


Abstract
Transcriptional silencing of tumor suppressor genes by DNA methylation occurs in cancer cell lines and in human tumors. This has led to the pursuit of DNA methyltransferase inhibition as a drug target. 5-Aza-2′-deoxycytidine [5-aza-CdR (decitabine)], a potent inhibitor of DNA methyltransferase, is a drug currently in clinical trials for the treatment of solid tumors and leukemia. The efficacy of 5-aza-CdR may be related to the induction of methylation-silenced tumor suppressor genes, genomic hypomethylation, and/or enzyme-DNA adduct formation. Here, we test the hypothesis that 5-aza-CdR treatment is perceived as DNA damage, as assessed by the activation of the tumor suppressor p53. We show that 1) colon tumor cell lines expressing wild-type p53 are more sensitive to 5-aza-CdR mediated growth arrest and cytotoxicity; 2) the response to 5-aza-CdR treatment includes the induction and activation of wild-type but not mutant p53 protein; and 3) the induction of the downstream p53 target gene p21 is partially p53-dependent. The induction of p53 protein after 5-aza-CdR treatment did not correlate with an increase in p53 transcripts, indicating that hypomethylation at the p53 promoter does not account for the p53 response. It is relevant that 5-aza-CdR has shown the greatest promise in clinical trials for the treatment of chronic myelogenous leukemia, a malignancy in which functional p53 is often retained. Our data raise the hypothesis that p53 activation may contribute to the clinical efficacy and/or toxicity of 5-aza-CdR.
Methylation of cytosines within CpG dinucleotides is associated with transcriptional silencing during mammalian development and tumorigenesis (Bird, 1996). The identification of methylation silencing as an alternative mechanism for tumor suppressor inactivation has raised interest in DNA methylation as a target for therapeutic intervention (Bender et al., 1998). The importance of methylation in tumor development is supported by studies demonstrating that pharmacological and/or genetic targeting of DNA methyltransferase I restricts tumor growth in mouse and human systems (Laird et al., 1995;Ramchandani et al., 1997). One DNA methyltransferase inhibitor undergoing clinical evaluation is 5-aza-2′-deoxycytidine (5-aza-CdR; clinical name, decitabine), a cytidine analog that sequesters DNA methyltransferase after its incorporation into genomic DNA (Momparler, 1985). Clinical trials evaluating 5-aza-CdR as a cancer chemotherapeutic have shown promise for the treatment of leukemia but less utility against solid tumors (Kantarjian et al., 1997; Momparler et al., 1997; Schwartsmann et al., 1997; Thibault et al., 1998; Sacchi et al., 1999; Wijermans et al., 2000).
The basis of the clinical efficacy and toxicity of 5-aza-CdR is unclear, but may involve gene reactivation. In vitro studies have demonstrated that methylation-regulated genes are released from silencing after treatment of tumor cells with 5-aza-CdR. These genes include the tumor suppressors p16, E-cadherin, and hMLH1 (Gonzalez-Zulueta et al., 1995; Yoshiura et al., 1995; Herman et al., 1998). The re-expression of these genes after 5-aza-CdR treatment correlated with cell growth inhibition, re-establishment of cell-cell adhesion, and renewed ability for mismatch repair, respectively (Yoshiura et al., 1995; Bender et al., 1998; Herman et al., 1998). A second important mediator of the biological activity of 5-aza-CdR is the formation of enzyme-DNA adducts (Ferguson et al., 1997). Supporting this model is the observation that mouse embryonic stem cell sensitivity to 5-aza-CdR is directly proportional to DNA methyltransferase I expression level (Juttermann et al., 1994). The formation of enzyme-DNA adducts also accounts for the toxicity of 5-aza-CdR in some breast cancer cell lines (Ferguson et al., 1997). The relative contribution of gene reactivation and enzyme-DNA adduct formation to the efficacy and toxicity of 5-aza-CdR in vivo is an important unresolved question.
Based on reports that cellular sensitivity to 5-aza-CdR correlates with the level of formation of covalent enzyme-DNA adducts (Juttermann et al., 1994; Ferguson et al., 1997), we hypothesized that cellular toxicity caused by 5-aza-CdR treatment could result in part from the induction of the tumor suppressor protein p53 as a cellular response to DNA damage. Here we show that 5-aza-CdR treatment in tissue-cultured colon tumor cell lines leads to the induction and activation of wild-type p53 and that this correlates with an increased sensitivity to 5-aza-CdR-mediated toxicity. Therefore, our data raise the possibility that p53 status may affect clinical responses to 5-aza-CdR treatment.
Materials and Methods
Cell Culture and Drug Treatments.
HCT116 and HT29 colon adenocarcinoma cell lines were cultured in McCoy's media supplemented with 10% fetal calf serum. RKO and SW480 cell lines were cultured in Dulbecco's minimal essential medium supplemented with 10% fetal calf serum. HCT116 cells with a targeted deletion of p53 (Bunz et al., 1998) or p21 (Waldman et al., 1995) were kindly provided by Bert Vogelstein (Johns Hopkins University, Baltimore, MD.). Cytidine compounds were solubilized in PBS. Cells were treated with 5-aza-CdR or 6-aza-CdR (Sigma, St. Louis, MO) 24 h after passage. Cells were subcultured at equivalent cell densities 2 days after treatment. For all concentration curve experiments, assays were conducted and/or extracts were harvested 5 days after treatment. Time course experiments used 1 μM 5-aza-CdR.
Cell Viability Assays.
All cell viability assays were performed in quadruplicate wells of 24-well plates 5 days after drug treatment. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium (MTT) (Calbiochem, San Diego, CA) reduction by mitochondria was used to assess drug-induced cell growth inhibition and cytotoxicity (Green et al., 1984). Cells were incubated with 250 μg/ml MTT in culture media for 45 min at 37°C. After incubation, media was removed and cellular material was solubilized with 0.1 M HCl in isopropanol. MTT absorbance was read at 570 nM with a KC4 microplate reader (Bio-Tek Instruments, Winooski, VT).
RNA Extraction and Northern Blotting.
RNA was extracted using TRIZOL reagent (Life Technologies, Rockville, MD). Total cellular RNA (5 μg) was fractionated through formaldehyde containing 1.2% agarose gels and transferred onto Hybond-N nylon membranes (Amersham Pharmacia Biotech, Uppsala, Sweden). [α-32P]CTP labeled probes were generated by the random-primed method using the RTS Radprime DNA labeling system (Life Technologies). Hybridizations with labeled p53, p21, and GAPDH probes were carried out using Rapid-hyb buffer (Amersham Pharmacia Biotech) according to the manufacturer's instructions. Bands were detected by PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and BioMax film (Eastman Kodak, Rochester, NY).
RT-PCR.
RT-PCR of p53 and GAPDH transcripts was carried out on cDNAs prepared from 5 μg of total RNA using Superscript II reverse transcriptase (Life Technologies) according to the manufacturer's instructions. PCR amplifications were performed with Taqpolymerase (Life Technologies) according to the manufacturer's instructions. Amplification primers were purchased from Stratagene (La Jolla, CA). PCR products were separated on 1.2% agarose gels, stained with ethidium bromide and photographed.
Protein Extraction and Western Blotting.
Whole cell extracts were generated by lysis in a buffer containing 1% Triton X-100, 25 mM Tris, pH 7.4, 150 mM NaCl, 1 mM CaCl2, 1 mM phenylmethanesulfonyl fluoride, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Nuclear and Cytosolic extracts were harvested as described previously (Wang et al., 1995). All protein extracts were quantified using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA). Equivalent concentrations of each protein sample were fractionated through 4 to 20% Tris-glycine gradient gels (Novex, San Diego, CA) and then transferred to polyvinylidene difluoride membranes (Amersham Pharmacia Biotech). The DO-1 monoclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used for detection of p53. The H-164 rabbit polyclonal antibody (Santa Cruz Biotechnology) was used for detection of p21. Proteins of interest were detected using horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibodies (Life Technologies) and chemiluminescence using the Renaissance reagent kit (PerkinElmer Life Science Products, Boston, MA).
Cell Cycle Analysis.
Cells were prepared for cell cycle analysis by first resuspending cells in ice-cold PBS, and then fixing by drop-wise addition of ice-cold 100% methanol. Next, cells were stained in a solution containing 50 μg/ml propidium iodide (Sigma, St. Louis, MO) and 100 U/ml RNase A in PBS. Linear fluorescence signals (area and width) were assessed on a Becton Dickinson FACScan flow cytometer with dye excitation by 15-mw, 488-nm laser light. Data were stored as list mode files of at least 20,000 single cell events for subsequent off-line analysis using Modfit and WinList software (Verity Software, Topsham, ME). DNA cell cycle analysis was accomplished using the DIP_N2 and DIP_N3 algorithms in Modfit.
Results
Colon Cancer Cell Lines Expressing Wild-Type p53 Are More Sensitive to 5-Aza-CdR-Induced Toxicity Than Are Mutant p53-Expressing Cell Lines.
If cells perceive enzyme-DNA adduct formation and/or genomic DNA hypomethylation as DNA damage, then 5-aza-CdR treatment may result in the activation of p53. It follows that if p53 is activated upon 5-aza-CdR treatment, then tumor cell lines differing in p53 status might show corresponding differences in sensitivity to 5-aza-CdR-induced toxicity. To test this hypothesis, we treated two colon cancer cell lines that express wild-type p53 (HCT116 and RKO) and two lines expressing mutant p53 (SW480 and HT29) with 5-aza-CdR and measured cell responses 5 days after treatment (Fig.1A). We observed that cell viability after 5-aza-CdR treatment is most severely effected at this time point (data not shown), presumably because toxicity requires both the incorporation of 5-aza-CdR into genomic DNA and time for the alteration of DNA methylation patterns and/or the accumulation of enzyme-DNA adducts. In addition, the kinetics of the induction of methylation-silenced genes and cell growth inhibition after 5-aza-CdR treatment is consistent with this time frame of treatment (Bender et al., 1998; Karpf et al., 1999). To measure the cellular response to 5-aza-CdR, we chose to analyze mitochondria function by MTT absorbance (Green et al., 1984). The MTT assay was chosen because it measures both cytostatic and cytotoxic responses to drug treatment and thus provides an accurate overall measure of drug sensitivity. We found that HCT116 and RKO cells are more sensitive to 5-aza-CdR-induced toxicity than are the mutant p53-containing cell lines (average p53 line IC50 ≅ 0.28 μM versus average p53 mutant line IC50 ≅ 1.0 μM). In evaluating the data from Fig. 1A, it is worth noting that the HT29 and SW480 cell lines are mismatch repair-proficient whereas the HCT116 and RKO cells are mismatch repair-deficient. Because mismatch repair proficiency has been shown to sensitize cancer cells to various DNA damaging chemotherapeutics, it was possible that 5-aza-CdR sensitivity might reflect this difference. However, our results imply that p53 status plays a more crucial role in 5-aza-CdR sensitivity because the two mismatch repair-proficient cell lines are actually less sensitive to 5-aza-CdR (Fig. 1A). We cannot exclude the possibility that other factors (for example, differential incorporation of 5-aza-CdR into genomic DNA) could account for the sensitivity difference. However, these initial results prompted us to investigate in more detail the potential role of p53 in the cellular response to 5-aza-CdR treatment.

Sensitivity of colon cancer cell lines to 5-aza-CdR-mediated toxicity correlates with p53 status. A, Two mutant p53 cell lines (SW480 and HT29) and two wild-type p53 cell lines (HCT116 and RKO) were treated with various concentrations of 5-aza-CdR. Two days after treatment, cells were subcultured at equivalent cell density into quadruplicate wells of a 24-well plate. Three days later (5 days after 5-aza-CdR treatment), the cellular response was measured by MTT assay. The data were normalized by subtraction of background staining and are expressed as the percentage of control staining of each cell line. Error bars indicate + or − 1 S.E. B, the four cell lines used in A were treated with various concentrations of 5-aza-CdR and total cellular protein was harvested 5 days after treatment. Proteins were quantified by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE. p53 was detected by Western blot analysis.
Wild-Type p53 Is Induced and Activated by 5-Aza-CdR Treatment.
We next examined whether p53 protein was induced after 5-aza-CdR treatment. DNA damage induces p53 post-translationally by reducing the rate of p53 protein degradation (Lakin and Jackson, 1999). Because cellular replication is required for the incorporation of 5-aza-CdR, alteration of genomic methylation, accumulation of enzyme-DNA adducts, and the appearance of cellular toxicity, we first analyzed 5-aza-CdR-treated cells for p53 induction after 5 days of treatment. Western blot analysis of total cellular protein revealed that 5-aza-CdR induces wild-type, but not mutant, p53 protein (Fig. 1B). Induction of p53 protein was characterized by a ∼4-fold increase in p53 protein after treatment with 1 μM 5-aza-CdR in both HCT116 and RKO cells. Because the activation of p53 in response to DNA damage involves the translocation of p53 into the nucleus (Lain et al., 1999; Liang and Clarke, 1999), we next measured the level of wild-type p53 protein in the nuclei of 5-aza-CdR-treated cells. We observed a concentration-dependent increase in total nuclear p53 protein in both HCT116 and RKO cells (Fig. 2A). In HCT116 but not RKO cells, activation of p53 was characterized by the appearance, in the nucleus, of a 50-kDa cleavage product of p53. This p53 cleavage product has been previously observed in cells responding to DNA damage, and cross reacts with the DO1 antibody used here (Okorokov et al., 1997; Okorokov and Milner, 1997).

Activation of p53 and induction of p21 after 5-aza-CdR treatment. A, HCT116 and RKO cells were treated with various concentrations of 5-aza-CdR and cytosolic and nuclear proteins were isolated 5 days after treatment. Proteins were quantified by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE. p53 was detected by Western blot analysis. B, HCT116 and RKO were treated with 1 μM 5-aza-CdR and cytosolic and nuclear protein extracts were isolated at different time points after treatment. Proteins were quantified by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE. p53 was detected by Western blot analysis. C, The four cell lines used in Fig.1 were treated with various concentrations of 5-aza-CdR and total cellular protein was isolated 5 days after treatment. Proteins were quantified by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE. p21 was detected by Western blot analysis. C, cytosol; N, nuclear; h, hours; arrow, p53 cleavage product p50.
In addition to our observation that the activation of p53 by 5-aza-CdR is concentration dependent, we also observed that nuclear p53 induction is time dependent (Fig. 2B). The time dependence of the response is manifested by the appearance of the p50 band at maximum levels 5 days after 5-aza-CdR treatment of HCT116 cells, and maximum induction of nuclear p53 4 days after treatment of RKO cells. Therefore, the kinetics of p53 induction after 5-aza-CdR treatment is consistent with the drug's dependence on cellular replication.
p21 Induction after 5-Aza-CdR Treatment Is Restricted to Cell Lines Expressing Wild-Type p53.
p53 Activation after cellular DNA damage is accompanied by the induction of the p53 target gene p21 (el-Deiry et al., 1994). To test whether 5-aza-CdR treatment results in the induction of p21, we analyzed p21 protein levels in whole-cell extracts from treated cells. We observed a concentration-dependent induction of p21 protein after 5-aza-CdR treatment in both the HCT116 and RKO cell lines, which express wild-type p53, whereas in the SW480 and HT29 cell lines, p21 levels were unchanged (Fig. 2C). The induction of p21 protein correlated with the induction of p21 mRNA (data not shown and Fig. 5B). These results suggest that p21 induction after 5-aza-CdR treatment is p53-dependent.

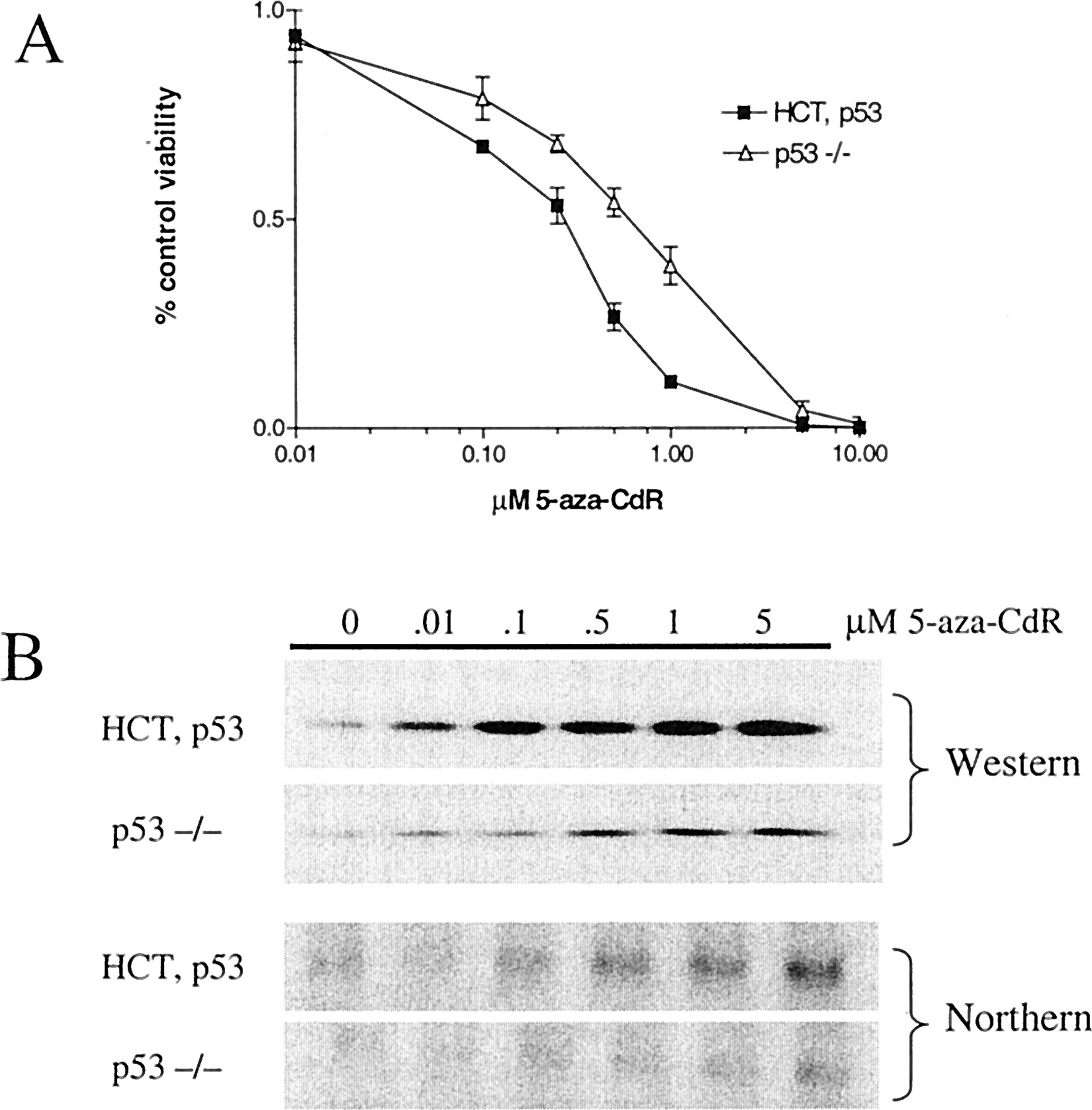
p53-null HCT116 cells are less sensitive to 5-aza-CdR-induced cell growth inhibition and toxicity, and display reduced levels of p21 induction. A, HCT116 parental cells (HCT, p53) and p53-null HCT116 cells (p53 −/−) were treated with various concentrations of 5-aza-CdR and the cellular response was assessed 5 days after treatment by MTT assay. The data were normalized by subtraction of background staining and are expressed as the percentage of control staining of each cell line. Error bars indicate ± 1 S.D. B, HCT, p53 and p53−/− cells were treated with various concentrations of 5-aza-CdR and total cellular protein and RNA was isolated 5 days after treatment. p21 was detected by Western blot (top) and Northern blot (bottom) analysis. For the Western analysis, cellular proteins were quantitated by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE.
6-Aza-CdR Treatment Does Not Result in Cellular Toxicity or p53 Activation.
We next examined whether cellular toxicity and p53 activation after 5-aza-CdR treatment are a consequence of the specific inhibition of DNA methyltransferase. If these phenomena are a result of nonspecific effects related to the incorporation of foreign nucleotide, then we would expect that treatment with similar concentrations of 6-aza-2′-deoxycytidine (6-aza-CdR) would elicit the same cellular effects. 6-aza-CdR is identical to 5-aza-CdR except that the nitrogen substitution is at the 6-position of the cytosine base. This modification renders the nucleotide insusceptible to nucleophilic attack by the cysteine thioate of DNA methyltransferase; therefore, 6-aza-CdR is unable to sequester the enzyme and target it for degradation. The enzymes required for 6-aza-CdR incorporation into genomic DNA should be identical to those required for 5-aza-CdR incorporation (Momparler, 1985). However, 6-aza-CdR may be inaccessible to methylation by DNA methyltransferase; thus, cellular treatment with this drug could result in passive genomic hypomethylation at high levels of incorporation. We treated HCT116 cells with various concentrations of 5-aza-CdR and 6-aza-CdR in parallel and compared cellular toxicity and p53 activation 5 days after treatment. Figure3A shows that 6-aza-CdR treatment is not toxic to HCT116 cells at concentrations at which 5-aza-CdR has a dramatic effect. The same correlation holds for p53 activation and p21 induction (Fig. 3, B and C). At the highest concentration of 6-aza-CdR tested, 5 μM, some cellular toxicity, p53 activation, and p21 induction was observed. This may result from effects unrelated to inhibition of DNA methyltransferase. Alternatively, high levels of 6-aza-CdR incorporation could lead to hypomethylation for the reason described above.

6-aza-CdR treatment does not lead to cellular toxicity, p53 activation, or p21 induction at equimolar concentrations as 5-aza-CdR. A, HCT116 cells were treated in parallel with various concentrations of 6-aza-CdR or 5-aza-CdR. Cell number and viability were measured 5 days after treatment by MTT assay. B, HCT116 cells were treated as in A and 5 days after treatment, cytosolic and nuclear protein extracts were isolated. Proteins were quantified by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE. p53 was detected by Western blot analysis. C, HCT116 cells were treated as above and 5 days after treatment, whole-cell protein extracts were isolated. Proteins were quantified by Bradford analysis and equivalent concentrations of proteins were fractionated by SDS-PAGE. p21 was detected by Western blot analysis. Error bars indicate + 1 S.D. Arrow indicates p53 cleavage product p50.
p53 Induction after 5-Aza-CdR Treatment Is Post-Transcriptional.
Induction of p53 after 5-aza-CdR treatment could result from either a classical p53 response to DNA damage (protein stabilization), or by release of the p53 gene from methylation silencing. Methylation-regulated expression of the p53 gene has been reported (Pogribny et al., 2000), as has in vivo methylation silencing of the p53 homolog p73 (Kawano et al., 1999). Therefore, we tested whether 5-aza-CdR treatment of HCT116 and RKO cells resulted in an altered expression of p53 mRNA. We did not observe an induction of p53 mRNA levels in HCT116 or RKO cells after treatment with various concentrations of 5-aza-CdR, as measured by Northern blot analysis (Fig. 4A). We then examined the expression level of p53 mRNA more closely by selecting two samples (0 and 1.0 μM) for analysis by RT-PCR. Using this method, we were unable to detect any increase in p53 mRNA levels after 5-aza-CdR treatment of either cell type (Fig. 4B). These data support the model that 5-aza-CdR-mediated induction of p53 protein is part of a classical response to DNA damage and is not caused by hypomethylation at the p53 promoter.
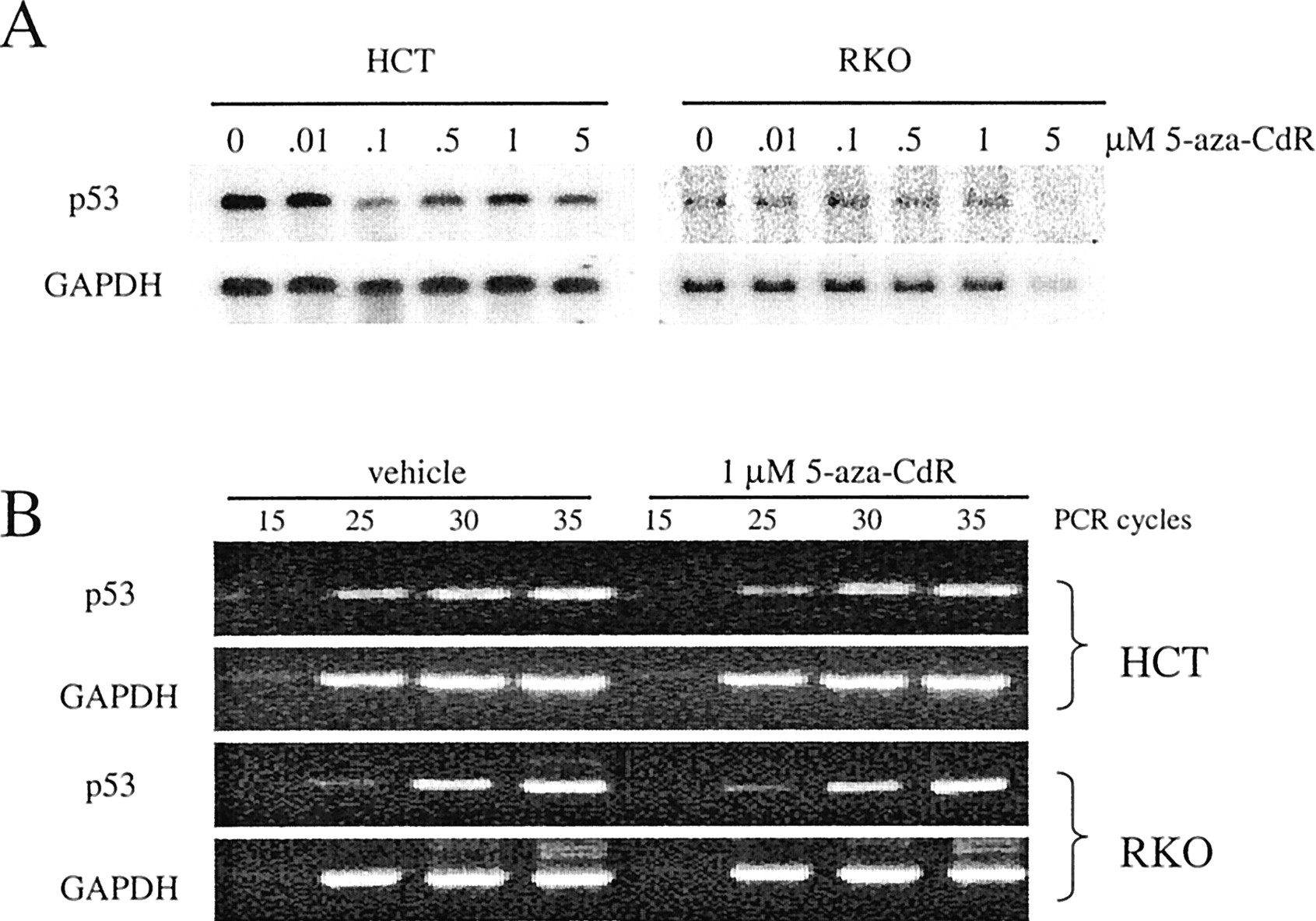
p53 mRNA is not induced after 5-aza-CdR treatment. A, HCT116 and RKO cells were treated with various concentrations of 5-aza-CdR and RNA was isolated 5 days after treatment and used for Northern blot detection of p53. GAPDH was used to assess RNA input. B, HCT116 and RKO cells were treated with vehicle or 1.0 μM 5-aza-CdR and RNA was isolated 5 days after treatment and used for RT-PCR detection of p53. GAPDH was amplified in parallel to confirm equivalent cDNA input.
p53-Null HCT116 Cells Are Less Sensitive to 5-Aza-CdR Induced Cellular Toxicity and Show Reduced Levels of p21 Induction
We used HCT116 cells with a targeted deletion of the p53 gene to analyze the contribution of p53 to the response of tumor cells to 5-aza-CdR treatment (Bunz et al., 1998). Northern and Western blot analysis of p53 confirmed the genotype of the knockout cells (data not shown). For comparison of the response of these cells to 5-aza-CdR treatment, we measured cellular responsiveness by MTT assay 5 days after treatment. Consistent with our initial observations using the four colon cancer cell lines (Fig. 1), the p53-null HCT116 cells were less sensitive to 5-aza-CdR-induced cellular toxicity than were the parental HCT116 cell line (Fig.5A). Next, we examined whether abrogation of p53 in HCT116 cells affects the level of induction of p21 after 5-aza-CdR treatment. Our results indicate that the induction of p21 protein by 5-aza-CdR treatment has a p53-dependent component, because the magnitude of induction was reduced in the knockout cells (Fig. 5B). There was approximately an 8-fold induction of p21 protein in the parental cells compared with a 3-fold induction of p21 protein in the p53-null cells. In addition, a greater concentration (approximately 10-fold) of 5-aza-CdR was required for detectable p21 induction in the p53 knockout cells. The magnitude of induction of p21 mRNA after 5-aza-CdR treatment in the p53 knockout cells was similarly reduced (Fig. 5B).
p21-Null HCT116 Cells Show a Reduced Level of G1 Arrest and an Increased Level of Cytotoxicity after 5-Aza-CdR Treatment.
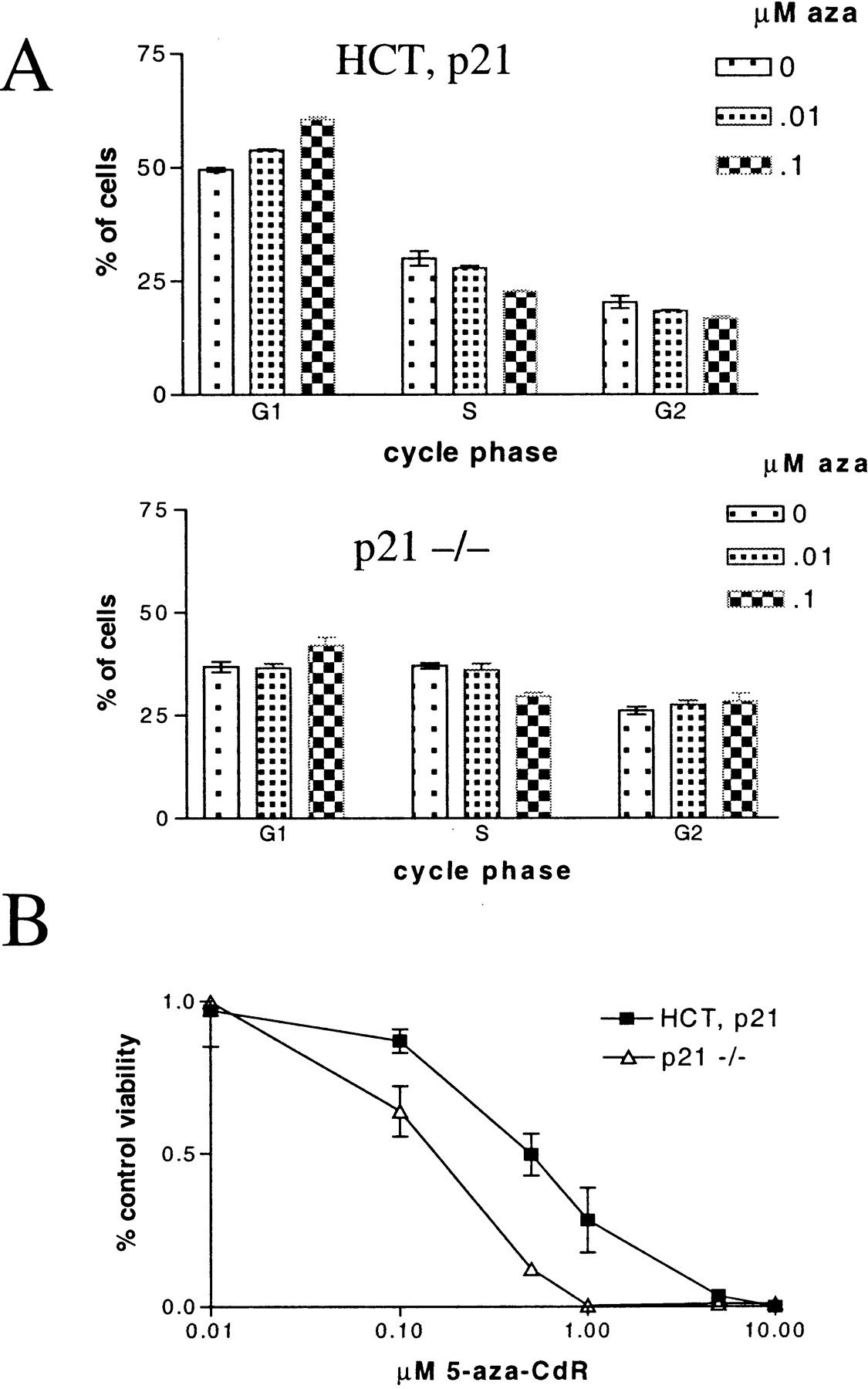
We examined the effect of p21 abrogation (in a wild-type p53 background) on the response of cells to 5-aza-CdR treatment. For this aim, we used HCT116 cells containing a targeted deletion of p21 (Waldman et al., 1995). Northern and Western blot analysis of p21 confirmed the genotype of the knockout cells (data not shown). p21 plays a critical role in determining whether p53 activation leads to growth arrest or apoptosis (Bunz et al., 1999; Tian et al., 2000). Based on our data that implicates p53 activation in the responsiveness of tumor cells to 5-aza-CdR, it follows that p21 abrogation in a wild-type p53 background should impact whether 5-aza-CdR treatment leads to either growth arrest or cytotoxicity. As expected, we observed that p21-null HCT116 cells showed reduced levels of G1 arrest after 5-aza-CdR treatment (Fig.6A). After treatment with 0.01 μM 5-aza-CdR, only the wild-type cells showed a G1increase or S-phase decline. After treatment with 0.1 μM 5-aza-CdR, the wild-type cells showed an increased proportion of cells in G1 with a concomitant decline in S and G2 proportions. The p21-null cells responded to treatment with 0.1 μM 5-aza-CdR with a smaller increase in G1 and a slight rise in the G2 proportion. When the response of the cells was measured by MTT assay, we found that the p21-null HCT116 cells were dramatically more sensitive to 5-aza-CdR–induced cytotoxicity (Fig.6B). Taken together, these data indicate that p21 abrogation in a wild-type p53 background shifts the cellular response to 5-aza-CdR treatment from G1 arrest to cell death.

p21-null HCT116 cells are less sensitive to 5-aza-CdR induced G1 arrest and are more sensitive to 5-aza-CdR induced cytotoxicity. A, HCT, p21, and p21−/− cells were treated with 0, 0.01, and 0.1 μM 5-aza-CdR and cells were harvested for cell-cycle analysis 5 days after treatment. Error bars indicate ± 1 S.D. B, HCT, p21, and p21−/− cells were treated with various concentrations of 5-aza-CdR and the cellular response was assessed 5 days after treatment by MTT assay. The data were normalized by subtraction of background staining and are expressed as the percentage of control of each cell line. Error bars indicate ± 1 S.D.
Discussion
The activation of p53 after treatment of tumor cells with chemotherapeutic agents is well known and correlates with responsiveness to some agents (Bunz et al., 1999; Lakin and Jackson, 1999). This observation is consistent with the fact that many chemotherapeutics are DNA-damaging agents and that p53 plays a critical role in the induction of cellular growth arrest and apoptosis in response to DNA damage (Lakin and Jackson, 1999). However, for drugs that target DNA methylation, the common view is that the cellular response is dictated by the reactivation of specific tumor-suppressor genes. The novelty of the current study is that we provide a new factor, p53 status, that has a significant impact on the cancer cell sensitivity to DNA methyltransferase inhibition by 5-aza-CdR.
We observed that 5-aza-CdR treatment results in the induction and activation of p53 protein in a manner consistent with the classical p53 response to DNA damage (Lakin and Jackson, 1999). Because 5-aza-CdR (decitabine) is under evaluation for the treatment of solid tumors and leukemia (Kantarjian et al., 1997; Momparler et al., 1997; Schwartsmann et al., 1997; Thibault et al., 1998; Sacchi et al., 1999; Wijermans et al., 2000), the discovery that p53 status affects the response of tumor cells to this drug may prove relevant for interpretation of both beneficial and toxic clinical responses. In support of this hypothesis is the observation that p53 mutations are rarely found in chronic myelogenous leukemia (Peller et al., 1998; Wang et al., 1998), the malignancy for which decitabine has proved the most beneficial (Sacchi et al., 1999). Therefore, it is possible that activation of p53 could account, in part, for the clinical benefits seen in leukemia. Similarly, p53 activation could contribute to the myelosuppression that has limited the success of decitabine therapy in solid tumors (Momparler et al., 1997). These scenarios leave open the question of whether the actions of decitabine as a DNA methyltransferase inhibitor have been fully tested clinically and raises the need for trials that simultaneously assess p53 activation and global methylation levels in target tissues. Likewise, the development of novel inhibitors for DNA methyltransferase, or agents that relieve transcriptional repression at methylated promoters, could provide a more focused test for reactivation of methylation-silenced genes as a therapeutic target.
Two independent lines of evidence suggest that p53 induction is specifically related to the inhibition of DNA methyltransferase by 5-aza-CdR: 1) the activation of p53 and cellular responsiveness after 5-aza-CdR treatment is time-dependent and is maximal only after 4 or 5 days of treatment, and 2) 6-aza-CdR treatment did not induce either cell toxicity or p53 activation at similar molar concentrations. Interestingly, nuclear p53 induction differs in the two cell types we studied. In RKO cells, the level of nuclear p53 is greatly enhanced in the absence of the formation of p53 cleavage products; this is considered a typical p53 response. However, in HCT116 cells, activation of p53 was characterized by the nuclear appearance of the p53 cleavage product p50. The appearance of p50 has been found to result from direct interaction of p53 at sites of DNA damage in some systems (Okorokov et al., 1997; Okorokov and Milner, 1997). For example, treatment of myeloblastic leukemia cells with the DNA-damaging chemotherapeutics doxorubicin or cisplatin induces the formation of p50 (Okorokov et al., 1997). The reason that 5-aza-CdR treatment results in distinct modes of wild-type p53 activation in the cell types studied here is unknown, but both observations support the contention that wild-type p53 is activated in response to 5-aza-CdR treatment.
5-aza-CdR-induced growth inhibition can result from the release of methylation silencing of cell cycle regulatory genes such as p16 (Bender et al., 1998). In addition, enzyme-DNA adduct formation can account for cellular toxicity after treatment with 5-aza-CdR (Juttermann et al., 1994; Ferguson et al., 1997). Because p53 mRNA levels were not induced by 5-aza-CdR treatment, the p53 protein induction we observed did not result from the release of methylation silencing of the p53 gene. Instead, our data imply that p53 induction resulted because 5-aza-CdR incorporation leads to DNA damage. The induction of p53 could be the result of either a direct response to the enzyme-DNA adducts or an indirect response to global DNA hypomethylation. In a recent study, it was shown that treatment of tumor cells with oligonucleotide-based substrate inhibitors of DNA methyltransferase does not lead to p53 activation (Milutinovic et al., 2000). This observation supports the notion that the 5-aza-CdR-mediated induction of p53 we observed is the result of enzyme-DNA adduct formation and not genomic hypomethylation.
Our data indicate that the induction of p21 by 5-aza-CdR has both p53 dependent and independent components. We observed that the induction of p21 protein after 5-aza-CdR treatment is dramatic in two cell lines expressing wild-type p53 but absent in two different cell lines expressing mutant p53. We also observed that p21 induction occurred at reduced levels in HCT116 cells with a targeted deletion of p53. However, p21 induction in HCT116 cells reached a maximum at lower concentrations of 5-aza-CdR than did p53 induction (Fig. 2). This result may reflect the fact that both transcriptional and post-transcriptional mechanisms of p21 induction after DNA methyltransferase inhibition can occur. For example, it has been demonstrated that p21 is induced after treatment of tumor cells with either antisense oligonucleotides directed against DNA methyltransferase I (Fournel et al., 1999) or oligonucleotide-based substrate inhibitors of DNA methyltransferase I (Milutinovic et al., 2000). Regardless of the mechanism of p21 induction after 5-aza-CdR treatment, we found that 5-aza-CdR mediated toxicity is influenced by cellular p21 status. This observation is consistent with a role for p53 activation in 5-aza-CdR-mediated cytotoxicity, in that p21 disruption is known to shift p53 dependent responses from growth arrest to apoptosis (Bunz et al., 1999; Tian et al., 2000). However, it is also possible that p21 disruption alters 5-aza-CdR-mediated cytotoxicity in a manner that is independent of p53 function.
In summary, our data indicate that p53 activation may contribute to the clinical efficacy and/or toxicity associated with decitabine treatment. It will be interesting to investigate the relationship between p53 status and other potential genetic determinants of drug sensitivity, including mismatch repair status. For example, we found an inverse relationship between mismatch repair proficiency and 5-aza-CdR sensitivity in this study. It is conceivable that the mismatch repair pathway influences responses to 5-aza-CdR by, for example, cross talk with the p53 pathway. Further studies are necessary to dissect these molecular relationships. Given the growing interest in therapies that modulate transcription, our findings warrant further basic and clinical investigations evaluating the mechanistic basis for p53 activation and its relationship to genomic hypomethylation and gene silencing within target tissues.
Acknowledgments
We thank Dr. Bert Vogelstein (Johns Hopkins University, Baltimore, MD) for generously providing the HCT116 cell lines with a homozygous disruption of the p53 and p21 genes. We thank the Huntsman Cancer Center Flow Cytometry Core Facility for assistance with the DNA content analysis.
Footnotes
- Received November 2, 2000.
- Accepted January 3, 2001.
-
Send reprint requests to: Dr. David A. Jones, Division of Molecular Pharmacology, Huntsman Cancer Institute, 2000 Circle of Hope, University of Utah, Salt Lake City, UT 84112. E-mail:david.jones{at}hci.utah.edu
-
This work was supported by a Postdoctoral Fellowship PF-99–151-01-CDD from the American Cancer Society (to A.R.K.) and by National Institutes of Health P01-CA73992 (to D.A.J.).
-
This work was previously presented under the title “Impact of p53 status on the response of tumor cells to 5-aza-2′-deoxycytide treatment” at the American Association of Cancer Research Annual Meeting, San Francisco, CA, 1–5 April, 2000.
Abbreviations
- 5-aza-CdR
- 5-aza-2′-deoxycytidine
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
- 6-aza-CdR
- 6-aza-2′-deoxycytidine
- PAGE
- polyacrylamide gel electrophoresis
- GAPDH
- glyceraldehyde-3 phosphate dehydrogenase
- RT-PCR
- reverse transcription-polymerase chain reaction
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}