


Abstract
Presynaptic dopamine D2 receptors (D2Rs) regulate dopamine transporter (DAT) activity in the brain. A potential mechanism was suggested by the observations that somatodendritic D2R activation produces hyperpolarization and the velocity of DAT expressed in Xenopus laevis oocytes varies with changes in membrane potential. To investigate whether D2R regulation of DAT function is voltage-dependent, we coexpressed the long isoform of the human (h) D2R and the hDAT in oocytes. Most DAT substrates fully activate D2Rs at concentrations used to measure uptake. Thus, DAT function was compared under conditions of maximal D2R activation (0.1–10 μM DA) or maximal D2R blockade (DA + 1 μM (−)-sulpiride). D2R activation significantly increased [3H]DA uptake into unclamped oocytes expressing relatively lower velocities. Uptake measured with a saturating concentration of DA suggested a D2R-induced increase in V max. The D2R-mediated enhancement of DA uptake was not associated with changes in resting membrane potential and was abolished by pertussis toxin pretreatment. Furthermore, in voltage-clamped oocytes, D2R activation enhanced both DA uptake and DAT-mediated steady-state currents by as much as 70%. Activation of D2Rs resulted in a 59% increase in cell surface binding of the cocaine analog [3H]WIN 35,428; this effect was also abolished by pertussis toxin pretreatment. Saturation experiments confirmed that D2R activation was associated with an increased B max and unchangedK i for [3H]WIN 35,428. These results suggest that D2R-induced up-regulation of DAT activity occurs via a voltage-independent mechanism that depends on Gi/o activation and a rapid increase in expression of functional DAT molecules at the cell surface.
The dopamine transporter (DAT) belongs to a large family of neurotransmitter and amino acid transporters that are related functionally by their requirement for extracellular Na+ and Cl− (Uhl and Hartig, 1992; Amara and Kuhar, 1993). DAT couples the translocation of dopamine (DA) or other substrates to the driving force of these ions down their electrochemical gradients. This transporter, which is exclusively expressed in DA neurons (Ciliax et al., 1995; Kuhar et al., 1998), plays a critical role in regulating synaptic concentrations of DA (Giros et al., 1996). DAT-mediated reaccumulation of DA into the presynaptic terminals terminates the synaptic actions of DA at its receptors, thus regulating the intensity, duration, and extent of dopaminergic neurotransmission. Thus, presynaptic elements such as receptors or other regulatory proteins that alter or regulate DAT function could have a profound impact on DA-mediated pre- and postsynaptic signaling.
Two DA receptor subtypes were originally proposed based on their ability to either stimulate (D1R) or inhibit (D2R) adenylyl cyclase activity (Kebabian and Calne, 1979). To date, five distinct DA receptors have been cloned that fall into two classes: “D1-like” (D1Rs and D5Rs) and “D2-like” (D2Rs, D3Rs, and D4Rs) receptors (see Sokoloff and Schwartz, 1995). Two isoforms of the D2R have been identified, which are encoded by a single gene and differentially spliced to include (long form) or exclude (short form) 29 amino acids within the third intracellular loop of the receptor (Giros et al., 1989; Monsma et al., 1989). Inhibitory D2-like autoreceptors localized on DA neuronal terminals in brain modulate DA synthesis (Tissari et al., 1983; Wachtel et al., 1989) and release (see Starke et al., 1989). Also, D2-like autoreceptors localized on the soma and dendrites of DA neurons inhibit impulse flow by activating G protein-coupled inwardly rectifying potassium channels (GIRKs), thereby hyperpolarizing the neurons (Lacey, 1993; White, 1996). Compelling evidence that D2-like autoreceptors are D2Rs comes from reports that autoreceptor functions are lost in D2R-deficient mice (Mercuri et al., 1997; L'Hirondel et al., 1998), but not in D3R-deficient mice (Koeltzow et al., 1998).
There is growing evidence that D2Rs also participate in the presynaptic regulation of DAT activity (Meiergerd et al., 1993; Parsons et al., 1993; Cass and Gerhardt, 1994; Rothblat and Schneider, 1997; Dickinson et al., 1999; Hoffman et al., 1999). In vivo studies showed that chronic cocaine-induced increases in striatal DA uptake were attenuated by pretreatment with a selective D2R antagonist (Parsons et al., 1993). Similarly, acute or chronic administration of the D2R antagonist haloperidol reduces DA transport into striatal tissue in vitro, and local administration of haloperidol reduces DA uptake in vivo (Meiergerd et al., 1993; Rothblat and Schneider, 1997). In vivo DA clearance is also decreased in striatum, nucleus accumbens, and prefrontal cortex by the selective D2R antagonist raclopride but not by the selective D1R antagonist SCH-23390 (Cass and Gerhardt, 1994). In striatal suspensions, agonist activation of D2Rs increases DA uptake velocity, an effect blocked by a selective D2R antagonist (Meiergerd et al., 1993); these results provide the best evidence that the D2R-mediated effects on DA uptake occur via presynaptic autoreceptors. Consistent with the idea that activation of D2Rs increases DAT activity, in vivo DA clearance is reduced in D2R-deficient mice (Dickinson et al., 1999). Thus, D2Rs regulate DAT function, and this regulation could represent an important mechanism by which dopaminergic neurotransmission is modulated.
The molecular mechanism by which presynaptic D2Rs regulate DAT function is unknown. However, the activity of human (h) DAT expressed in Xenopus laevis oocytes is sensitive to changes in membrane potential (Sonders et al., 1997). Using electrophysiological and electrochemical techniques, Hoffman et al. (1999) recently concluded that DAT activity in rat substantia nigra is also voltage-dependent. These observations, along with the observation that presynaptic D2Rs can alter membrane potential through activation of GIRKs (Lacey, 1993), suggest one mechanism by which D2Rs could regulate DAT function (i.e., changes in membrane potential). Investigating this type of mechanistic question is difficult in native systems, because of the numerous regulatory proteins and neural networks that must be controlled. To address this question in a simpler model system in which membrane potential could be monitored and controlled, we coexpressed the long isoform of the hD2R and the hDAT in oocytes. We examined D2R regulation of DAT activity by measuring both DAT-mediated substrate uptake and currents and DAT expression by measuring cell surface binding of [3H]WIN 35,428. We found that D2R activation enhances DAT function by increasing the number of cell surface transporters via a Gi/o-dependent mechanism and that this effect was independent of changes in membrane potential.
Experimental Procedures
Materials.
cDNAs encoding the hDAT and the long isoform of the hD2R were provided by M. S. Sonders and S. G. Amara (Vollum Institute, Oregon Health Sciences University, Portland, OR), and D. K. Grandy (Dept. of Physiology & Pharmacology, Oregon Health Sciences University), respectively. cDNA encoding the rat GIRK1 (Kir3.1) was provided by H. A. Lester (California Institute of Technology, Pasadena, CA). Mazindol, GBR 12909, (−)-sulpiride, apomorphine, DA, and pertussis toxin (PTX) were purchased from RBI/Sigma (St. Louis, MO). [3H]DA (3,4-[7-3H]dihydroxyphenylethylamine; specific activity, 21.7 Ci/mmol), [3H](−)-sulpiride (specific activity, 73.7 Ci/mmol), and [3H]WIN 35,428 (2β-carbomethoxy-3β-(4-fluorophenyl)[3H]tropane; specific activity, 86.0 Ci/mmol) were purchased from PerkinElmer Life Sciences, Boston, MA. WIN 35,428 was obtained from the National Institute on Drug Abuse (Bethesda, MD). All other drugs were purchased from Sigma Chemical Co.
cRNA Preparation and Oocyte Expression.
Capped cRNAs were transcribed from linearized plasmids using standard in vitro transcription reactions (mMessage mMachine; Ambion, Austin, TX). Stage V or VI X. laevis oocytes were manually defolliculated, injected with water-diluted cRNA (∼10 ng/oocyte), and maintained at room temperature in frog Ringers' buffer (FRB), containing 96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 5 mM HEPES, pH 7.5, supplemented with 2.5 mM sodium pyruvate, 0.5 mM theophylline, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamicin. Oocytes were either coinjected with a mixture of DAT and D2R cRNAs on the day of oocyte isolation or injected with DAT cRNA on the day of isolation, followed 2 days later by D2R cRNA injection. Initially to confirm D2R function, voltage-clamp experiments were conducted on oocytes coexpressing D2R and GIRK1 (mixed and injected concurrently). GIRK1-mediated inward potassium currents were measured in FRB buffer containing 50 mM extracellular KCl. D2R agonists activated GIRK1-mediated currents in a manner consistent with previous reports using the oocyte expression system (Werner et al., 1996; Nelson et al., 1997), and these currents were reversibly blocked by the D2R antagonist (−)-sulpiride (1 μM).
DA Uptake Assays.
For assays using [3H]DA, oocytes were incubated in 0.5 ml of FRB containing 100 nM [3H]DA for 10 min at room temperature. The oocytes were then washed three times in 5 ml of ice-cold FRB and solubilized in 0.25 ml of 2% SDS. [3H]DA accumulation into individual oocytes was quantified by liquid scintillation spectroscopy. Nonspecific uptake was <2% of total uptake and was determined by incubating water-injected oocytes or cRNA-injected oocytes in the presence of a saturating concentration of mazindol (10 μM).
The dependence of DAT-mediated DA uptake on membrane potential was examined in individual oocytes coexpressing DAT and D2R cRNAs. In these experiments, oocytes were placed in a recording chamber as described below for two-electrode, voltage-clamp studies, and then superfused with FRB at approximately 4 ml/min. FRB containing 10 μM DA in the absence (D2R-activated) or presence of 1 μM (−)-sulpiride (D2R-blocked) was applied for 3 min at room temperature. Membrane potential was monitored continuously. In addition, voltage-clamped (−60 mV) DA uptake was measured in oocytes from the same batch to give a direct within-batch comparison between unclamped and voltage-clamped DA uptake. After 3 min of DA exposure, the electrodes were withdrawn, and oocytes were quickly removed from the bath and washed three times in 5 ml of ice-cold FRB. The oocytes were homogenized by sonication in 2 mM perchloric acid, and the DA content of individual oocytes was quantified by HPLC with electrochemical detection. The nominal detection limit for DA was 0.5 pg/injection. DA was not detected in water-injected oocytes or DAT/D2R expressing oocytes that were not exposed to DA (data not shown).
[3H](−)-Sulpiride Binding Assays
Whole-cell D2R binding was performed in 0.5 ml of FRB buffer containing 5 nM [3H](−)-sulpiride for 15 min at room temperature. Binding was terminated by washing the oocytes three times in 5 ml of ice-cold FRB. Radioactivity was quantified by liquid scintillation spectroscopy. Nonspecific binding was defined in the presence of 1 μM (−)-sulpiride or in water-injected oocytes.
[3H]WIN 35,428 Binding Assays.
Indirect saturation radioligand binding to intact oocytes was performed in 0.5 ml of FRB containing 4 nM [3H]WIN 35,428 and unlabeled WIN 35,428 (final concentrations of 5 nM to 1 μM) at room temperature for 20 min. Binding was terminated by washing three times in 5 ml of ice-cold FRB. Radioactivity was quantified by liquid scintillation spectroscopy. Nonspecific binding was defined in the presence of 1 μM GBR 12909 or in water-injected oocytes.B max and K ivalues were estimated by nonlinear regression analysis. To determine the time course for the D2R-mediated up-regulation of DAT, specific binding of 4 nM [3H]WIN 35,428 was carried out for 15 min at 0°C after first preincubating oocytes in DA (100 nM) or apomorphine (1 μM) for 0, 5, 30, or 60 min at room temperature.
PTX Treatment.
Oocytes were incubated in 0.5 μg/ml PTX in FRB for 24 h before assay (Nelson et al., 1997).
Two-Electrode Voltage-Clamp Recording.
Electrophysiological recordings from oocytes were performed at room temperature using glass microelectrodes filled with 3 M KCl solution and an active bath probe. Oocytes were superfused with FRB buffer at approximately 4 ml/min (bath volume, 0.5 ml). A Warner OC-725B amplifier (Warner Instruments, Hamden, CT) was used with a DigiData 1200 interface. pClamp software (Axon Instruments, Foster City, CA) was used to control command voltage parameters, data acquisition, and data analysis. MacLab software and a MacLab/2e interface (AD Instruments, Milford, MA) were also used to simultaneously monitor and record electrophysiological experiments. Analog signals were low-pass-filtered at 100 Hz and digitized at 2048 Hz.
Electrophysiological recording from DAT-expressing oocytes has been described in detail previously (Sonders et al., 1997). Briefly, oocytes were superfused with FRB buffer, and membrane potential was clamped at −60 mV. DAT-mediated currents were examined using a voltage jump protocol in which steady-state currents were measured at each of 17 command voltages (10-mV increments from −120 to +40 mV). Membrane potential was held at each voltage for 400 ms. Drug-induced DAT-mediated currents were determined off-line by subtracting control currents from those induced by various drug conditions (I drug −I control, by convention). The resulting subtractive currents were then plotted as a function of membrane potential, thus generating current-voltage (I-V) curves for each drug response.
Results
Effects of D2R Activation on DAT-Mediated DA Uptake.
In initial experiments, the effect of D2R activation on 100 nM [3H]DA uptake was examined in oocytes coexpressing DATs and D2Rs. Because most DAT substrates are D2R agonists, [3H]DA uptake was measured under conditions of complete D2R blockade (100 nM [3H]DA in the presence of 1 μM (−)-sulpiride; D2R-blocked) or maximal D2R activation (100 nM [3H]DA in the absence of (−)-sulpiride; D2R-activated). In these experiments, the effects of D2R activation on [3H]DA uptake depended on DAT expression level as assessed by measuring the uptake of 100 nM [3H]DA. In oocytes expressing relatively low DA uptake velocities (<2 fmol/s/oocyte), [3H]DA uptake increased significantly by 22% in response to D2R activation (Fig.1A). In contrast, in batches of oocytes expressing relatively higher velocities (>3 fmol/s/oocyte), D2R activation produced no consistent effects on [3H]DA uptake (Fig. 1B). Whole-cell D2R number, as measured by 5 nM [3H](−)-sulpiride binding, ranged from 1.8 to 4.6 fmol/oocyte. However, no correlation was observed between D2R expression level and D2R regulation of DAT activity.

Effects of D2R activation on [3H]DA accumulation into oocytes coexpressing DAT and D2R proteins. [3H]DA accumulation into individual oocytes was determined under conditions of full D2R blockade (presence of 1 μM (−)-sulpiride; D2R Blocked) or maximal D2R activation by 100 nM [3H]DA (D2R Activated). A, oocytes expressing low DA uptake velocities (<2.0 fmol/s/oocyte;N = 12–21 oocytes/group from three oocyte batches; *p < 0.05, single factor ANOVA followed by Newman-Keul's test). B, oocytes expressing relatively higher DA uptake velocities (>3.0 fmol/s/oocyte; N = 16–28 oocytes/group from four oocyte batches).
Experiments were conducted to determine whether the D2R-dependent increase in DAT activity was associated with D2R-mediated changes in membrane potential. DAT and D2R expression levels, again measured with 100 nM [3H]DA uptake and 5 nM [3H](−)-sulpiride binding, respectively, were similar to those reported above in Fig. 1A. In these experiments, accumulation of a DAT-saturating concentration of unlabeled DA (10 μM) was also measured in individual oocytes under D2R-blocked versus D2R-activated conditions while simultaneously monitoring membrane potential. In addition, before DA application, apomorphine (1 μM) was applied so that membrane potential could be monitored under D2R-activated conditions in the absence of substrate translocation. After washout of apomorphine, under D2R-blocked conditions, uptake of 10 μM DA was approximately 44 fmol/s/oocyte. D2R activation increased DA uptake by approximately 50% (Fig.2A). However, the D2R-mediated increase in uptake was not associated with changes in membrane potential (D2R-blocked = −31 ± 2 mV; D2R-activated = −33 ± 2 mV). The D2R-mediated increase in DA uptake was abolished in oocytes pretreated with PTX (bath applied; 0.5 μg/ml; data not shown), suggesting that activation of Gi/oproteins is involved. Voltage-clamped DA uptake experiments were also conducted to address the voltage dependence of the D2R-mediated increase in DA uptake. In oocytes with membrane potential clamped at −60 mV, uptake of 10 μM DA was approximately 45 fmol/s/oocyte (Fig. 2B). D2R activation increased DAT-mediated DA uptake by approximately 70%. Thus, because membrane potential was clamped at −60 mV, the D2R-mediated increase in DA uptake did not require changes in membrane potential.

D2R-mediated increases in DA uptake were independent of changes in membrane potential. Accumulation of unlabeled DA (10 μM) was measured in oocytes coexpressing DAT and D2R under D2R-blocked (presence of 1 μM (−)-sulpiride) versus D2R-activated (10 μM DA) conditions. A, D2R activation increased DA uptake without changing resting membrane potential (RMP) in unclamped oocytes. RMP values were obtained 1 min after the start of DA application (N = 24–30 oocytes/group from three oocyte batches; *p < 0.05, single factor ANOVA followed by Newman-Keul's test). B, D2R activation increased DA uptake in oocytes with membrane potential voltage-clamped at −60 mV (N = 32–36 oocytes/group from three oocyte batches; *p < 0.05, single factor ANOVA followed by Newman-Keul's test).
Effects of D2R Activation on DAT-Mediated Currents.
Two-electrode, voltage-clamp experiments were performed with oocytes coexpressing DAT and D2R proteins to determine whether D2R activation alters DAT-mediated ionic conductances. DAT expression levels in the oocytes used in these experiments were similar to those in Fig. 1A. In DAT-expressing oocytes voltage-clamped at −60 mV, DA (10 μM) induced low nA inward currents that returned to baseline after substrate washout (Fig. 3A). Neither DA (10 μM) nor (−)-sulpiride (1 μM) had effects on water-injected controls (Fig. 3B). In oocytes coexpressing D2R and DAT proteins under D2R-blocked conditions, 10 μM DA induced inward currents that were similar to those observed in oocytes expressing only DAT (Fig. 3, A and C). Sulpiride alone had no effect on baseline currents (Fig. 3C). After a 10-min washout period, DA-induced D2R activation enhanced DAT-mediated inward currents significantly compared with D2R-blocked conditions (Fig. 3D).
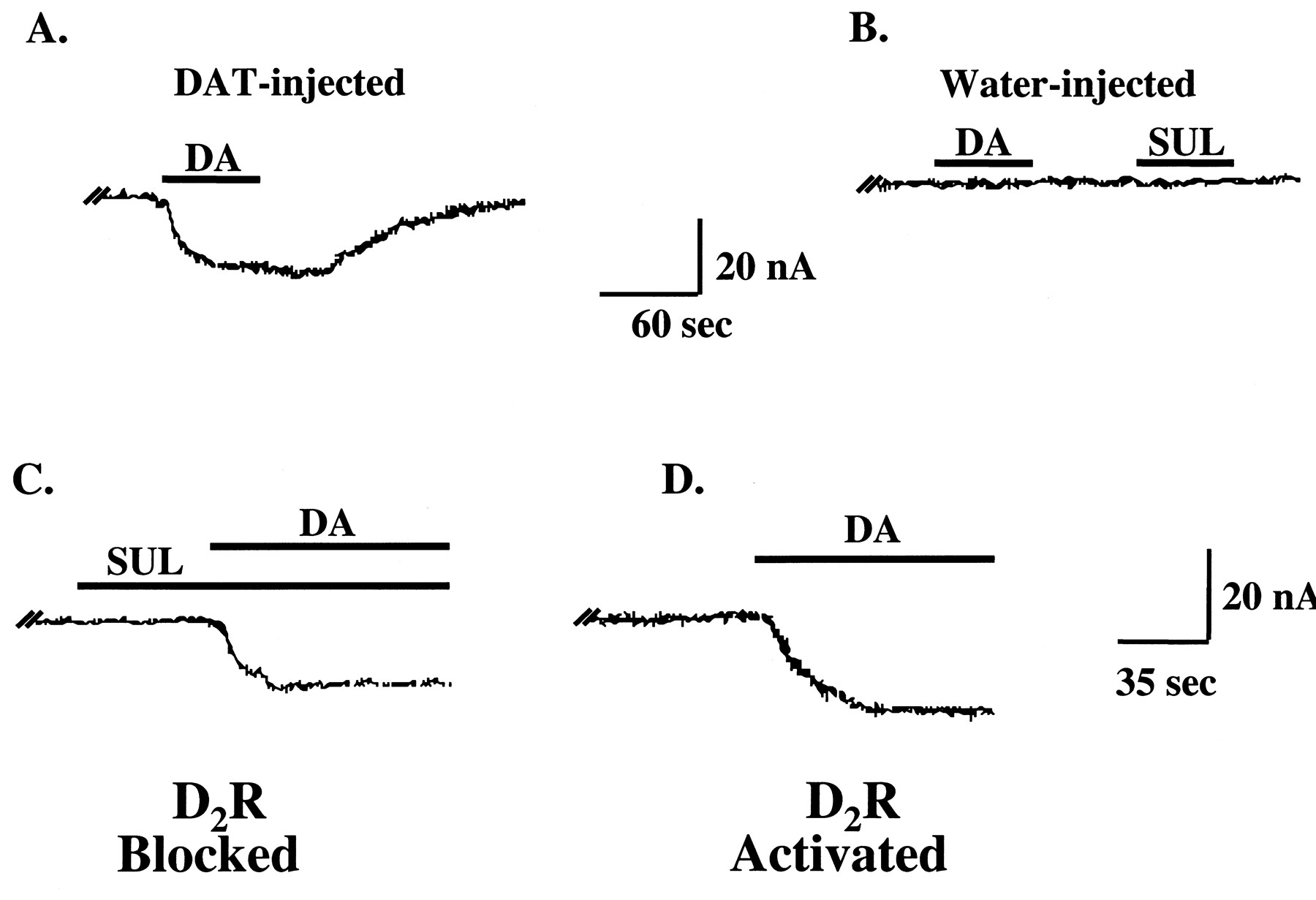
D2R regulation of DAT-mediated currents in voltage-clamped (−60 mV) oocytes coexpressing D2R and DAT proteins. A, representative trace from an oocyte injected with DAT cRNA demonstrating a DAT-mediated inward current produced by 10 μM DA. B, neither DA (10 μM) nor (−)-sulpiride (1 μM) had effects in water-injected control oocytes. C, representative trace from an oocyte coexpressing DAT and D2R proteins showing a DAT-mediated inward current produced by 10 μM DA in the presence of 1 μM (−)-sulpiride (D2R-blocked condition). Note that exposure to (−)-sulpiride alone produced no change in current. D, trace from the same oocyte shown in C after a 10-min wash with FRB. D2R activation by 10 μM DA increased the DAT-mediated inward current (D2R-activated condition).
Using a voltage-jump protocol, steady-state currents were recorded over a range of holding potentials. Consistent with previous findings (Sonders et al., 1997), at least two distinct currents were mediated by DAT (see Fig. 4A). The first was a “transport-associated” inward current, which was induced by DAT substrates and was predominant at hyperpolarized holding potentials (−20 to −120 mV). The second was a constitutively active inward leak current, which was blocked by DAT substrates and apparent at more depolarized potentials (−10 to +40 mV). Similar to DA uptake assay conditions, DAT-mediated currents were compared under D2R-blocked versus D2R-activated conditions (Fig. 4A). Subtractive currents (I drug −I control) were plotted as a function of holding potential, and the current-voltage (I-V) relationships were compared. D2R activation by 10 μM DA enhanced inward-directed, transport-associated currents across a wide range of hyperpolarized potentials (−20 to −120 mV). Similar results were obtained when tyramine (10 μM) was used to induce DAT-mediated currents either in the absence (1 μM (−)-sulpiride) or the presence of D2R activation (100 nM apomorphine; data not shown). Figure 4B shows the mean data for the effect of D2R activation on DAT-mediated currents at holding potentials of −90 or +20 mV. On average, at a holding potential of −90 mV, transport-associated currents were increased by 70% by D2R activation (Fig. 4B). The block of the leak current by DAT substrates was apparent at more depolarized potentials (−20 to +40 mV). Although D2R activation had modest effects on the leak current at depolarized potentials as high as 20 mV, the robust increases in DAT-mediated currents at hyperpolarized potentials suggests that the primary effect of D2R activation is on transport-associated ionic conductances. When the effect of D2R activation on DAT-mediated currents was expressed as the percentage change from control (D2R-blocked) and plotted function of holding potential, the D2R-induced increase in DAT-mediated currents was relatively constant across a wide range of hyperpolarized potentials (−20 to −120 mV; Fig. 4C). At the most depolarized potentials, where the leak current predominates, currents were reduced slightly by D2R activation (Fig. 4C).

Effects of D2R activation on DAT-mediated currents in oocytes coexpressing D2R and DAT proteins. A, representative data from a single oocyte demonstrating enhanced substrate-associated currents by D2R activation. The I-V plot shows steady-state subtractive currents as a function of membrane potential in response to instantaneous jumps in voltage (−120 to +40 mV; 10-mV increments) from a holding potential of −60 mV. DA-induced DAT-mediated currents were obtained under D2R-blocked (1 μM (−)-sulpiride) versus D2R-activated (10 μM DA) conditions. Boxes indicate potentials for which mean data are shown in panel B. B, DAT-mediated currents were increased significantly by D2R activation at hyperpolarized holding potentials (−90 mV shown; represents transport-associated current) but not at depolarized potentials (+20 mV shown; represents the block of tonic leak current by DAT substrates). Mean values ± S.E.M. are shown for N = 14 oocytes from four batches (*p < 0.05 single factor ANOVA). C, effect of D2R activation on DAT-mediated currents expressed as the percentage change from control and plotted as a function of holding potential. The effect of D2R activation on the substrate-associated current, observed at negative holding potentials, was voltage-independent.
D2R Activation Increased Cell Surface Binding of [3H]WIN 35,428.
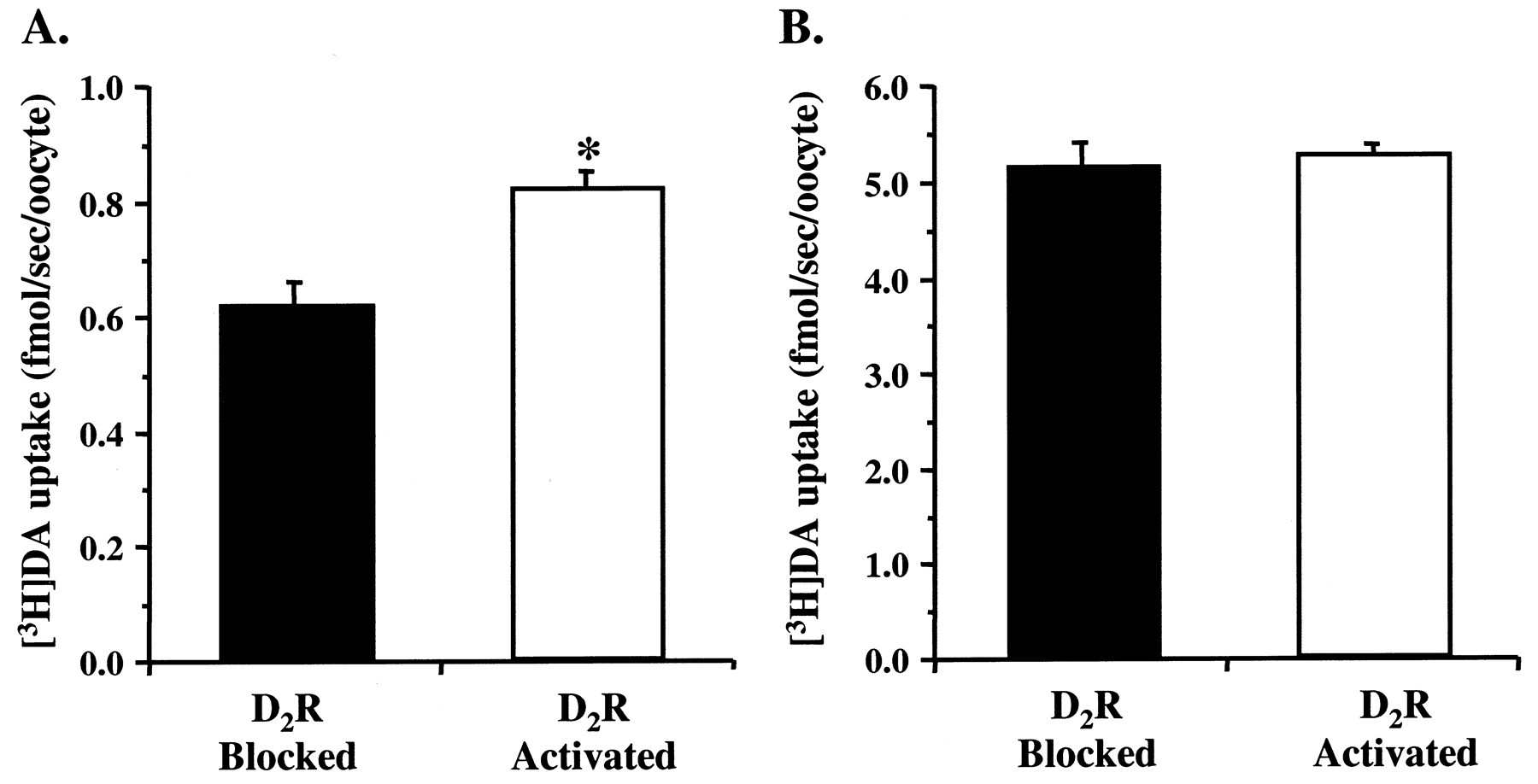
A potential mechanism for the D2R-mediated increases in DAT function might involve an increase in the number of DAT molecules trafficked to the cell surface. To address this question, the effects of D2R activation on whole-cell binding of the cocaine analog [3H]WIN 35,428 was determined. Indirect saturation curves using an approximateK D concentration of [3H]WIN 35,428 (4 nM) were generated in the absence (1 μM apomorphine in the presence of 1 μM sulpiride) or presence (1 μM apomorphine) of D2R activation (Fig. 5A). D2R activation resulted in a 60% increase inB max values (D2R-blocked = 26 ± 3.2 fmol/oocyte; D2R-activated = 42 ± 2.6;N = 3–5 oocytes/group from three batches). The increase in [3H]WIN 35,428 binding was not associated with a concomitant change in K ivalues (D2R-blocked = 26 ± 10.3 nM; D2R-activated = 18 ± 0.9) for WIN 35,428. The effect of PTX pretreatment was also tested on [3H]WIN 35,428 binding under D2R-blocked versus D2R-activated conditions (Fig. 5B). Activation of D2Rs by 1 μM apomorphine resulted in a 59% increase in specific binding of [3H]WIN 35,428. Similar to the results for DA uptake, the D2R-mediated increase in [3H]WIN 35,428 binding was abolished by 24 h pretreatment with PTX (Fig. 5B). The time course of the up-regulation of specific 4 nM [3H]WIN 35,428 binding was determined after activating D2Rs with DA (100 nM) or apomorphine (1 μM) for various times (5, 30, or 60 min). [3H]WIN 35,428 binding increased maximally by 40% after 5 min of D2R activation and remained relatively constant for up to 60 min (Fig. 5C). Furthermore, activation of D2Rs by either DA or apomorphine increased [3H]WIN 35,428 binding to the same extent.

Effects of D2R activation on whole-cell binding of [3H]WIN 35,428 to DAT in oocytes coexpressing D2R and DAT proteins. A, D2R activation increased the number of cell surface DAT binding sites. Indirect saturation curves were generated with 4 nM [3H]WIN 35,428 at room temperature under D2R-blocked (1 μM apomorphine in the presence of 1 μM (−)-sulpiride) versus D2R-activated conditions (1 μM apomorphine). Curve fitting revealed that D2R activation increasedB max without alteringK i (see Results). Mean values ± S.E.M. are shown for N = 3–5 oocytes/group from three batches. B, the D2 R-mediated increase in specific binding of 4 nM [3H]WIN 35,428 was abolished by PTX pretreatment. GBR 12909 (1 μM) or uninjected oocytes were used to define nonspecific binding. Mean values ± S.E.M. are shown forN = 10–16 oocytes from two batches (**p < 0.01 single factor ANOVA). PTX was bath applied (0.5 μg/ml) for 24 h. Mean values ± S.E.M. are shown for N = 6–8 oocytes from a single batch. C, the D2R-mediated increase in [3H]WIN 35,428 binding was maximal after 5 min of receptor activation. D2Rs were activated by either DA (100 nM) or apomorphine (1 μM) for 5, 30, and 60 min at room temperature. After each period of activation, specific [3H]WIN 35,428 binding (4 nM) was determined at 0°C. The effects of DA and apomorphine were identical, and the data were combined for illustration. Mean values ± S.E.M. are shown for N = 5 to 6 oocytes/group from four batches.
Discussion
There is growing evidence that D2Rs regulate brain DAT activity. However, the complexities of intact neuronal preparations make investigations into the molecular mechanisms involved difficult. In the present study, we used the X. laevisoocyte expression system to coexpress the hD2R (long isoform) and the hDAT to address the hypothesis that D2Rs regulate DAT activity by altering membrane potential. D2R activation enhanced DAT-mediated substrate translocation, as well as ionic conductances associated with substrate translocation. However, the D2R-mediated increases in DAT activity were independent of changes in membrane potential. D2R activation also increased the number of whole-cell binding sites detected with [3H]WIN 35,428, suggesting that D2R activation enhances DAT activity by increasing the cell surface expression of DAT. These D2R-mediated effects likely depend on Gi/o signaling, because pretreatment with PTX abolished both the enhanced function and cell surface binding of DAT.
It has been established previously that, when expressed individually in oocytes, DAT or D2R proteins exhibit their expected activities and pharmacological profiles. For example, the translocation kinetics of the hDAT expressed in oocytes has been characterized in detail (Sonders et al., 1997) and found to be consistent with those observed in mammalian cell lines expressing hDAT (Giros et al., 1992; Eshleman et al., 1995). We observed similar results in oocytes coexpressing hDAT and hD2R proteins. Initially, we confirmed D2R function by coexpressing hD2Rs with rat GIRK1 (Kir3.1). Agonist stimulation of D2R-activated GIRK1-mediated potassium currents in a manner consistent with previous reports (Werner et al., 1996; Nelson et al., 1997). The D2R-activated potassium currents were completely and reversibly blocked by the D2R antagonist (−)-sulpiride. Thus, in most subsequent experiments, D2R expression was confirmed using [3H](−)-sulpiride binding, rather than GIRK1 coexpression.
Because most DAT substrates are agonists at D2Rs, D2Rs were already maximally activated in the presence of DA (here either 100 nM [3H]DA or 10 μM unlabeled DA). Thus, baseline DAT activity was measured in the presence of DAT substrate and 1 μM (−)-sulpiride. In oocytes expressing lower DAT velocities (<2 fmol/s/oocyte, measured with 100 nM [3H]DA), uptake was enhanced significantly by D2R activation. These results demonstrate that hDAT activity can be regulated by hD2Rs and are consistent with reports suggesting that D2R ligands modulate rodent brain DAT function (Meiergerd et al., 1993;Parsons et al., 1993; Cass and Gerhardt, 1994; Rothblat and Schneider, 1997; Dickinson et al., 1999; Hoffman et al., 1999). However, it is unclear why a D2R-mediated increase in DA uptake did not occur when DAT velocities were relatively higher (>3 fmol/s/oocyte, measured with 100 nM [3H]DA). It is unlikely that substrate depletion reduced the effect of D2R activation on DA uptake in these experiments, because identical results were obtained using a saturating concentration of DA (10 μM; data not shown). Regulation of the γ-aminobutyric acid (GABA) transporter GAT1 by phorbol esters has been shown to depend on the level of protein expression (Quick et al., 1997). Thus, it is possible that, in oocytes expressing a high number of DAT molecules, the mechanism(s) and/or signaling cascades linking D2Rs and DATs may be limiting, thereby saturating DAT or D2R protein-protein associations and leading to diminished regulatory potential. However, the magnitude of D2R-activated [3H]DA uptake was not correlated with the level of D2R expression as assessed by [3H](−)-sulpiride binding.
DA uptake into DAT-expressing oocytes has been shown to be voltage-dependent (Sonders et al., 1997). Consistent with this conclusion, Hoffman et al. (1999) found that activation of D2Rs or GABAB receptors in rat substantia nigra hyperpolarizes DA neurons and increases DAT-mediated DA clearance. Thus, a potential mechanism underlying the D2R-mediated increase in DA uptake would be a D2R-dependent change in the resting membrane potential. In brain, activation of D2Rs hyperpolarizes membrane potential through activation of an inwardly rectifying potassium conductance (Lacey, 1993). The extent to whichX. laevis oocytes express endogenous potassium channels is not precisely known. However, we did not observe D2R-mediated changes in membrane potential (see below), suggesting that the effects of D2R activation on DAT function were not mediated by coupling of D2Rs to endogenous potassium channels. Furthermore, in a limited number of experiments, we observed no differences between D2R-mediated increases in [3H]DA uptake in oocytes coexpressing D2Rs, DATs, and GIRK1 channels or coexpressing only D2Rs and DATs (R. D. Mayfield and N. R. Zahniser, unpublished observations).
Several lines of evidence suggest that the D2R-mediated increases in DAT activity were independent of changes in membrane potential. The effects of D2R activation on membrane potential were measured directly while concurrently measuring DA uptake. Membrane potential was not changed in response to D2R activation; however, DA uptake was increased by as much as 50%. Also, in voltage-clamped uptake experiments, D2R activation increased DAT-mediated DA uptake significantly. If D2R-mediated increases in DA uptake depended on changes in membrane potential, D2R activation would not have regulated DAT activity, because membrane potential was held constant. Together, these findings and those discussed below argue strongly against D2R regulation of membrane potential as a mechanism underlying the enhanced DAT activity observed in these experiments.
The results from experiments measuring DAT-associated currents also argue that D2R activation of DAT is voltage-independent. DAT substrates induce transport-associated currents, which are Na+- and voltage-dependent (Sonders et al., 1997; Sitte et al., 1998). In voltage-clamped oocytes coexpressing D2R and DAT, we found that activation of D2Rs by DA significantly enhanced transport-associated inward currents. Similar to our findings with DA uptake, enhanced substrate-associated currents were only observed in oocytes expressing lower DAT velocities. The D2R-mediated increase in DAT currents were observed at holding potentials between −20 and −120 mV, and the current magnitude increased as a function of increasing hyperpolarization. Although steady-state currents at hyperpolarized potentials were voltage-dependent under both D2R-blocked and -activated conditions, the percentage increase in steady-state currents resulting from D2R activation was relatively constant over this range of holding potentials, equaling approximately 50%. This finding suggests that the increased transport-associated current induced by D2R activation is voltage-independent.
In contrast with the lack of effect of membrane potential on D2R-mediated enhancement of DAT function, pretreatment with PT abolished the effects of D2R activation on both DAT function and binding (see below). D2Rs couple to PTX-sensitive Gi/o proteins to inhibit adenylyl cyclase, voltage-gated calcium channels, and protein kinase C (PKC), as well as to activate GIRKs and mitogen-activated protein kinase (see Giambalvo and Wagner, 1994; Sokoloff and Schwartz, 1995; Watts et al., 1998). The long isoform of the D2R, which we investigated here, can selectively activate Go (Watts et al., 1998). However, it remains to be determined whether the D2R regulation of DAT involves Gi and/or Go proteins. Furthermore, D2R actions can be mediated by both Gα and Gβγ subunits (Watts et al., 1998; Choi et al., 1999). In primate brain the short isoform of the D2R predominates in DA neurons (Khan et al., 1998). Signal transduction mechanisms can differ between the long and short isoforms (Sokoloff and Schwartz, 1995; Choi et al., 1999). Nonetheless, our findings of enhanced DAT function, mediated by the long isoform of the D2R in the oocyte expression system, agrees with the observations of D2R-mediated up-regulation of DA clearance in rodent brain.
Recent results suggest that DAT trafficking plays a prominent role in the regulation of transporter activity. For example, activation of PKC by phorbol esters decreases DAT function by reducing the number of functional transporters on the cell surface (Zhang et al., 1997; Zhu et al., 1997; see Pristupa et al., 1998). Furthermore, the phorbol ester-induced loss of cell surface DAT is associated with increased endocytotic trafficking of the transporter (Daniels and Amara, 1999;Melikian and Buckley, 1999). Here, we used whole-cell [3H]WIN 35,428 binding to estimate the number of cell surface DATs. There are no hydrophilic radiolabeled DAT ligands available for detection of DAT exclusively at the cell surface. However, [3H]WIN 35,428 binding to DAT is negligible at low Na+ concentrations (Reith and Coffey, 1993), such as are present intracellularly in oocytes [6 mM (Barish, 1983)]. We found that D2R activation increased the number of cell surface DATs by 60%, as measured by [3H]WIN 35,428 binding. This D2R-mediated increase in [3H]WIN 35,428 binding was maximal after 5 min of receptor activation, suggesting that redistribution of DAT to the cell surface occurs rapidly. The rapid redistribution of DAT is also evident from electrophysiological studies showing enhanced DAT-mediated currents within 1 min of D2R activation. These results are consistent with a hypothesis that D2Rs regulate cell surface expression of DAT, thereby providing a rapid, local feedback mechanism for transporter regulation. Whether the net increase in cell surface transporters reflects reduced DAT endocytosis or increased trafficking to the cell surface remains to be determined.
In conclusion, the increase in the number of [3H]WIN 35,428 binding sites, together with the enhanced DAT uptake and currents, suggest strongly that the D2R-induced up-regulation of DAT function results from an increase in the number of functional DAT molecules expressed at the cell surface. D2R-mediated changes in membrane potential do not seem to be involved in this regulation of DAT function, whereas D2R-mediated changes in Gi/o signaling do. It is possible that common mechanisms are involved in receptor-mediated regulation of neurotransmitter transporters. Receptors that couple to PKC via G proteins alter the activity of the norepinephrine and GABA transporters by inducing a rapid redistribution of these transporters (Apparsundaram et al., 1998; Beckman et al., 1999). Whatever the molecular mechanism for D2R/DAT interactions, these findings support the idea that presynaptic D2Rs can alter not only DA neuronal firing, synthesis, and release, but also DA uptake.
Footnotes
- Received April 21, 2000.
- Accepted September 21, 2000.
-
Send reprint requests to: Dr. R. Dayne Mayfield, Institute for Cellular and Molecular Biology, College of Natural Sciences, University of Texas, 2500 Speedway, MBB 1.124, Austin, TX 78712. E-mail: dayne.mayfield{at}mail.utexas.edu
-
This work was supported by National Institutes of Health Grants DA04216 and DA00174.
Abbreviations
- DAT
- dopamine transporter
- D2R
- dopamine D2 receptor
- DA
- dopamine (3,4,7-dihydroxyphenylethylamine)
- FRB
- frog Ringers' buffer
- GIRK
- G protein-coupled inwardly rectifying potassium channel
- h
- human
- PKC
- protein kinase C
- PTX
- pertussis toxin
- RMP
- resting membrane potential
- WIN 35,428
- 2β-carbomethoxy-3β-(4-fluorophenyl)tropane
- GABA
- γ-aminobutyric acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}