Giα-subunits (Arai et al., 1997; Neptune and Bourne, 1997), RGS3 may be among the most potent of the RGS proteins in inhibiting Gi-linked signaling pathways. Thus, RGS3 emerges as a potent inhibitor of both Giα and Gqα signaling. Its effectiveness as an inhibitor of Gq-signaling likely arises from both its Gqα GAP activity and its ability to inhibit signaling by GTP-bound Gqα and G11α. The function of the extended N terminus of RGS3 remains unknown; however, it is unlikely to account for the superiority of RGS3 in inhibiting Gi- and Gq-linked signaling pathways. Based on the signaling and cellular localization studies the N terminus may have a role in regulating the access of RGS3 to cellular membranes.
Acknowledgments
We thank Gaye Lynn Wilson and Kathy Harrison for technical assistance, Dr. Wen Jinn Chan for performing the initial GqαQ209L and G11αQ209L experiments, Mary Rust for editorial assistance, and Dr. Anthony S. Fauci for his support.
Footnotes
- Received September 3, 1999.
- Accepted June 12, 2000.
-
Send reprint requests to: Dr. John H. Kehrl, National Institutes of Health, Bldg. 10 Rm. 11B13 Center Dr. MSC 1876, Bethesda, MD 20892.
-
This work was supported in part by a grant from the Deutscher Akademischer Austauschdienst (A.S.) and the Fogarty International Center, National Institutes of Health (Bethesda, MD).
-
1 Present address: Department of Integrative Biology, Pharmacology and Physiology, University of Texas-Houston Medical School, 6431 Fannin, MSB 4.109, Houston, TX 77225.
-
2 Present address: Department of Pharmacology/Toxicology, University of Western Ontario, Medical Sciences Building, London, Ontario, N6A5C1 Canada.
Abbreviations
- GPCR
- G protein-coupled receptors
- GAP
- GTPase-activating proteins
- RGS
- regulators of G protein signaling
- IP3
- inositol triphosphate
- GnRH
- gonadotropin-releasing hormone
- PCR
- polymerase chain reaction
- CREB
- cAMP-response element binding
- GST
- glutathione S-transferase
- DMEM
- Dulbecco's modified Eagle's medium
- FCS
- fetal calf serum
- LPA
- lysophosphatidic acid
- HA
- hemagglutinin
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}