


Abstract
GABA transporters control extracellular GABA levels by coupling transmitter uptake to the sodium and chloride cotransport. The rat brain GABA transporter GAT1 and other members of this family are regulated by direct interactions with syntaxin 1A, a protein involved in vesicle docking and in the regulation of several ion channels and transporters. We have shown previously that syntaxin 1A exerts its effects on GAT1 by decreasing the net uptake of GABA and its associated ions through interactions with aspartic acid residues in the N-terminal tail of GAT1. This reduction in net uptake could be caused by many steps in the transport cycle, including substrate binding, substrate flux, substrate efflux, or reorientation of the unliganded transporter. To address this question, we performed GABA flux assays, measured flux- and efflux-associated ion currents, and assessed GABA exchange in multiple experimental systems expressing syntaxin 1A and wild-type GAT1 or GAT1 mutants. Syntaxin 1A caused similar reductions in forward and reverse transport that did not involve changes in apparent transport affinities for sodium, chloride, or GABA. The syntaxin 1A-mediated reduction in GABA flux and efflux was mimicked by mutations in GAT1 at the syntaxin 1A binding site. The binding site GAT1 mutant also caused a reduction in exchange. These data suggest that syntaxin 1A exerts its effects, directly or indirectly, on GAT1 function through interactions with GAT1's N-terminal tail and that the inhibition occurs at a step in the translocation process after substrate binding but which involves both unidirectional transport and transmitter exchange.
GABA is the major inhibitory neurotransmitter in the central nervous system. Its action is terminated by GABA transporters, integral membrane proteins located on neurons and glia. Recent evidence suggests that the function of GABA transporters, and the function of other transporter family members, is altered by a variety of mechanisms (for review, see Blakely and Bauman, 2000; Robinson, 2002). The various forms of regulation affect transporter function in two ways: altering the number of transporters on the cell surface or altering the transport process directly. Transporter function is affected through interactions with the plasma membrane protein syntaxin 1A. The transporters for GABA (Beckman et al., 1998), glycine (Geerlings et al., 2000), serotonin (Haase et al., 2001; Quick, 2002a), and norepinephrine (Sung et al., 2003) all show syntaxin-mediated transport regulation.
Syntaxin 1A was originally characterized as a component of the machinery involved in transmitter release (Bennett et al., 1993; Söllner et al., 1993), and is a key player in the SNARE hypothesis of vesicle trafficking and fusion (Südhof, 1995). Syntaxin 1A directly interacts and functionally regulates several excitability proteins, including Ca2+ channels (Sheng et al., 1994; Bezprozvanny et al., 1995), cystic fibrosis transmembrane regulator Cl- channels (Naren et al., 1997), K+ channels (Fili et al., 2001), and epithelial Na+ channels (Qi et al., 1999; Saxena et al., 1999). The mechanisms of the effects of syntaxin 1A on these channels include changes in protein trafficking and unitary channel properties. For example, syntaxin 1A promotes slow inactivation gating of calcium channels (Bezprozvanny et al., 1995). For GAT1, syntaxin 1A inhibits GABA uptake in hippocampal neurons that endogenously express both proteins, and in heterologous systems in which both proteins are expressed. Electrophysiological approaches in oocytes expressing GAT1 and syntaxin 1A reveal that the net effect of syntaxin 1A expression is an approximate 4-fold reduction in GAT1 turnover rates (Deken et al., 2000), the turnover rate being the net rate at which GABA and associated ions are translocated across the membrane. However, the functional and structural mechanisms by which syntaxin 1A achieves this reduction in net turnover rates remains unknown.
An important question in the field of neurotransmitter transport is how transporters mediate the translocation of substrates, and a number of structural models have been proposed for this process (Hilgemann and Lu, 1999; DeFelice et al., 2001). Functional schemes (Hilgemann and Lu, 1999) assume that GAT1 exists in a conformation in which it can bind two Na+, one Cl-, and a GABA molecule extracellularly. This binding then induces a conformational change in which these molecules are translocated to the cytoplasmic face and released. The unliganded transporter then reorients its substrate binding sites to the extracellular milieu. [This reduced model ignores many interesting features such as the order of substrate loading and unloading (Hilgemann and Lu, 1999), and the evidence that Cl- may be exchanged (Loo et al., 2000).] The inhibition of GAT1 turnover rates by syntaxin 1A could be caused by effects at any of these steps. Also of interest are the domains of the transporter that relate to these various steps. It has been shown that specific residues in the N-terminal cytoplasmic tail of GAT1 probably exert their effects on transport by regulating the transporter reorientation step (Bennett et al., 2000). These residues are in the region in which syntaxin 1A binds GAT1 (Deken et al., 2000), thus raising the possibility that syntaxin 1A inhibits GAT1 function through a similar mechanism. Thus, we wished to address the following questions: Is the inhibition of transport of syntaxin 1A mediated by changes in substrate binding, translocation, or transporter reorientation? Are the effects of syntaxin 1A mimicked by mutations of GAT1 residues at the site of syntaxin 1A binding?
Materials and Methods
Reagents. Immunoblotting reagents and [3H]GABA were obtained from Amersham Biosciences Inc. (Piscataway, NJ). Botulinum toxin C1 (BoTx/C1) was obtained from Roche Diagnostics (Indianapolis, IN). Biotinylation reagents were obtained from Pierce Chemical (Rockford, IL). GAT1 antibody 346J was obtained from Dr. Nicholas Brecha (University of California, Los Angeles, CA); isoform-specific polyclonal syntaxin antibodies were generated as described previously (Naren et al., 1997). GAT1 mutants at or near the syntaxin 1A binding site (Deken et al., 2000) were generated and sequenced in both directions to verify proper mutagenesis. All other reagents were obtained from Sigma-Aldrich (St. Louis, MO).
Xenopus laevis Oocyte Preparation and cRNA Injection.X. laevis oocytes were prepared as described previously (Quick and Lester, 1994). Briefly, oocytes were defolliculated in collagenase A and maintained at 18°C in ND96 (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, 5 mM HEPES, and 1.8 mM CaCl2) supplemented with 2.5 mM sodium pyruvate, 50 μg/ml gentamycin, and 5% horse serum. The medium was changed daily. Expression was found to be maximal 4 to 7 days after injection; all experiments were carried out during that time.
Cell Culture. Primary hippocampal cultures were prepared from postnatal days 0 to 3 rats by mincing tissue in α minimal essential medium supplemented with cysteine, glucose, and 100 U of papain. Tissue was incubated for 20 min at 37°C followed by gentle trituration, dilution, and plating onto poly-l-lysine-coated glass coverslips. To obtain pure neuronal cultures, mixed cultures were treated for 48 h with 10 μM cytosine arabinoside; treatment was initiated beginning 24 h after plating.
[3H]GABA Flux Assays. To measure GABA uptake in non-voltage-clamped oocytes, individual oocytes were placed in tubes or multiwell plates in ND96. At timed intervals, GABA was added at various concentrations; the concentration of [3H]GABA was 40 nM. The reaction was terminated by rapid removal of the oocyte followed by six washes in ice-cold ND96. Subsequently, the oocyte was solubilized in 10% SDS at 60°C for 2 h and [3H]GABA uptake was determined by liquid scintillation spectrometry. For all uptake experiments, specific uptake at each concentration was determined by subtracting total uptake from nonspecific uptake measured in sister oocytes from the same oocyte batch. For hippocampal neurons, cells were rinsed twice in 1× Hanks' balanced salt solution (HBSS) and allowed to equilibrate for 10 min in the final wash. Buffer was then exchanged with control HBSS or drug-containing HBSS; preincubation times were 10 to 30 min. [3H]GABA was added to initiate the assay. The final [3H]GABA concentration was 30 nM, and assay times were 10 to 60 min. The assay was terminated by rapidly rinsing the cells three times with 1× HBSS, followed by solubilization in 300 μl of 0.001 to 0.005% SDS at 37°C for 2 h. To assess [3H]GABA efflux in hippocampal neurons, cells were preloaded by incubating the cultures for 4 h in 300 nM [3H]GABA. The cells were washed twice in HBSS and then incubated in 250 μl of HBSS for 10 min. The amount of [3H]GABA present in the incubation buffer and the amount remaining sequestered in cells was quantified. Under control conditions, approximately 4% of the preloaded GABA was released during the assay. Statistical analyses of the uptake data were performed using SPSS (SPSS Science, Chicago, IL). Two-sample comparisons were made using t tests; multiple comparisons were made using one-way analyses of variance followed by Tukey's honestly significant difference post hoc test.
Electrophysiology. Two-electrode voltage clamp was performed using a GeneClamp amplifier and Axoscope 6.0 software (Axon Instruments, Union City, CA). Data were digitized at 10 Hz and stored on disk. Simultaneous chart-recorder traces were also made. Control recording buffer was ND96. To minimize liquid junction potentials, all anion substitution experiments under voltage-clamp were performed using a 3 M KCl agar bridge. Currents were recorded under continuous perfusion using gravity flow. The flow rate was approximately 12 ml/min; bath exchange was complete in 2 s. GABA efflux measurements were performed by injecting GABA into oocytes under voltage clamp. For concentration estimates, the volume of the oocyte was assumed to be 500 nl.
Measurements of GAT1 charge movements were as described previously (Quick, 2002b). Charge movements were measured during a 500-ms voltage step from -40 to -100 mV. Each oocyte was tested in the presence and absence of SKF89976A to isolate (by subtraction) the charge movements associated with the presence of GAT1 in the plasma membrane. Capacitative transients generated by these jumps were integrated to yield the amount of charge movement in and out of the oocyte's membrane field. Surface transporter number was calculated from the equation N = Qmax/qsδ, where N is the number of transporters/oocyte, Qmax is the total charge movement, q is the elementary charge, and zδ is the sum total of the distance that all charges move within the membrane field. The empirical value of zδ for GAT1 transporter is approximately 1.0 (Mager et al., 1993). Surface transporter number was divided into peak steady-state current, measured at saturating GABA concentrations, to yield the transport rate. This rate was also assessed by measuring the time constant (estimated by single exponential fits) of the transient relaxations of voltage jumps (-40 to -100 mV) performed in the presence of 100 μM GABA (Deken et al., 2000).
Reconstitution and Exchange Assays. Reconstitution of GAT1 into liposomes and subsequent exchange assays were performed as described previously (Bennett et al., 2000). cDNAs encoding wild-type or mutant GAT1 were transfected into human embryonic kidney 293 cells. Cells were rinsed twice in saline and then taken up in 20 μl of PBS. The cell suspension was mixed with liposomes (4 μmol of asolectin, 0.7 μmol of brain phospholipids) and 0.9% cholate in a final volume of 220 μl. The reconstitution columns were equilibrated with 5 mM Tris-SO4, pH 7.4, 1 mM MgSO4, 0.5 mM EDTA, and 1% glycerol. For the exchange reaction, the spin column contained an internal solution consisting of 120 mM NaCl, 20 mM NaPi, pH 7.4, 5 mM Tris-SO4, 0.5 mM EDTA, 1 mM MgSO4, 1% glycerol, and 10 mM unlabeled GABA; for control experiments, GABA was excluded from the spin column. The external solution contained 120 mM NaCl, 20 mM NaPi, pH 7.4, 5 mM Tris-SO4, 0.5 mM EDTA, 1 mM MgSO4, 1% glycerol, and 100 nM [3H]GABA. Assays were performed for 2 min.
Biotinylation and Immunoprecipitation. Surface biotinylation experiments were performed as described previously (Whitworth and Quick, 2001). Briefly, the cells were rinsed twice with 37°C PBS/Ca2+/Mg2+ (138 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 9.6 mM Na2HPO4, 1 mM MgCl2, and 0.1 mM CaCl2, pH 7.4). The cells were next incubated with 2 ml of a solution containing 1 mg/ml sulfo-N-hydroxysuccinimidobiotin in PBS/Ca2+/Mg2+ for 20 min at 4°C with gentle shaking. The biotinylation solution was removed by two washes in PBS/Ca2+/Mg2+ plus 100 mM glycine and quenched in this solution by incubating the cells at 4°C for 45 min with gentle shaking. The cells were lysed with 1 ml of radioimmunoprecipitation assay buffer (100 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 250 μM phenylmethylsulfonyl fluoride) at 4°C for 60 min. The cell lysates were centrifuged at 20,000g and 4°C for 60 min. The supernatant fractions (300 μl) were incubated with an equal volume of Immunopure immobilized monomeric Avidin beads at room temperature for 60 min. The beads were washed three times with radioimmunoprecipitation assay buffer and adsorbed proteins were eluted with SDS sample buffer (62.5 mM Tris-Cl, pH 6.8, 2% SDS, and 100 mM β-mercaptoethanol) at room temperature for 30 min. The product was washed in buffer, and run on a 10% acrylamide gel. Protein was transferred to a nitrocellulose membrane and visualized using enhanced chemiluminescence reagents. Anti-actin antibodies were used to verify that the biotinylated fraction was not staining intracellular proteins.
Results
Treatment of dissociated primary hippocampal cultures, which endogenously express GAT1 and syntaxin 1A, with BoTx/C1, an endotoxin that specifically cleaves the plasma membrane SNARE protein syntaxin 1A (Blasi et al., 1993), caused an increase in the amount of GABA taken up by these cultures (Fig. 1A). This effect was probably mediated through the predominantly neuronal GABA transporter GAT1 because SKF89976A, an antagonist with high selectivity for GAT1, almost completely inhibited GABA uptake. Treatment of parallel cultures with botulinum neurotoxin B, an endotoxin that specifically cleaves the synaptic vesicle SNARE protein vesicle-associated membrane protein (synaptobrevin; Schiavo et al., 1992, had no effect on GABA uptake. The effect of BoTx/C1 was concentration-dependent (Fig. 1B) with a maximal dose (1 ng/ml and above) causing a 2-fold increase in uptake. These results replicate previous findings (Deken et al., 2000) and further support the idea that syntaxin 1A is a negative regulator of GABA uptake.

Botulinum toxin C1 regulates GABA flux independent of surface GAT1 levels. A, BoTx/C1 up-regulates GABA uptake in neuronal cultures. Drug and toxin concentrations are shown below the abscissa and were applied to cultures 15 min before assay. Data are from four separate experiments, three wells/condition/experiment. GABA uptake under control conditions ranged from 513 to 787 fmol/min/mg of protein. B, BoTx/C1 regulation of uptake is concentration-dependent. Experiments at different toxin concentrations were performed as in A. C, saturation analysis of BoTx/C1-mediated changes in transport. Uptake assays at various GABA concentrations were performed on cultures incubated in control medium (○) or medium containing 1 ng/ml BoTx/C1 (•). Data shown are from one experiment, six wells/concentration; comparable results were obtained in two other experiments. D, BoTx/C1 decreases steady-state surface GAT1 levels. Hippocampal cultures were preincubated for 15 min before biotinylation in control medium (untreated) or medium containing 1 ng/ml BoTx/C1. Representative immunoblot shows GAT1 immunoreactivity in total cell lysates and surface (biotinylated) fractions. Data plotted in the graph are from densitometry measurements made from three separate experiments and plotted as a percent of total immunoreactivity.
Changes in uptake induced by BoTx/C1 could be caused by alterations in the turnover rate of individual transporters or by altering the number of functional transporters. Thus, substrate dose-response curves were used to determine both the maximum transport capacity (Vmax) and GABA affinity (Km) of BoTx/C1-treated cultures (Fig. 1C). Eadie-Hofstee transformations (not shown) of the saturation data from three experiments revealed Vmax values of 144 ± 22 fmol/min/mg protein (untreated cells) and 321 ± 28 fmol/min/mg protein (BoTx/C1-treated cells). Km values, which were not significantly affected by BoTx/C1, were 5.2 ± 1.4 and 4.7 ± 1.9 μM for untreated and BoTx/C1-treated cultures, respectively. These data show that BoTx/C1 reduces GABA uptake approximately 2-fold across all GABA concentrations. These same cultures in which uptake was performed were subjected to surface biotinylation labeling and Western blot analysis to determine the role of syntaxin 1A on the subcellular distribution of GAT1 protein (Fig. 1D). In untreated control cultures, approximately 50% of GAT1 immunoreactivity was found in the biotinylated (surface) fraction. Interestingly, in BoTx/C1-treated cultures, the amount of surface GAT1 immunoreactivity was reduced by half, suggesting that syntaxin 1A is a positive regulator of GAT1 expression. Given that GABA uptake is reduced 2-fold and that surface protein levels are increased by 2-fold, these data indicate (assuming that the surface fraction of GAT1 represents the population of functional transporters) that syntaxin 1A is responsible for a reduction in GABA uptake of approximately 4-fold for a given number of GAT1 proteins. That is, syntaxin 1A causes a 4-fold reduction in the rate at which GAT1 transports GABA.
To examine in more detail the role of syntaxin 1A in regulating GAT1, we expressed recombinant GAT1 and syntaxin 1A in X. laevis oocytes and examined GABA-mediated responses electrophysiologically using two-electrode voltage clamp (Fig. 2). Oocytes expressing both proteins produced inward currents in response to superfusion of GABA at negative holding potentials (Fig. 2A, ○), which were sodium-dependent and blocked by SKF89976A (data not shown). These oocytes were then injected with BoTx/C1 and remeasured 15 min later under the same conditions (Fig. 2A, •). In oocytes coexpressing GAT1 and syntaxin 1A, BoTx/C1 caused an approximate 2-fold, voltage-independent increase in GABA-induced current mediated by the transporter, comparable with the 2-fold increase seen in hippocampal cultures treated with BoTx/C1.

Botulinum toxin C1 regulates GABA flux in X. laevis oocytes expressing GAT1 and syntaxin 1A. A, BoTx/C1 regulation of GABA-mediated currents is voltage independent. Representative current-voltage plot from one oocyte measured before and 20 min after injection of 50 nl of 50 ng/ml BoTx/C1. The GABA concentration was 100 μM. B, BoTx/C1 reverses syntaxin 1A-mediated reductions in GAT1 turnover rates. Turnover rates were calculated by dividing peak GABA-mediated currents by transporter number estimated from voltage-jump experiments performed as described under Materials and Methods. Data are from at least nine oocytes per condition. C, BoTx/C1 does not alter Qmax in oocytes expressing GAT1 alone. Individual oocytes expressing GAT1 were subjected to voltage-jump analysis before and 10 min after injection of 50 nl of 50 ng/ml BoTx/C1. Lines on the plot connect data obtained from the same oocyte.
We subjected these same oocytes (before and after BoTx/C1 treatment) to voltage-jump experiments in the absence of GABA to obtain an estimate of the number of functional transporters. The rationale for these experiments and verification of the method has been covered in detail previously (Deken et al., 2000; Quick, 2002b). Dividing whole-cell GABA-induced currents by the number of transporters in the same oocyte yields the turnover number (i.e., the rate of transporter cycling). In oocytes expressing GAT1, under our experimental conditions, the turnover rate was approximately 7 s-1 (Fig. 2B, ○; Mager et al., 1993; Deken et al., 2000). In oocytes expressing GAT1 and syntaxin 1A, the turnover rate was approximately 4-fold less, and BoTx/C1 reversed this inhibition in a dose-dependent manner (Fig. 2B, •). This change in rates was probably not a result of nonspecific effects of BoTx/C1 on GAT1 charge movements because oocytes expressing GAT1 alone showed comparable maximal charge movements before and after injection with BoTx/C1 (Fig. 2C). The 4-fold reduction in rates seen in oocytes injected with GAT1 and syntaxin 1A was comparable with that seen in hippocampal neurons, suggesting that the oocyte was serving as a good model for syntaxin 1A-mediated inhibition of GAT1.
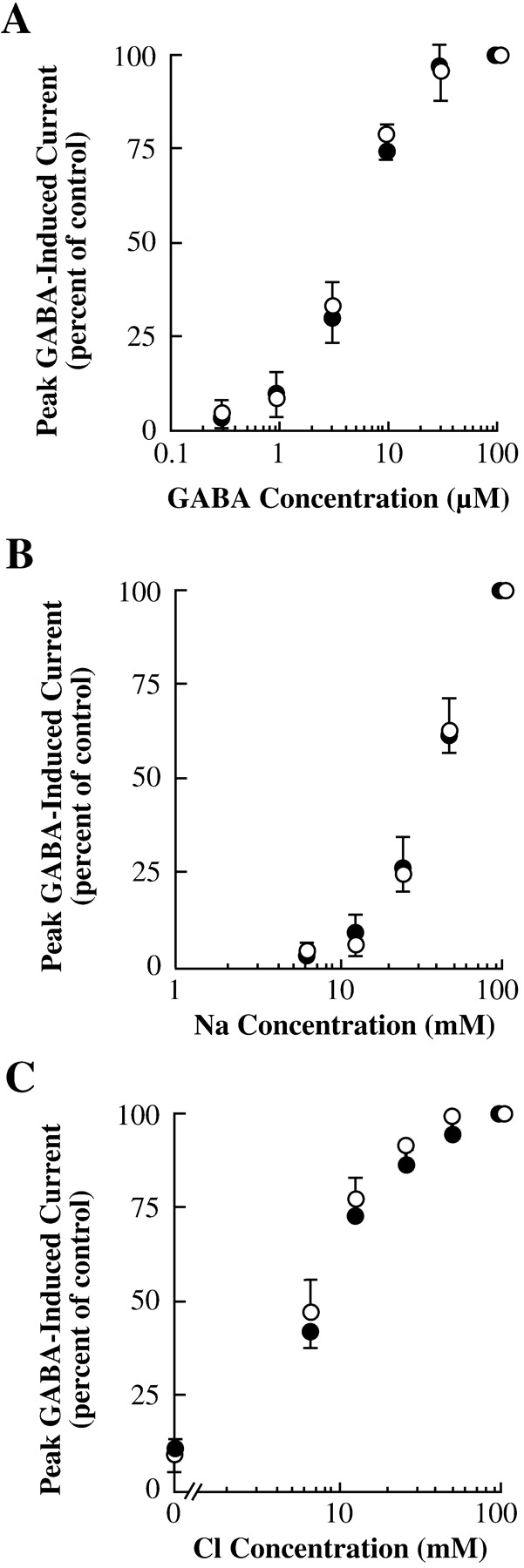
This reduction in net transport rate could be caused by effects on many different steps in the transport cycle, including substrate binding, substrate flux, reverse transport, or reorientation of the unliganded transporter. Forward transport by GAT1 requires the binding and subsequent translocation of Na+, Cl-, and GABA; thus, syntaxin 1A could be exerting its effects by shifting the apparent affinity for any of the transporter's requisite substrates. To examine this possibility, we measured GABA-induced currents in oocytes expressing GAT1 and syntaxin 1A at various substrate concentrations (Fig. 3). In oocytes expressing GAT1 alone (○) or GAT1 plus syntaxin 1A (•), there was no difference in the concentrations of GABA (Fig. 3A), Na+ (Fig. 3B), or Cl- (Fig. 3C) required to elicit GABA-mediated inward ion flux. These data suggest that syntaxin 1A affects GAT1-mediated forward transport at a step distinct from substrate binding.

Syntaxin 1A inhibition of GAT1 inward flux is independent of substrate concentrations. Each oocyte expressing GAT1 alone (○) or GAT1 plus syntaxin 1A (•) was measured (holding potential, -80 mV). Experiments were performed using various concentrations of GABA (A), Na+ (B), and Cl- (C). In B and C, to maintain proper osmolarity, Na+ was replaced by choline and Cl- was replaced by acetate, respectively. The GABA concentration for these experiments was 100 μM. Data are from at least six oocytes per data point.
The reduction in rates of transport, but not in relative apparent binding affinities, suggested alterations at the translocation step or in reorientation of the unbound transporter. Thus, we next examined reverse transport in the presence of syntaxin 1A (Fig. 4). First, we examined efflux of [3H]GABA that was preloaded into hippocampal cultures (Fig. 4A). As seen in the flux experiments (Fig. 1A), incubation of these preloaded cultures with BoTx/C1 induced an approximate 2-fold increase in efflux that was SKF89976A-sensitive; botulinum neurotoxin B had no effect on efflux.

Syntaxin 1A inhibits GABA-mediated efflux. A, BoTx/C1 up-regulates [3H]GABA efflux in hippocampal neurons. Drug and toxin concentrations are shown below the abscissa. Toxins were applied to cultures 15 min before assay; SKF89976A was present only during the assay. B, raw data trace of GABA-mediated outward currents. Oocytes were injected with GAT1 and syntaxin 1A cRNAs 3 to 5 days before assay. Fifty nanoliters of 50 ng/ml BoTx/C1 was injected into the oocyte 30 min before assay. To assay efflux, oocytes were bathed in an extracellular medium in which 95% of the Na+ was replaced by choline. The assay was initiated by injection (left arrow) of approximately 1/10 oocyte volume of 10 mM GABA and 800 mM NaCl, and currents were assessed at 40 mV. To verify specificity, efflux was terminated (right arrow) by superfusion of 100 μM extracellular SKF89976A. C, GABA-induced outward currents are reduced by syntaxin 1A. Representative current-voltage plot from one oocyte measured before and 20 min after injection of 50 nl of 10 ng/ml BoTx/C1. D, BoTx/C1 reverses syntaxin 1A-mediated reductions in GAT1 turnover rates. Efflux turnover rates were calculated by dividing peak GABA-mediated outward currents by transporter number estimated from voltage-jump experiments performed as described under Materials and Methods. Data are from at least 12 oocytes per condition.
We also took advantage of the fact that we can inject substrates into the oocyte while the oocyte is voltage-clamped. An example of one such efflux experiment is shown in Fig. 4B. To enhance efflux, the oocyte was voltage-clamped at positive holding potentials, bathed in saline in which almost all extracellular Na+ was replaced by choline, and injected with NaCl and GABA to final estimated concentrations of approximately 80 and 1 mM, respectively. We confirmed that the specificity of the outward currents was caused by efflux through the transporter by adding SKF89976A to the bath, which eliminated the efflux current. In oocytes expressing GAT1 alone, outward currents were unaffected by injection of BoTx/C1. In oocytes coinjected with GAT1 and syntaxin 1A cRNA, there was a significant reduction in efflux compared with oocytes injected with GAT1 alone. More importantly, injection of BoTx/C1 caused a significant increase in efflux. As with GABA flux, syntaxin 1A caused an approximate 2-fold decrease in GABA efflux that was voltage-independent (Fig. 4C). Measurement of charge movements (as in Fig. 2B) in addition to the efflux current allowed us to estimate GAT1 turnover rates (Fig. 4D). As with flux, syntaxin 1A induced an approximate 4-fold reduction in rates of GABA translocation that could be reversed by injection of BoTx/C1. Together, these data show that syntaxin 1A comparably inhibits GABA flux and efflux through GAT1.
To determine whether the syntaxin 1A-mediated reduction in efflux was a result of changes in the apparent affinity of substrate binding to the intracellularly facing transporter, we measured GABA-induced efflux currents after injection of various concentrations of GABA (Fig. 5A) and Na+ (Fig. 5B). There was significant variability in the results, probably because of variability in injecting substrates into the oocyte, and the fact that each oocyte yielded only one datum. However, the results suggested that there was little difference in apparent substrate affinities for oocytes expressing GAT1 alone or with syntaxin 1A.

Syntaxin 1A inhibition of GAT1 outward flux is independent of substrate concentrations. Each oocyte expressing GAT1 alone (○) or GAT1 plus syntaxin 1A (•) was measured. To assay efflux, oocytes were bathed in an extracellular medium in which 95% of the Na+ was replaced by choline. The assay was initiated by injection of approximately 1/10 oocyte volume of various 10× concentrations of GABA (A) or NaCl (B). Currents were assessed at 40 mV. In B, to maintain proper osmolarity, Na+ was replaced by choline. The GABA concentration for the data in B was approximately 1 mM. Data are from at least eight oocytes per data point.
We next tested the hypothesis that the reduction in both flux and efflux was caused by effects at the syntaxin 1A binding site in the N-terminal tail of GAT1; that is, we examined whether the effects of syntaxin 1A were mimicked by mutation of the aspartic acid residues in GAT1 that mediate syntaxin binding (Deken et al., 2000). If this were true, then a GAT1 construct lacking these residues, in the absence of syntaxin 1A, would also show reduced inward and outward fluxes. Results from this experiment, expressed as turnover rates, are shown in Fig. 6. As shown previously in Fig. 2, oocytes expressing wild-type GAT1 showed reduced turnover rates for forward transport when syntaxin 1A was coexpressed (Fig. 6A). And as shown previously in Fig. 4, oocytes expressing wild-type GAT1 showed reduced turnover rates for reverse transport when syntaxin 1A was coexpressed (Fig. 6B). Similar results were seen for the control construct 2A-GAT1 (G39A, L41A), which served as a negative control for effects of mutagenesis near the syntaxin 1A binding site on GAT1.

Mutations of GAT1 at the syntaxin 1A binding site reduce both flux and efflux turnover rates. Oocytes expressing various GAT1 constructs, with or without coexpression of syntaxin 1A, were examined for inward (A) and outward (B) GABA-mediated currents. Inward currents were assessed at -80 mV using 100 μM GABA. Outward currents were assessed at 40 mV using the procedure described in Fig. 4. Turnover rates were calculated by dividing peak GABA-mediated currents by transporter number estimated from voltage-jump experiments performed as described under Materials and Methods. Data are from at least seven oocytes per condition.
For the syntaxin 1A binding site mutant construct 3D-GAT1 (D40A, D43A, D45A), inward turnover rates were reduced, as shown previously (Deken et al., 2000). There was no further effect on inward flux with syntaxin 1A coexpression, consistent with the fact that syntaxin 1A binding to GAT1 is almost completely eliminated in this construct. The 3D-GAT1 construct also showed reduced efflux, which was, as expected, unaffected by syntaxin 1A coexpression. These data suggest that mutation of the syntaxin 1A binding site on GAT1 are sufficient to mimic the syntaxin 1A effects on forward and reverse transport.
The data to this point showed that both forward and reverse GAT1-mediated transport were equally reduced by either coexpression of syntaxin 1A or by mutations in GAT1 at the syntaxin 1A binding site. These reductions were probably not a result of changes in apparent affinities for substrate binding and raised the likelihood that the reduction was due either to a slowing of the translocation step after substrate binding or to a slowing of the return of the unloaded transporter to its opposite facing conformation. To distinguish between these two steps, we performed GABA exchange experiments (Fig. 7). The rationale for this idea comes from experiments in which mutation of specific residues in GAT1 was shown to affect the reorientation step, thus altering unilateral GABA flux but not substrate exchange (i.e., the unloaded reorientation step is not required for exchange) (Bennett et al., 2000).

Mutations of GAT1 at the syntaxin 1A binding site reduce GABA exchange. Proteoliposomes were prepared from human embryonic kidney-293 cells expressing wild-type or mutant GAT1. A, characterization of exchange measured for wild-type GAT1. Assays were performed in the presence of various external solutions containing [3H]GABA (abscissa), and in the absence (▪) or presence (□) of internal unlabeled GABA. B, GABA exchange measured for various GAT1 constructs. Assays were performed in the presence of external NaCl and [3H]GABA, and internal unlabeled GABA. Data are plotted relative to exchange measured for wild-type GAT1.
To verify that we could accurately assess GABA exchange, liposomes prepared with wild-type GAT1 were examined (Fig. 7A). The liposomes were prepared with or without unlabeled GABA present intracellularly; [3H]GABA was present extracellularly. Significant [3H]GABA uptake into liposomes only occurred when NaCl was present extracellularly and unlabeled GABA was present intracellularly. Because the NaCl concentration was equal on each side of the membrane, this accumulation of intracellular [3H]GABA lacked the driving force necessary for forward flux; the requirement for intracellular GABA in mediating this accumulation makes it likely that it was caused by exchange (Bennett et al., 2000). The replacement of extracellular Na+ or Cl- with choline or acetate, respectively, eliminated the [3H]GABA accumulation.
We then examined exchange in liposomes prepared from various GAT1 constructs (Fig. 7B). All constructs were examined in the presence of extracellular NaCl and [3H]GABA, and in the presence of intracellular unlabeled GABA. Compared with wild-type GAT1, 3D-GAT1 showed reduced exchange. This reduction in exchange was also distinct from exchange seen in the GAT1 R44K construct, which was shown previously to have reduced flux but normal exchange (Bennett et al., 2000). Together, these data suggest that either coexpression of syntaxin 1A or mutations in GAT1 at the syntaxin 1A binding site cause comparable reductions in GABA flux, efflux, and exchange.
Discussion
Neurotransmitter transporter function is subject to control in multiple ways (for review, see Blakely and Bauman, 2000; Robinson, 2002). Recent work has led to an appreciation of transporters as components of protein complexes and the roles that these complexes play in transporter regulation. Amine transporters interact with protein phosphatase 2A, which provides a signaling complex for kinase-mediated changes in transporter function (Bauman et al., 2000). Amine transporters also interact with the scaffold protein PICK1, which results in an increase in transporter uptake capacity (Torres et al., 2001). Such changes in function correlate with changes in expression of transporters on the cell surface, suggesting that protein-protein interactions influence transporter trafficking. Changes in subcellular transporter distribution also occur because of interactions of transporters with syntaxin 1A (Beckman et al., 1998; Geerlings et al., 2000; Haase et al., 2001; Quick, 2002a; Sung et al., 2003). Given the fact that transporter structure must map in some way onto the process of substrate transport, direct protein-protein interactions also raise the formal possibility that these transporter interacting proteins not only affect transporter trafficking but also substrate translocation. This aspect of regulation has been less well elucidated. Although syntaxin 1A is known to reduce net GABA flux through GAT1 (Deken et al., 2000), such inhibition could occur at any of several steps in the transport cycle. In the present experiments, we show that GABA flux, efflux, and exchange are all reduced by syntaxin 1A or by mutations in the N-terminal of GAT1 at the syntaxin 1A site of interaction.
At which step(s) in the transport cycle does syntaxin 1A exert its effects? Given that flux and efflux were affected by syntaxin 1A but apparent substrate affinities for these processes remained unchanged, it suggests that the effects are unrelated to Na+, Cl-, or GABA binding on either side of the membrane. GABA exchange was also reduced, which suggests that reorientation of the unliganded transporter is not necessarily affected, because exchange reactions bypass this step. The most parsimonious explanation is that syntaxin 1A exerts its effects by inhibiting substrate translocation, a step common to all three affected processes. One possibility is that syntaxin 1A acts to hinder conformational changes that are required for transport after substrate binding. Alternatively, syntaxin 1A could be exerting effects on multiple steps in the transport process but which our outcome measures are inadequate to resolve. For example, any effect on a reorientation step would conceivably be masked by an additional effect on a substrate translocation step. The fact that mutations near the syntaxin 1A binding site on GAT1 affect the reorientation step (Bennett et al., 2000) make it quite possible that syntaxin 1A affects this step as well. Also unresolved is the manner of syntaxin 1A inhibition. The 80% reduction in GABA transport could be due either to partial inhibition of all transporters, or complete inhibition of 80% of the transporters. The latter result would suggest that syntaxin 1A interactions place GAT1 in an inactive transporter state or rather that syntaxin 1A favors binding to GAT1 in its inactive conformation.
The focus of the present experiments has been on the role that syntaxin 1A plays in regulating GAT1-mediated substrate translocation rates. Thus, the role of syntaxin 1A in regulating transporter expression was either not an important variable (e.g., in determining apparent substrate affinities) or was accounted for (e.g., in calculating the turnover number). However, as part of the SNARE complex syntaxin 1A regulates membrane trafficking (Südhof, 1995), and its role in regulating GAT1 trafficking in oocytes (Quick et al., 1997), in mammalian cell lines (Beckman et al., 1998), and in hippocampal neurons (Horton and Quick, 2001) has been documented in detail previously. Consistent with its role in enhancing membrane fusion events, syntaxin 1A is a positive regulator of GAT1 surface expression in neurons (Horton and Quick, 2001). Thus, it is unlikely that the decrease in transport rates seen in the present study is a result of the effects of syntaxin 1A on membrane trafficking, because these effects oppose one another. The physiological relevance of these opposing effects are unknown. One possibility is that these two processes are temporally distinct and thus are not truly in opposition. Another possibility is that neurons use syntaxin 1A to increase surface GAT1 expression but keep the transporter functionally suppressed until a time when removal of GABA becomes a priority. This latter hypothesis is consistent with data showing that substrates of the transporter reverse syntaxin 1A inhibition of transport (Quick, 2002b).
Syntaxin 1A directly binds GAT1 (Deken et al., 2000), and mutations at this binding site in GAT1 mimic the effects of syntaxin 1A on transport. However, this does not prove that a direct interaction is responsible for the inhibition of GAT1 turnover rates. Syntaxin 1A is a highly interactive component of multimeric brain complexes (Südhof, 1995; Rothman, 1996), and thus its role in inhibiting transport could very well be mediated indirectly. Some support for direct interactions mediating changes in GAT1 translocation rates would come from identifying unique residues in syntaxin 1A that bind GAT1. We are presently addressing this issue with the goal of creating syntaxin 1A constructs that cannot bind GAT1 but function normally in all other interactions. This approach is similar to that done for identifying syntaxin 1A constructs that cannot bind cystic fibrosis transmembrane regulator, but show wild-type binding to SNARE complexes and calcium channels (Ganeshan et al., 2003). Regardless of whether the effects of syntaxin 1A on GAT1 are direct or indirect, this form of regulation is intriguing because of syntaxin 1A's ability to be influenced by multiple molecules and binding partners, and because of its role in transmitter release. Thus cells, through syntaxin 1A, would be able to coordinate aspects of both transmitter release and clearance.
Because the cloning of GAT1 a decade ago much has been learned regarding how functional aspects of the transporter map onto its structure. The transporter probably has 12 transmembrane domains with the amino and carboxyl termini oriented intracellularly. The functional role of the C-terminal tail is unclear because deletion of this tail has minimal effects on transport (Mabjeesh and Kanner, 1992). GABA binding is affected in GAT1 mutants altered at position Y140 (in the third transmembrane domain; Bismith et al., 1997) and in the extracellular loops in the second half of the transporter (Tamura et al., 1995). Ion substrate binding is probably mediated by residues in the first half of the transporter, especially at or near the first transmembrane domain: mutations to residues W68 and R69 have effects on affinities for ion transport (Pantanowitz et al., 1993; Mager et al., 1996). The N-terminal tail of GAT1 is also critical, and serves multiple roles. Deletion of the first 42 amino acids has little effect on function (Mabjeesh and Kanner, 1992). Residue R44 seems to play a role in the reorientation of the transporter to the extracellular milieu in the absence of substrates (Bennett et al., 2000). This region is also crucial for mediating syntaxin binding (Deken et al., 2000). The present results show that, at least in the presence of syntaxin 1A, this region of the transporter also plays a role in regulating forward and reverse substrate transport.
There are multiple potential reasons for syntaxin 1A inhibition of GAT1 flux and exchange. It is possible that the reduction in transport rates per se can alter GABAergic transmission; such a mechanism of regulation has been suggested for glutamatergic transmission (Mennerick et al., 1999). However, the slow turnover rate of GAT1, and its further reduction by syntaxin 1A, would seem to indicate that this form of regulation might occur only at synapses involving slow transmission (e.g., signals mediated by GABAB receptors). Transporter regulation of GABAergic synaptic transmission is more likely to have been caused by the transporter providing GABA binding sites that buffer transmitter from GABA receptors. Our previous data suggest that syntaxin 1A may play a role in this regard by increasing the number of transporters on the cell surface (Horton and Quick, 2001); our present data show that syntaxin 1A is not affecting the ability of GABA to bind to its sites on the transporter. We think that it is more likely that the regulation by syntaxin 1A of translocation plays a role in the ability of the transporter to modulate ambient transmitter concentrations, which might be important in controlling spillover to neighboring GABAergic terminals (Ichinose and Lukasiewicz, 2002).
Footnotes
-
This work was supported in part by National Institutes of Health grant MH61468.
-
ABBREVIATIONS: SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; BoTx/C1, botulinum toxin C1; PBS, phosphate-buffered saline; SKF89976A, 4,4-diphenyl-3-butenylnipecotic acid; HBSS, Hanks' balanced salt solution.
- Received May 20, 2003.
- Accepted July 2, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}