


Abstract
Vesnarinone, a cardiotonic agent, blocks IKr and, unlike other IKr blockers, produces a frequency-dependent prolongation of action potential duration (APD). To elucidate the mechanisms, we studied the effects of vesnarinone on HERG, the cloned human IKr channel, heterologously expressed inXenopus laevis oocytes. Vesnarinone caused a concentration-dependent inhibition of HERG currents with an IC50 value of 17.7 ± 2.5 μM at 0 mV (n = 6). When HERG was coexpressed with the β-subunit MiRP1, a similar potency for block was measured (IC50: 15.0 ± 3.0 μM at 0 mV, n= 5). Tonic block of the HERG channel current was minimal (<5% at 30 μM, n = 5). The rate of onset of block and the steady-state value for block of current were not significantly different for test potentials ranging from −40 to +40 mV [time constant (τ) = 372 ± 76 ms at +40 mV,n = 4]. Recovery from block at −60, −90, and −120 mV was not significantly different (τ = 8.5 ± 1.5 s at −90 mV, n = 4). Vesnarinone produced similar effects on inactivation-removed mutant (G628C/S631C) HERG channels. The IC50 value was 10.7 ± 3.7 μM at 0 mV (n = 5), and the onset and recovery from block of current findings were similar to those of wild-type HERG. Amino acids important for the binding of vesnarinone were identified using alanine-scanning mutagenesis of residues believed to line the inner cavity of the HERG channel. Six important residues were identified, including G648, F656, and V659 located in the S6 domain and T623, S624, and V625 located at the base of the pore helix. These residues are similar but not identical to those determined previously for MK-499, an antiarrhythmic drug. In conclusion, vesnarinone preferentially blocks open HERG channels, with little effect on channels in the rested or inactivated state. These actions may contribute to the favorable frequency-dependent prolongation in APD.
Vesnarinone inhibits phosphodiesterase (PDE) and has been used as a cardiotonic agent to treat chronic heart failure (Inoue et al., 1987; Asanoi et al., 1989; Feldman et al., 1993). Although PDE inhibitors greatly improve left ventricular function, most drugs with this activity have failed to decrease mortality in patients with heart failure. Vesnarinone is an exception and was reported to improve mortality rates when administered orally at a dose lower than that required to inhibit PDE or to have a positive inotropic action (Taira et al., 1984). Besides inhibiting PDE, vesnarinone inhibits potassium channels and prolongs the action potential duration (APD) of cardiomyocytes (Toyama et al., 1997; Yang et al., 1997). This additional action may contribute to the ability of vesnarinone to improve cardiac contractility.
We demonstrated previously that vesnarinone prolongs the APD of rabbit ventricular myocytes in a frequency-dependent manner (Toyama et al., 1997). This action was associated with frequency-dependent inhibition of the rapid delayed rectifier potassium channel, IKr. Recently, Katayama et al. (2000) reported that vesnarinone blocks HERG channels, but not the KvLQT1/minK current. These investigators found that the onset of HERG current inhibition by vesnarinone was faster and more prominent as the membrane potential became progressively depolarized. Channel block was modeled by a first-order reaction that was independent of channel gating (Katayama et al., 2000).
Channel inactivation or coexpression of HERG with MiRP1 subunits can modulate the potency of drug block of HERG. C-type inactivation of HERG channels can be removed through the simultaneous introduction of two point mutations (G628C, S631C) (Smith et al., 1996). Inactivation-removed HERG mutant channels are far less sensitive to block by most IKr blockers (Wang et al., 1996,1997; Ficker et al., 1998; Lees-Miller et al., 2000). It was unlikely, given the study by Katayama et al. (2000), that vesnarinone block of HERG channels would be affected by the removal of inactivation, but this possibility has not been tested experimentally. The effect of MiRP1 on the block of HERG channels by vesnarinone has also not been investigated.
We recently used alanine-scanning mutagenesis of HERG to identify the specific binding site for the potent blocker MK-499 (Mitcheson et al., 2000a). Mutation to Ala of specific residues that line the inner cavity of the HERG channel reduced the potency of block and defined the high-affinity binding site for the drug. Recovery from the block of HERG channels by MK-499, as with many other drugs, is extremely slow. Vesnarinone is much less potent than MK-499, and recovery from channel block is much faster. Thus, we were interested to determine whether vesnarinone interacted with the same residues that line the inner cavity of HERG as we demonstrated previously for MK-499.
In this study, we use the oocyte expression system and a two-microelectrode voltage-clamp technique to investigate the voltage- and time-dependence of block, the role of channel inactivation, and the effect of MiRP1 coexpression on the potency of vesnarinone block of HERG channels. In addition, we used Ala-scanning mutagenesis to define the structural basis of HERG channel block by vesnarinone.
Materials and Methods
Site-Directed Mutagenesis.
Mutations were introduced into the HERG K+ channel (Warmke and Ganetzky, 1994) with the use of site-directed mutagenesis as described previously (Sanguinetti and Xu, 1999). The cDNA expression construct in the pSP64 transcription vector (Promega, Madison, WI) and synthesis of cRNA were conducted as described previously (Sanguinetti and Xu, 1999). MiRP1 cDNA was kindly provided by S. A. N. Goldstein (Yale University, New Haven, CT).
cRNA Injection and Voltage Clamp of Oocytes.
Isolation and maintenance of Xenopus laevis oocytes and injection with cRNA were performed as described previously (Stuhmer, 1992). X. laevis frogs were anesthetized by immersion in 0.2% tricane (Sigma, St. Louis, MO) for 15 to 30 min. Ovarian lobes were removed and digested with 2 mg/ml type IA collagenase (Sigma) in Ca2+-free ND96 solution for 1.5 h to remove follicle cells. Stage V and VI oocytes were injected with 10 ng of cRNA encoding wild-type (WT) HERG or G628C:S631C HERG. For HERG-MiRP1 coexpression studies, 2 ng of MiRP1 cRNA was coinjected with 10 ng of HERG cRNA. To study IKs, KVLQT1 (5 ng) and minK(1 ng) cRNA were coinjected into oocytes. Oocytes were cultured in Barth's solution supplemented with 50 μg/l gentamicin and 1 mM pyruvate at 18°C.
Currents were recorded with a Dagan TEV-200 amplifier (Dagan, Minneapolis, MN) using standard two-microelectrode voltage-clamp techniques (Stuhmer, 1992) 2 to 4 days after injection of cRNA. Currents were recorded at room temperature (22–24°C). Glass microelectrodes were filled with 3 M KCl, and their tips were broken to obtain a resistance of 0.5 to 1.0 MΩ. To attenuate endogenous chloride currents, Cl− was replaced with 2-(N-morpholino)ethanesulfonic acid (MES) in the external solution that contained 96 mM NaMES, 2 mM KMES, 2 mM CaMES2, 5 mM HEPES, and 1 mM MgCl2, adjusted to pH 7.6 with methanesulfonic acid. Voltage commands were generated using pCLAMP software (version 6.0.4; Axon Instruments, Burlingame, CA). Specific voltage-clamp protocols used to elicit currents are described underResults.
Vesnarinone was obtained from Otsuka Pharmaceutical Co. (Tokyo, Japan). A 30 mM stock solution was prepared in dimethyl sulfoxide and diluted with extracellular solution to the desired final concentrations immediately before each experiment.
Data Analysis.
Data are presented as mean ± S.E.M. unless otherwise specified. Statistical comparisons between the different experimental groups were obtained using analysis of variance. Differences were considered significant at p < 0.05. Concentration-response relationships were fit to the Hill equation to determine the drug concentration required for 50% inhibition (IC50). A nonlinear least-squares curve-fitting program (Clampfit 6.0.4) was used to analyze the kinetics of current deactivation.
Results
Effects of Vesnarinone on Oocytes Expressing HERG or HERG + MiRP1 Subunits.
The effects of vesnarinone on currents induced by the expression of HERG alone and with MiRP1 were determined. Currents were elicited during 2-s depolarizing pulses to potentials ranging from −80 to +50 mV from a holding potential of −90 mV. Tail currents were elicited by return of the membrane potential to −80 mV after each voltage step. Vesnarinone (10 μM) caused a similar decrease in the amplitude of outward currents during depolarization and tail current on repolarization in oocytes expressing HERG or HERG + MiRP1 subunits. The decreases in time-dependent current and tail current at 0 mV, respectively, were 35.0 ± 10.2% (n = 6) and 30.4 ± 12.5% for HERG alone and 37.7 ± 11.0% (n = 6) and 32.2 ± 12.5% for HERG with MiRP1. There were also nonsignificant differences in the time-dependent kinetics (Fig. 1A). The drug concentration required to block the current by 50% (IC50) was determined from a plot of the peak time-dependent current recorded during a pulse to +0 mV versus drug concentration. The IC50 value for HERG was 17.7 ± 2.5 μM (n = 6). The IC50 value for HERG/MiRP1 was 15.0 ± 3.0 μM (n = 5), which was not significantly different (p > 0.5) from HERG alone (Fig. 1B).

Effects of vesnarinone on HERG ± MiRP1 channel current and KvLQT1/minK channel current in X. laevisoocytes. A, representative HERG and HERG/MiRP1 channel currents recorded from oocytes before and after application of 10 μM vesnarinone. Voltage-clamp protocol is shown above current traces. B, concentration-response relationship for block of current recorded at the end of a 2-s depolarizing pulse to 0 mV for HERG (○) or HERG/MiRP1 (●). IC50 values were 17.7 ± 2.5 μM (n = 6) for HERG and 15.0 ± 3.0 μM (n = 5) for HERG/MiRP1. C, representative current traces recorded from oocytes expressing KvLQT1 + minK subunits before and after application of 300 μM vesnarinone. D, I-V relationship for KvLQT1/minK currents measured at the end of a 7.5-s depolarization step (n = 5). Data were obtained before (○) and after (●) 300 μM vesnarinone.
KvLQT1/minK Currents.
The effect of vesnarinone on the KvLQT1/minK current was examined. From a holding potential of −90 mV, 7.5-s pulses were applied to potentials ranging from −80 mV to +30 mV. Tail currents were elicited by return of the membrane potential to −80 mV. Vesnarinone at 300 μM affected neither the slowly activating outward current elicited during depolarization nor the tail currents at −80 mV (Fig. 1C). Similar results were obtained in five oocytes (Fig.1D). Thus, vesnarinone blocks only one of the two major types of delayed rectifier K+ currents expressed in ventricular myocytes.
Voltage Dependence of Vesnarinone Block of HERG Channels.
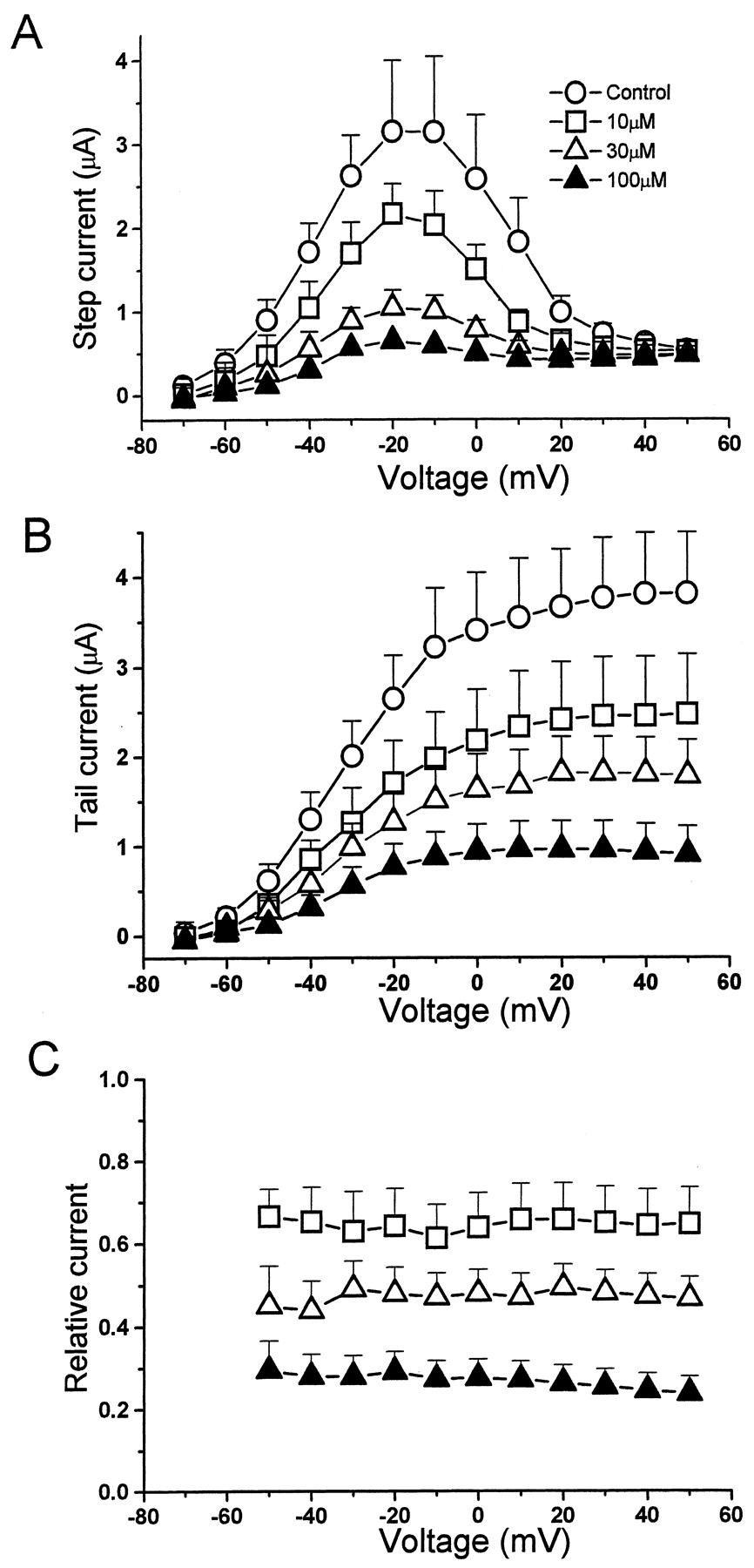
The effect of vesnarinone on currents recorded at voltages ranging from −70 to +50 mV are shown in Fig. 2. The I-V relationship peaked at −20 mV both before and after exposure of oocytes to 10, 30, and 100 μM vesnarinone (Fig. 2A). Drug-induced inhibition of the current during a 2-s pulse (Fig. 2A) or peak tail current (Fig. 2B) was concentration-dependent; however, there was no voltage-dependent inhibition of current when quantified as relative tail current amplitude (Fig. 2C). The relative tail current over the voltage range of −50 to +50 mV varied from 0.61 to 0.64 at 10 μM, 0.46 to 0.48 at 30 μM, and 0.22 to 0.25 at 100 μM.

Block of HERG channel current by vesnarinone. A, HERG channel I-V relationship before (○) and after application of 10 μM (■), 30 μM (▵), and 100 μM (▴) vesnarinone (n = 5). B, I-V relationship for peak tail currents. C, voltage-dependence for block of HERG tail current by vesnarinone. The ratio of tail currents in the presence and absence of vesnarinone was used as a measure of relative current.
Time-Dependent Inhibition by Vesnarinone.
Time-dependent changes of HERG block by vesnarinone were evaluated by two protocols. First, an envelope of tail currents was used as a measure of onset of drug block. HERG tail currents were generated by applying depolarizing pulses from −40 to +40 mV in 20-mV steps of variable duration (50–1000 ms) from a holding potential of −90 mV. Tail currents elicited by repolarization to −80 mV were measured before and after application of vesnarinone at 30 and 100 μM. In the absence of drug, tail currents increased as the pulse duration was prolonged and reached a plateau level (Fig. 3A, left). In the presence of 100 μM vesnarinone, tail currents decreased as the pulse was lengthened (Fig. 3A, right). The ratio of tail current amplitudes measured in the presence and absence of drug was plotted as a function of pulse duration (Fig. 3A, bottom). The time course of decay of relative tail current was well fitted with a single exponential function. The onset of HERG block by 30 μM vesnarinone developed with a time constant (τ) of 372 ± 76 ms at +40 mV (n = 4). The time constants for onset of block (τonset) were faster at 100 μM (98 ± 33 ms at +40 mV, n = 4) than at 30 μM, but were voltage-independent (Fig. 3B), as were the steady-state values for block (Fig. 3C).
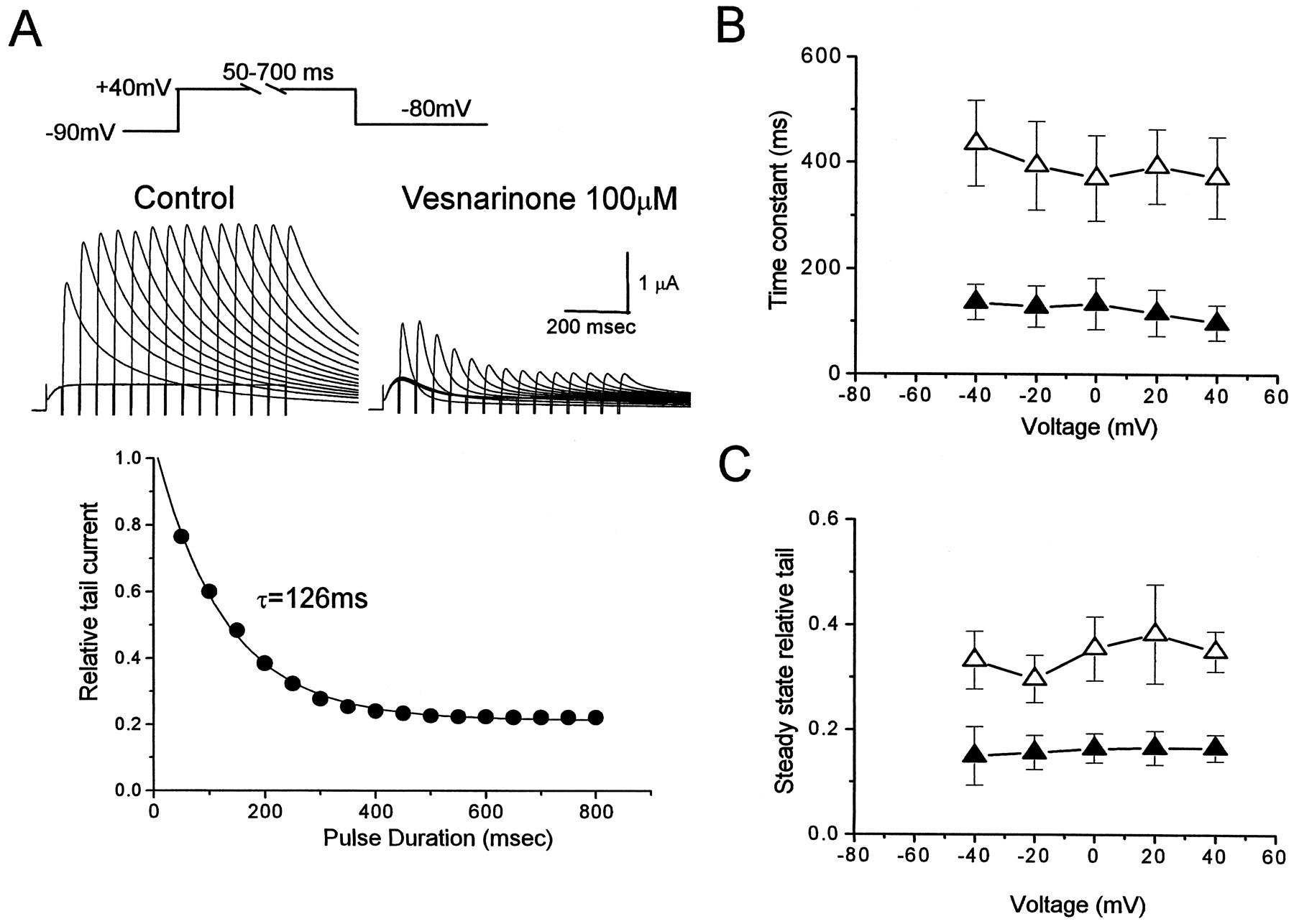
Onset of HERG channel current block by vesnarinone using an envelope of tails protocol. A, voltage-clamp pulse protocol and representative recordings of HERG current before and after exposure of an oocyte to 100 μM vesnarinone (top). Bottom, a plot of the onset of current block expressed as relative tail current. The time-dependent decay in relative tail current was fitted to a single exponential function with a time constant of 126 ms. The relative steady state value of current was 0.21. B, τonset for decay of relative tail current plotted as a function of voltage after application of 30 μM (▵) and 100 μM (▴) vesnarinone (n = 4). C, steady-state value of relative tail current (n = 4).
The time course for onset of drug block was also measured more directly by comparing the currents throughout a 2-s depolarizing pulse. Current traces in the presence of drug were subtracted by residual current traces after 20 μM E-4031 and divided by control current traces to obtain relative current amplitudes as shown in Fig.4, A and B. Although the amplitudes of currents were decreased in a concentration-dependent manner, the onset of block was similar for pulses to potentials ranging from −20 to +40 mV (Fig. 4B). The time constant varied from 350 ms to 410 ms at 30 μM and from 180 ms to 220 ms at 100 μM (Fig. 4C). Steady-state values for residual block varied from 0.40 to 0.46 at 30 μM and from 0.14 to 0.20 at 100 μM. These results provided additional evidence that the development of block during depolarization is not voltage-dependent. Tonic block of HERG channel current by vesnarinone was estimated by the initial value of the ratio (Fig. 4B) extrapolated by the exponential fitting. Tonic block was 0.6 ± 2.7% (n = 4) for 30 μM, and 5.4 ± 4.4% (n = 4) for 100 μM vesnarinone. This finding indicates that vesnarinone does not bind appreciably to HERG channels in the rested state.

Onset of HERG channel current block by vesnarinone assessed during depolarizing voltage steps. A, representative recordings of HERG currents elicited by 2-s pulses to −20 mV before and after 30 and100 μM vesnarinone. E-4031 at 20 μM was applied at the end of the experiment to completely block HERG current. B, onset of current block by 100 μM vesnarinone during depolarizing voltage steps to the indicated voltage. Residual current remaining after complete block of HERG with E-4031 was subtracted from currents before and after vesnarinone at 100 μM. Then, relative tail currents were estimated by the ratio of these currents. The time constants for development of block of HERG current was 345 ms at +20 mV, 376 ms at 0 mV, 438 ms at −20 mV, and 456 ms at −40 mV. C, time constants for the onset of HERG current block at different voltages for 30 μM (▵) and 100 μM (▴) vesnarinone (n = 5). D, steady-state value of the relative tail current determined from single exponential fit of relationships shown in B (n = 5).
Recovery from Block.
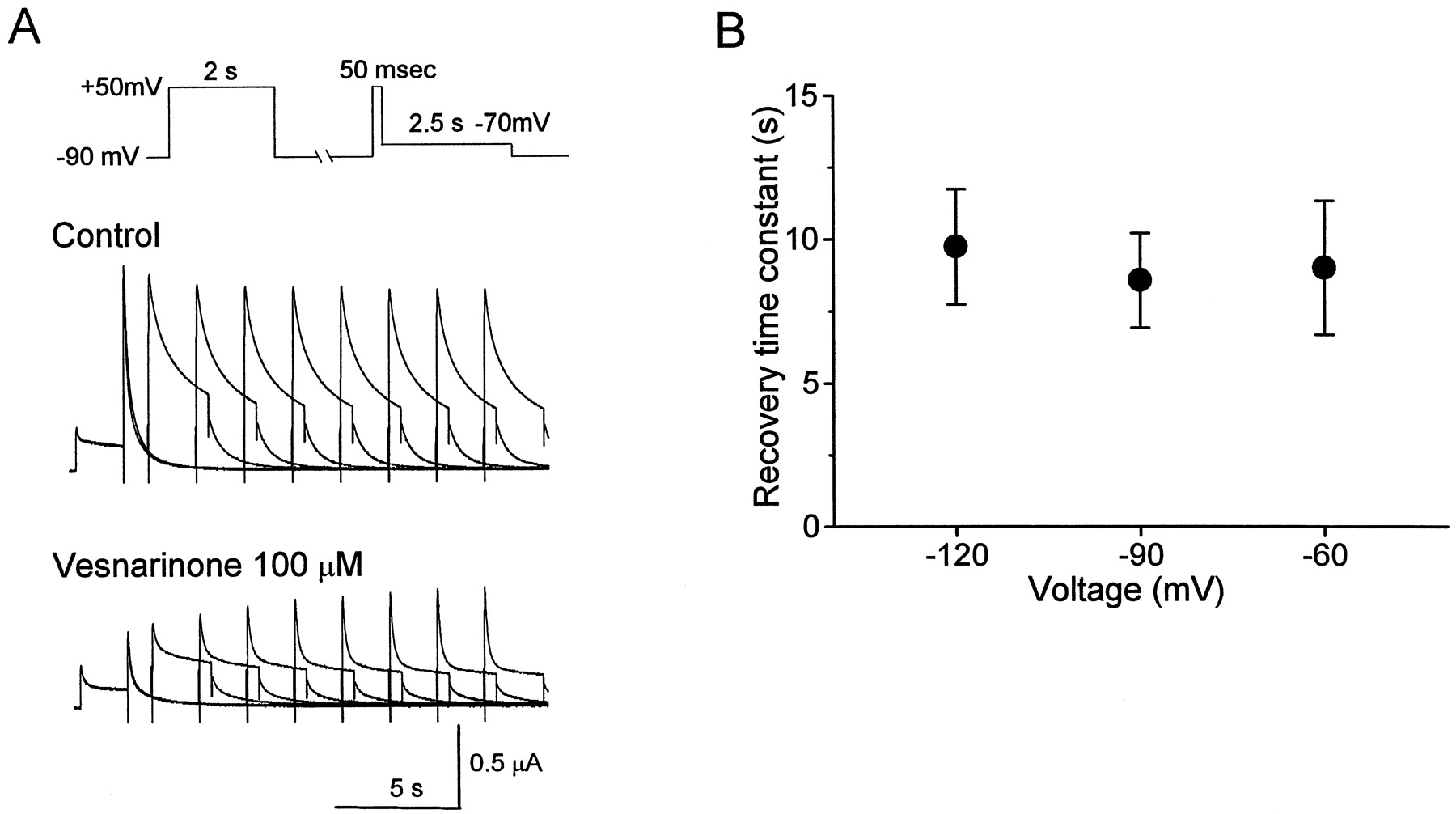
Recovery from HERG channel block by vesnarinone was evaluated at different membrane potentials using a paired-pulse protocol. A conditioning pulse to +50 mV was applied for 2 s to induce steady-state block. The potential was then clamped to −60, −90, or −120 mV for a variable time (1–29 s, in 2-s increments), followed by a 50-ms test pulse to +50 mV, then returned to −70 mV to assess recovery from block (Fig.5A). Before drug, the amplitude of the tail current was relatively unchanged as the duration of the conditioning pulse was prolonged (Fig. 5A, top). However, in the presence of 100 μM vesnarinone, the tail current amplitude was progressively increased with longer conditioning pulses and reached a steady state within 13 to 15 s (Fig. 5A, bottom). The recovery of the tail current was fitted by a single exponential function. The time constants (τrecov) varied little at the three potentials studied, from 9.1 ± 2.3 s at −60 mV (n = 4) to 8.6 ± 1.6 s at −90 mV (n = 4) to 9.7 ± 2.0 s at −120 mV (n = 4) (Fig. 5B). Thus, the recovery of HERG current from block by vesnarinone was not significantly affected by membrane voltage.

Recovery from block of HERG channel current by vesnarinone at hyperpolarized holding potentials. A, voltage-clamp pulse protocol and representative recordings for an oocyte in which the recovery was assessed at −90 mV. B, time constants for recovery from block of HERG current (n = 4).
Block of Inactivation-Removed Mutant HERG Channels.
The potency for many blockers is usually greater for WT HERG than inactivation-removed HERG mutant channels. Therefore, we determined the concentration-dependence for block of G628C/S631C HERG channel current by vesnarinone.
The outward G628C/S631C HERG channel current was elicited by the same protocol used to evaluate WT currents (Fig. 1). Unlike WT HERG, the mutant current elicited by depolarizing pulses was quite large as a result of the lack of C-type inactivation (Fig.6A). Because of a decrease in K+ selectivity (Smith et al., 1996), the tail current for mutant channels was very small at −70 mV. Application of 10 μM vesnarinone decreased the outward current measured during depolarization in a time-dependent manner (Fig. 6A). The IC50 value for current measured at the end of a 2-s pulse to 0 mV was 10.7 ± 3.7 μM (n = 5; Fig. 6B), which was slightly greater than the potency calculated for the WT HERG channel current. The inhibition of G628C/S631C HERG channel current was not voltage-dependent (Fig. 6 C and D). The relative current, defined as the current in drug divided by control currents, was decreased as the vesnarinone concentration increased and varied from 0.79 to 0.81 at 3 μM, from 0.41 to 0.48 at 10 μM, from 0.21 to 0.27 at 30 μM, and from 0.14 to 0.21 at 100 μM.

Block of inactivation-removed (G628C/S631C) HERG channel current by vesnarinone. A, voltage-clamp pulse protocol and representative currents recorded from oocytes before and after application of 10 μM vesnarinone. B, concentration-response relationship for block of current measured at the end of a 2-s depolarization to 0 mV. IC50 value was 10.7 ± 3.7 μM (n = 5). C, I-V relationship for currents measured at the end of a 2-s pulse to the indicated voltage before (○) and after 3 μM (▪), 10 μM (■), 30 μM (▵), and 100 μM (▴) vesnarinone (n = 5). D, block of step current by vesnarinone is not voltage-dependent. The ratio of step currents in the presence of vesnarinone relative to control currents in C was calculated and plotted again.
Because tail currents were too small to analyze, time-dependent changes of G628C/S631C HERG channel block by vesnarinone were evaluated during a 2-s depolarizing pulse (Fig. 7A). The kinetics and intensity of block of mutant channels were quite similar to that of WT HERG, again indicating that the absence of inactivation did not influence drug block. The relative current amplitude at any voltages from −30 to +50 mV decayed as pulse duration prolonged. The decay phase was fitted by a single exponential function. The time constant for block onset and steady-state block are shown in Figs. 7 B and C. The time constant was reduced at the higher drug concentration and varied from 170 to 318 ms at 30 μM and from 81 to 172 ms at 100 μM. The time constants were slightly increased at hyperpolarization. The steady-state value for block was also decreased at the higher concentrations and varied from 0.40 to 0.46 at 30 μM and from 0.14 to 0.20 at 100 μM. Recovery from block of G628C/S631C HERG current was evaluated at different hyperpolarizing levels using a paired-pulse protocol (Fig. 7D). A conditioning pulse to +50 mV was applied for 2 s to induce steady-state block. The potential was then clamped to −60, −90, or −120 mV for a period that was varied in 2-s increments from 1 to 29 s. A 500-ms test pulse followed the conditioning pulse to +50 mV. In the absence of drug, the current amplitude during the test pulse remained constant regardless of the duration of the conditioning pulse (data not shown). In the presence of 100 μM vesnarinone, current amplitude was progressively increased as the conditioning pulse was prolonged (Fig. 7D). The envelope of current amplitudes was fitted by a single exponential function that varied from 9.7 ± 1.1 s at −60 mV (n = 4), 11.9 ± 1.8 s at −90 mV (n = 4), and 12.1 ± 1.6 s at −120 mV (n = 4) (Fig. 7E). Thus, similar to WT HERG, the recovery of G628C/S631C HERG current block by vesnarinone was only weakly voltage-dependent.

Onset and recovery of G628C/S631C HERG channel current block by vesnarinone assessed during and after depolarizing voltage steps. A, representative recordings of G628C/S631C HERG currents elicited by 2-s pulses to +50 mV before and after 100 μM vesnarinone. E-4031 at 20 μM did not delete the time-dependent outward current. Therefore, relative step currents were estimated by the ratio of G628C/S631C HERG currents before and after application of vesnarinone. B, time constants for the onset of G628C/S631C HERG current block at different voltages for 30 μM (▵) and 100 μM (▴) vesnarinone (n = 5). *P< 0.05 versus values of 100 μM vesnarinone at +50 mV. C, steady-state value of the relative tail current determined from single exponential fit of relationships shown in B (n = 5). D, recovery from block of G628C/S631C HERG channel current by vesnarinone at hyperpolarized holding potentials. Voltage-clamp pulse protocol and representative recordings were shown for an oocyte where the recovery was assessed at three different voltages. E, time constants for recovery from block of G628C/S631C HERG current by 100 μM vesnarinone (n = 4).
Alanine-Scanning Mutagenesis to Define Binding Site.
Alanine-scanning mutagenesis was used to determine residues important for block of the HERG channel by vesnarinone as reported previously (Mitcheson et al., 2000a). We mutated to alanine individual residues of S6 (L646-Y667) and the few residues of the pore helix (L622-V625) predicted to line the channel cavity and inner pore regions (Fig.8A) based on homology with the solved crystal structure of the KcsA channel (Doyle et al., 1998). The sensitivity to block by a single concentration of vesnarinone (180 μM, 10 times IC50) was determined for each mutant channel. Currents were measured during repetitive 5-s depolarizing steps to 0 mV, applied at a frequency of 0.166 Hz. Vesnarinone at 180 μM caused approximately 85% inhibition of WT HERG current and was used to assess each mutant channel. Examples of WT and three mutant HERG channel currents recorded before and after block by vesnarinone had reached a steady-state level are shown in Fig. 8B. Note that V659A HERG channel currents differ from WT because they deactivate very slowly, and a proportion of the channels are open at the −90-mV holding potential (Mitcheson et al., 200a). Consequently, an instantaneous component of current is observed upon depolarization. As before, T623A and G648A HERG channels are more inactivated than other channels; therefore, as described previously, the currents conducted by these channels were measured using a 96 mM KMES extracellular solution to elicit larger currents (Mitcheson et al., 200a). Vesnarinone reduced the current magnitude of most mutant channels to an extent similar to that measured for WT HERG (Fig. 8C). However, three channels with Ala missense mutations located in the S6 domain (G648A, F656A, and V659A) and three channels with a mutation located in the base of the pore helix (T623A, S624A, and V625A) were less sensitive to block by 180 μM vesnarinone. However, in contrast to all other drugs that have been investigated, there was little effect of mutating Y652.
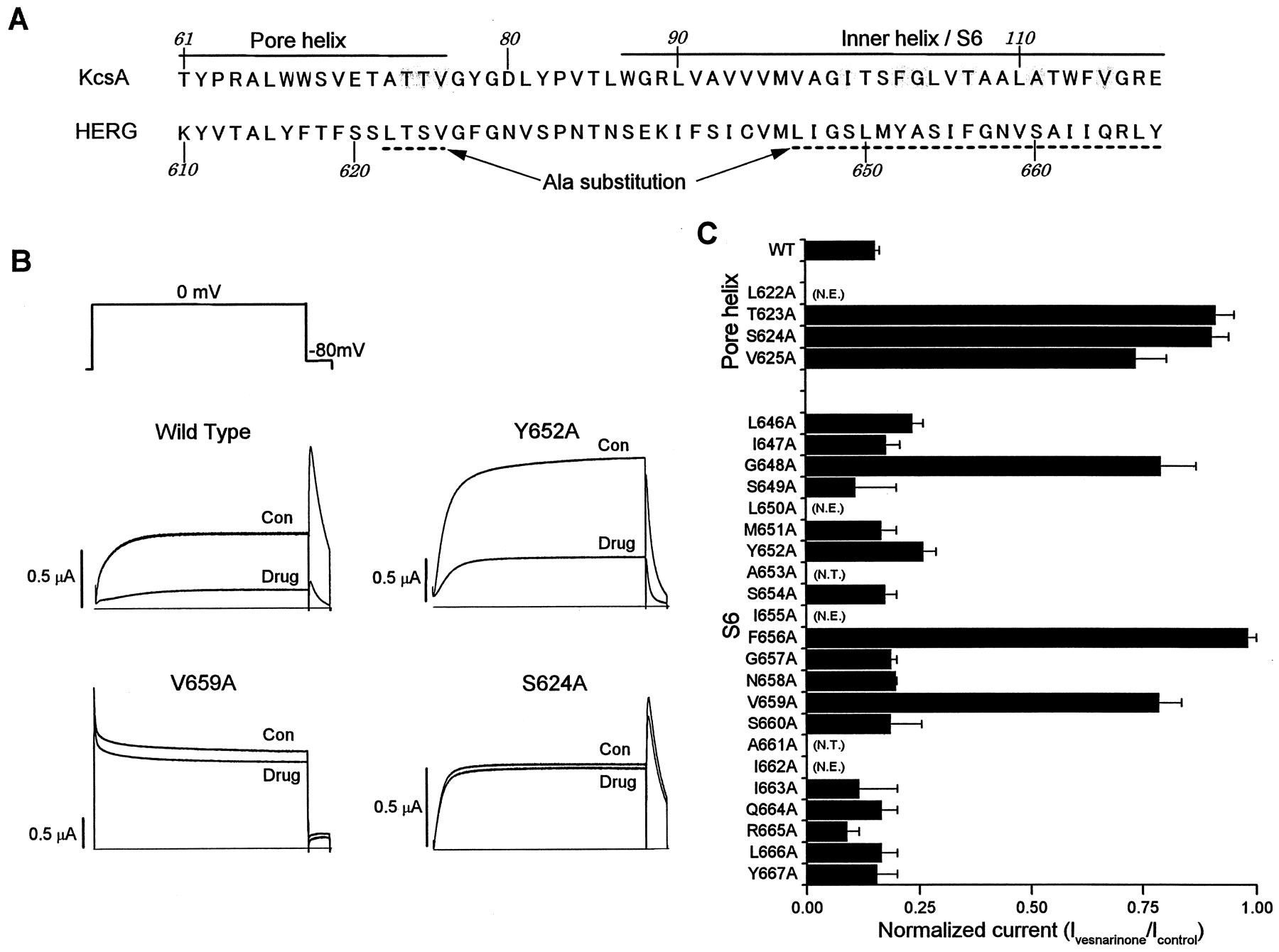
Alanine-scanning mutagenesis of HERG to define binding sites for vesnarinone. A, sequence of the pore helix and inner helix for the KcsA channel (Doyle et al., 1998) and the equivalent residues in the pore helix and S6 transmembrane domain of the HERG K+ channel. Shaded residues of KcsA are predicted to face the inside of the inner channel pore. The region of HERG analyzed by Ala-scanning mutagenesis is dot-underlined. B, block of WT and mutant HERG channel current in oocytes by vesnarinone. HERG channel currents recorded before (Con) and after (Drug) achieving steady-state block of current with 180 μM vesnarinone. From a holding potential of −90 mV, currents were elicited during 5-s pulses to 0 mV applied repetitively at 0.166 Hz. C, normalized current (Ivesnarinone/Icontrol) measured after steady-state block by 180 μM vesnarinone (n = 4 − 5; error bars, +S.E.M.). N.T., residues that were not tested; N.E., mutant channels that lacked functional expression.
Discussion
State-Dependent Block of HERG Channels by Vesnarinone.
Our findings suggest that vesnarinone preferentially blocks HERG channels in the open state. Recovery from block of the HERG current was readily achieved when the membrane was held for a sufficient time at a negative potential. There was no significant decrease in current magnitude if a single, short, depolarizing pulse was subsequently applied, indicating very low affinity of the drug for HERG channels in the rested state. However, a decrease of HERG channel current developed gradually during a maintained depolarization, indicating that for drug-induced block to occur, the channels must first open. Moreover, we found that the drug was equally effective in blocking WT and inactivation-removed (G628C/S631C) HERG channels. Most drugs that block HERG channels have been shown to be far less potent in blocking inactivation-removed channels compared with WT channels (Wang et al., 1996, 1997; Ficker et al., 1998; Lees-Miller et al., 2000).
The slight voltage-dependence of block could easily be explained by the requirement for open channels for efficient block by the drug. The relative lack of voltage-dependent block is in contrast to an earlier study (Katayama et al., 2000) that reported a significantly faster rate of HERG current block onset by vesnarinone at higher voltages when examined in HEK293 cells. Vesnarinone was also 17 times more potent in blocking HERG channel current in these mammalian cells (IC50 = 1.06 μM) than we measured in oocytes (IC50 = 17.7 μM). The greater potency of drug block in mammalian cells compared with oocytes is a common finding and may be related to the lipophilic yolk sac in oocytes that acts to sequester the drug.
The pK a value of vesnarinone is 2.86. At physiological pH, more than 99.99% of the drug is in the neutral form and could presumably diffuse through a lipophilic pathway to reach its binding site within the channel, irrespective of membrane potential or channel state. However, our findings suggest that vesnarinone preferentially blocks HERG channels in the open state. This implies either that access to its binding site occurs only when the activation gate is open or that channel activation causes a conformational change that reveals the high-affinity binding site for the drug. State-dependent access to the drug receptor site is unlikely because recovery from block occurs readily at a negative holding potential. This finding implies that the drug is not trapped by closure of the activation gate and can easily exit the binding site, even when the channel is closed. Recovery from block is another difference between vesnarinone and the more potent blockers of HERG channels such as the methanesulfonanilide class III antiarrhythmic agents (Carmeliet, 1992,1993).
MiRP1 Does Not Affect Block of HERG Channels by Vesnarinone.
Coexpression of MiRP1 with HERG subunits was reported to increase the rate of channel deactivation and increase the potency of channel block by E-4031, an experimental class III antiarrhythmic agent (Abbott et al., 1999). Unlike HERG alone, HERG/MiRP1 channels exhibited tonic block by E-4031. In addition to its affect on HERG, MiRP1 has also been shown to alter the gating of KvLQT1 channels (Tinel et al., 2000). Therefore, we determined whether MiRP1 might affect the block of HERG by vesnarinone. Under the conditions of our study, we found that MiRP1 did not alter the kinetics of HERG current, nor did it affect the potency of block by vesnarinone. Coexpression of MiRP1 with HERG was also recently reported not to affect channel block by dofetilide (Weerapura et al., 2000).
Binding Site for Vesnarinone Is within the Inner Cavity of the HERG Channel.
Alanine-scanning mutagenesis of the HERG subunit revealed that the binding site for vesnarinone shares important similarities with that previously defined for MK-499. Channels containing a single Ala mutation at any one of the three amino acids located at the base of the pore helix (T623, S624, and V625), or of three amino acids located in the S6 domain (G648, F656, and V659), were significantly less sensitive to block by vesnarinone than WT HERG channels. Given the sequence alignment with the KcsA channel, it is clear that all of these residues face the inner cavity of the HERG channel. Mutation of these same residues was also found to decrease the potency of MK-499, although the magnitude of effects was different. For example, mutation of T623 and S624 to Ala had less effect on block by MK-499 than it did for vesnarinone, and the V659A mutation reduced block by vesnarinone more than by MK-499. A striking difference between the two drugs was the finding that Y652 was an important binding site for MK-499 but not for vesnarinone. Y652 is one of two aromatic residues unique to the eag channel family that is important for high-affinity drug binding (Mitcheson et al., 2000a). The lack of interaction of vesnarinone with Y652 might be responsible, in part, for the reduced potency of this drug. Another reason for the decreased potency of vesnarinone might relate to the inability of the drug to be trapped by closure of the activation gate, which can occur with charged drugs such as MK-499 (Mitcheson et al., 2000b). Our previous study also indicated that unlike block by MK-499, block of HERG channels by terfenadine and cisapride were not much affected by mutation of either V625 or G648 to Ala. The finding that vesnarinone also seems to interact with G648 and with residues at the base of the pore helix indicates that such interaction is not unique to methanesulfonanilides, as speculated previously (Mitcheson et al., 2000a).
Clinical Implications.
Most potent blockers of IKr (e.g., dofetilide, E4031) prolong cardiac APD in a reverse-frequency–dependent manner: the faster the heart rate, the less the prolongation of APD (Carmeliet, 1992; Sedgwick et al., 1992; Jurkiewicz and Sanguinetti, 1993). The mechanism of this frequency profile is uncertain, but is not caused by frequency-dependent block of IKr (Jurkiewicz and Sanguinetti, 1993). Whatever the mechanism, it is commonly believed that reverse-frequency–dependent changes in APD may be proarrhythmic because of excessive prolongation of repolarization with bradycardia. A frequency-dependent lengthening of APD such as that caused by vesnarinone would theoretically provide protection against tachyarrhythmias and reduce the risk of proarrhythmia associated with bradycardia (Hondeghem and Snyders, 1990).
Acknowledgments
We gratefully acknowledge the technical assistance of Mayumi Hojo.
Footnotes
- Received March 9, 2001.
- Accepted April 30, 2001.
-
This study was supported by National Institutes of Health (NHLBI) Grant HL55236 (to M.C.S.), a Welcome Trust Fellowship (to J.S.M.), and a grant-in-aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture, Japan (to K.K.).
Abbreviations
- APD
- action potential duration
- PDE
- phosphodiesterase
- WT
- wild-type
- MES
- 2-(N-morpholino)ethanesulfonic acid
- τ
- time constant
- I-V
- current-voltage
- human ether-a-go-go-related gene
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}