


Abstract
Human constitutive androstane (or active) receptor (hCAR), a member of the nuclear receptor superfamily NR1I3, regulates the expression of several genes that are mainly involved in the metabolism of endogenous and xenobiotic compounds (e.g., CYP2B6, CYP3A4, and UGT1A1). We found four novel splice variants in the ligand-binding domain (LBD) of hCAR (NCBI reference sequence, NM_005122; designated SV0 herein). The variants designated SV1 and SV2 contained in-frame 12- and 15-base pair (bp) insertions, respectively. SV3 carried both of the insertions, and SV4 contained an in-frame 117-bp deletion. The insertion site of SV1 is located in the α6 helix of hCAR LBD, which makes up the ligand-binding cavity, and that of SV2 is located in the highly conserved loop between helices α8 and α9. SYBR Green real-time reverse transcription-polymerase chain reaction analysis of each splice variant revealed that the hepatic expression of SV2 was almost comparable with that of SV0 (approximately 40%), whereas other variants accounted for 6 to 10% of the total hCAR transcripts. In the reporter gene assays employing the phenobarbital-responsible enhancer module (PBREM) from CYP2B6 and UGT1A1 genes, the splice variants, except for SV1, were inactive, whereas SV1 transactivated the CYP2B6 PBREM but not the UGT1A1 PBREM reporter. A nuclear translocation assay in rat hepatocytes revealed that all the splice variants lack the responsiveness toward phenobarbital and 6-(4-chloropheny-l)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) in terms of the ligand-dependent nuclear translocation. Further characterization, such as the identification of specific ligands, will help elucidate physiological implication of these hCAR splice variants.
Human constitutive androstane (or active) receptor (hCAR, NR1I3) belongs to the nuclear receptor subfamily 1, group I, along with the vitamin D receptor (NR1I1) and pregnane X receptor (hPXR, NR1I2). hCAR forms a heterodimer with retinoid X receptor α (NR2B1) and subsequently binds to the direct repeat (DR)-4 motifs of the phenobarbital-responsive enhancer module (PBREM) located in the upstream region of the CYP2B6 and UGT1A1 genes (Sueyoshi et al., 1999; Sugatani et al., 2001). hCAR/retinoid X receptor heterodimer also binds to the DR4 and DR5 motifs in the CYP2C9 gene (Ferguson et al., 2002; Gerbal-Chaloin et al., 2002), and the DR3 and everted repeat-6 motifs in the CYP3A4 gene (Sueyoshi et al., 1999; Goodwin et al., 2002). The majority of these hCAR recognition sites are shared with both hPXR and human vitamin D receptor, indicating that these nuclear receptors make up a complex network for the regulation of the drug metabolizing enzymes, as recently reviewed by Pascussi et al. (2003).
In this article, we report four novel splice variants of hCAR identified in human liver. Alternative splice events have been reported for some of the nuclear receptors, including hPXR (Dotzlaw et al., 1999; Fukuen et al., 2002), hepatocyte nuclear factor (HNF) 4α (Chartier et al., 1994, Kritis et al., 1996), and peroxisome proliferator-activated receptor (PPAR) α (Gervois et al., 1999). A PPARα splice variant, lacking part of the hinge region and the entire ligand-binding domain (LBD), has been reported to represent 20 to 50% of total PPARα transcript in the liver, where it exhibits a dominant-negative activity (Gervois et al., 1999). HNF4α1 and its splice variant HNF4α2 have been shown to interact differently with coactivators, depending on the insertion of 10 amino acid residues at the C-terminal region (Sladek et al., 1999). For hPXR, a splice variant mRNA lacking 111 nucleotides in the LBD was detected in both normal and neoplastic human breast tissues (Dotzlaw et al., 1999). Recently, seven more hPXR splice variants have been identified (Fukuen et al., 2002), although their function remains to be elucidated. In this study, we quantitatively determined the hepatic expression levels of hCAR splice variants, and functionally characterized the variants in terms of ligand-dependent and ligand-independent transactivation of PBREM and intracellular localization.
Materials and Methods
Materials. 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime (CITCO) and phenobarbital sodium salt (PB) were purchased from BIOMOL Research Labs (Plymouth Meeting, PA) and Wako (Osaka, Japan), respectively. Human liver total RNA (of male subjects aged 24–64) was obtained from BioChain (Hayward, CA).
Construction of Reporter Plasmids. The luciferase reporter gene construct pCYP2B6/PBREM-TAL-Luc was prepared by annealing complementary oligonucleotides corresponding to human CYP2B6 PBREM (5′-gatcTGGTTCAGGAAAGTCCATGCTGCCACCTCTTCAGGGTCAGGAAAGTACAGT-3′ and 5′-gatcACTGTACTTTCCTGACCCTGAAGAGGTGGCAGCATGGACTTTCCTGAACCA-3′) (Sueyoshi et al., 1999), before insertion into the BglII site of pTAL-Luc plasmid (BD Biosciences Clontech, Palo Alto, CA), which contains a TATA-like promoter region from the Herpes simplex virus thymidine kinase promoter.
For construction of pUGT1A1/PBREM-TAL-Luc, a 290-bp fragment of human UGT1A1 PBREM (AF313454) was amplified by PCR using the following oligonucleotides: forward, 5′-cgacgcgTACACTAGTAAAGGTCACTCAATTCC-3′; reverse, 5′-gaagatctCCCTCTAGCCATTCTGGATC-3′. The amplified product was digested with MluI and BglII and cloned into the corresponding sites of the plasmid pTAL-Luc.
Gateway Recombinational Cloning of hCAR cDNA. Oligo(dT) primed cDNA was synthesized from 500 ng of human liver polyA+ RNA (OriGene Technologies, Rockville, MD) using a SuperScript first-strand synthesis system for RT-PCR (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. An aliquot of the first cDNA (2 μl) was used as a template for the attB adapter PCR using Pfx DNA polymerase (Invitrogen, Carlsbad, CA). To obtain the attB-flanked hCAR fragment, the following primers were used; 5′-AAAAAGCAGGCTCCACCATGGCCAGTAGGGAAGATGA-3′ and 5′-AGAAAGCTGGGTCTCAGCTGCAGATCTCCTGGAG-3′. Template-specific regions are underlined. An aliquot of the first PCR mixture (10 μl) was further amplified using attB1 adaptor primer (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCT-3′) and attB2 adaptor primer (5′-GGGGACCACTTTGTACAAGAAAGCTGGGT-3′). The resulting attB-flanked PCR product was cloned into pDONR 201 vector by the Gateway recombinational cloning technique. The insert was sequenced to verify identity using a BigDye terminator sequencing kit and an ABI 3700 sequencer (Applied Biosystems, Foster City, CA). Each hCAR variant was subcloned into the Gateway destination vectors pcDNA-DEST40 and pDEST17 (Invitrogen).
For construction of in-frame N-terminal fusions with EGFP, hCAR cDNAs were amplified by PCR from pcDNA-DEST40-hCAR expression vectors using the following primers: 5′-ATCTCGAGCTATGGCCAGTAGGGAAGATGAGCTGA-3′ and 5′-CAGAATTCGATCAGCTGCAGATCTCCTGGAGCA-3′ (the underlined sequences indicate XhoI and EcoRI sites, respectively). The resulting fragments were digested with XhoI and EcoRI, and inserted into the corresponding sites of pEGFP-C1 vector (BD Biosciences Clontech).
SYBR Green Real-Time RT-PCR Analysis of hCAR Splice Variants in Human Liver. The expression level of each splice variant of hCAR mRNA was determined by SYBR Green real-time RT-PCR. The following primers were used: a forward primer for SV0 and SV2, 5′-AGATGGAGCCCGTGTGGG-3′; a forward primer for SV1 and SV3, 5′-GAAGATGGAGCCCGTGTATCTC-3′; a forward primer for SV4, 5′-AGATGGAGCCCGTGACCG-3′; a reverse primer for SV0 and SV1, 5′-GGTAACTCCAGGTCGGTCAGG-3′; a reverse primer for SV2 and SV3, 5′-AACTCCAGGTCGGTCTGTAAGAT-3′; and a reverse primer for SV4, 5′-TGCTGGCCCTTGATGTAGCT-3′. Oligo(dT) primed cDNA was synthesized from 1 μg of human liver total RNA using a MultiScribe reverse transcriptase (Applied Biosystems) according to the manufacture's instructions. An aliquot of the cDNA (5 μl) was used as a template for SYBR Green real-time PCR. The transcript of each hCAR variant was quantified using PCR conditions described previously (Jinno et al., 2003), although in this study glyceraldehyde-3-phosphate-dehydrogenase was used as an internal control.
PBREM Reporter Gene Assay. HepG2 cells were transfected with the PBREM-TAL-Luc reporter plasmid (0.175 μg), pcDNA-DEST40 carrying one of the hCAR splice variants (0.175 μg) and phRL-Null vector as the internal control (0.05 μg, Promega, Madison, WI). Mock transfections were carried out in parallel, using pcDNA3.1 without an insert. All transfections were performed in 24-well plates using LipofectAMINE Plus reagent (Invitrogen) according to the manufacturer's instructions. Twenty-four hours after transfection, the cells were treated with CITCO (0.1–10 μM). The control cells were treated with vehicle [0.4% (v/v) dimethyl sulfoxide] alone. The luciferase activities after 24 h were measured using a dual luciferase assay system (Promega). The firefly luciferase activity was normalized with the Renilla reniformis luciferase activity in the same sample.
Expression of EGFP-Tagged hCAR Proteins in Primary Rat Hepatocytes. Rat hepatocytes were prepared from male Wistar rats (Charles River Japan, Kanagawa, Japan) by the two-step collagenase perfusion method described previously (Jinno et al., 1997), and plated at a density of 5 × 105 cells/well of a six-well plate in Williams' E medium (2 ml) supplemented with 5% fetal bovine serum, 10 nM insulin and 10 nM dexamethasone. After 24-h preincubation, the cells were transfected with 2.8 μg of pEGFP-hCAR expression plasmid and 0.4 μg of pDsRed2-Nuc (BD Biosciences Clontech), a nuclear localization vector, using LipofectAMINE 2000 (Invitrogen) according to the manufacture's instructions. Fluorescence microscopic observation of the EGFP-hCAR fusion and DsRed2 proteins was performed using an Eclips TE2000 inverted microscope equipped with GFP-B and G-2A filters (Nikon, Tokyo, Japan), and the images were captured by a cooled charge-coupled device camera (ORCA-ER; Hamamatsu Photonics, Shizuoka, Japan).
Data Analysis. Statistical analysis was carried out where appropriate by one-way analysis of variance (ANOVA) with Dunnett's post-hoc test using Prism 4.00 (GraphPad Software, San Diego, CA). Differences were considered significant when P < 0.01.
Results
Identification of hCAR Splice Variants. During the cloning of hCAR from human liver cDNA, we identified four novel alternative splice variants. The nucleotide sequence of hCAR, designated SV0 here, was identical to the NCBI reference sequence (NM_005122) except for a synonymous substitution, 540C→T (SnpID, rs2307424). As depicted in Fig. 1A, the variants designated SV1 and SV2 contained in-frame 12- and 15-bp insertions, respectively, in the LBD of hCAR. In addition, SV3 carried both of the insertions, whereas SV4 contained an in-frame 117-bp deletion between the insertion sites of SV1 and SV2. The human CAR gene is located on chromosome 1q23.1, spanning a genomic region of 8.5 kb, and is composed of nine exons (NCBI reference sequence NT_004668). Nucleotide sequence analysis revealed that SV1 was produced using an alternative splice acceptor site in the 5′-UTR of exon 7 (Fig. 1B). Similarly, SV2 is produced as a result of an alternative splice event in the 5′-UTR of exon 8. The 117-bp deletion in SV4 occurred by skipping exon 7. Interestingly, similar deletion variants in the LBD, lacking either 111 and 123 nucleotides because of exon-skipping, have been reported for human and mouse PXR, respectively (Kliewer et al., 1998; Dotzlaw et al., 1999), although the deletion sites in the LBD are dissimilar to hCAR SV4.

Alternative splice variants of hCAR found in the liver. A, nucleotide sequence alignment of hCAR splicing variants. The synonymous nucleotide substitution (540C→ T; SnpID, rs2307424) found in hCAR cDNAs is indicated in bold. B, schematic presentation of the alternative splicing events generating variant hCAR transcripts. SV1, SV2, and SV3 are produced using an alternative splice acceptor site in the 5′-UTRs of exon 7 (for SV1 and SV3) and exon 8 (for SV2 and SV3). SV4 is generated by skipping exon 7, resulting in an in-frame 117-bp deletion.
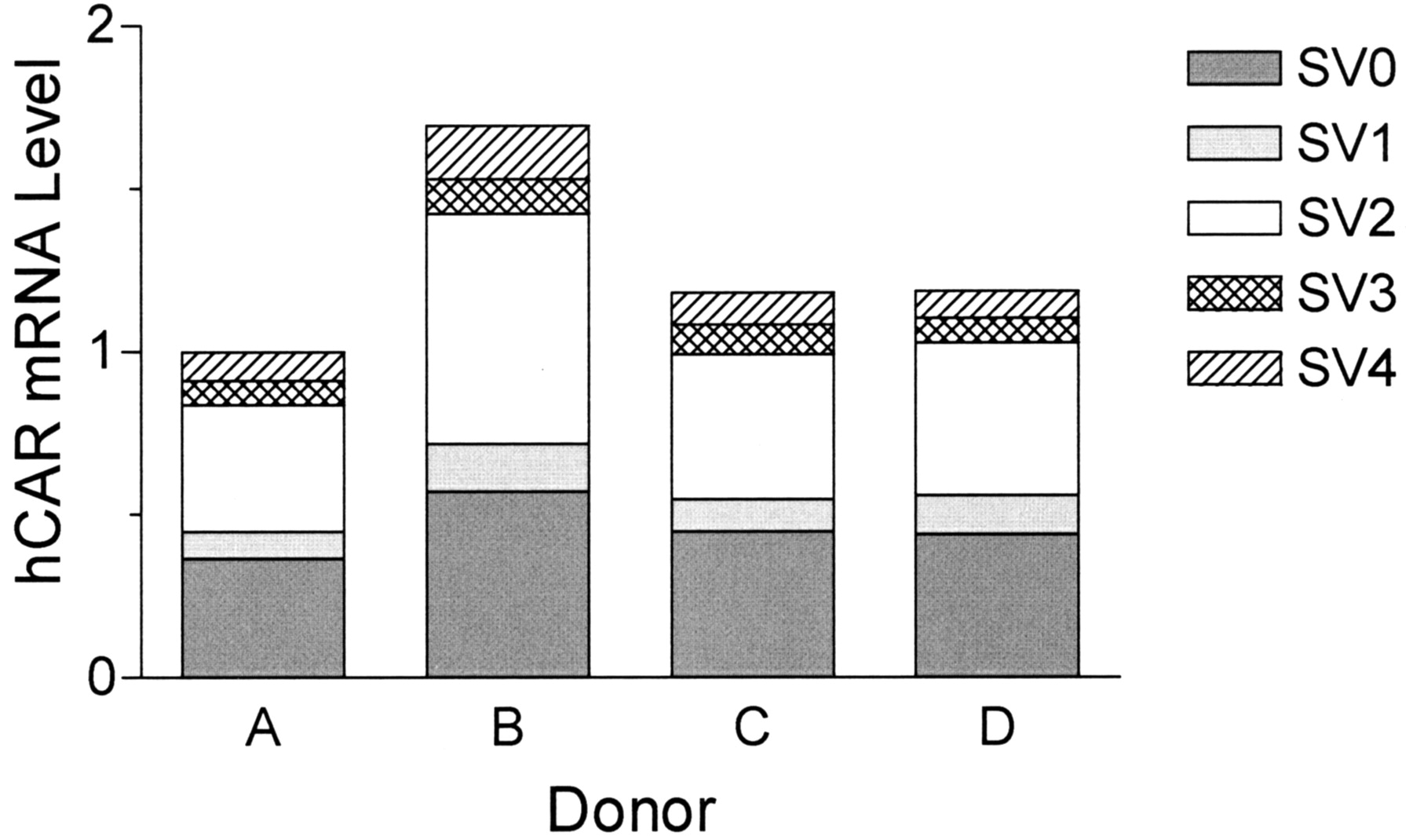
Hepatic Expression Levels of hCAR Splice Variants. To determine the hepatic expression levels of these hCAR splice variants, we developed a SYBR Green real-time RT-PCR method with splice variant-specific primer pairs. Specificity of the primer pairs for each splice variant was tested by using hCAR cDNA clones in pcDNA-DEST40 vector as template. A specific amplification of at least 500-fold was achieved in each case (data not shown). Interindividual variation of the total hCAR mRNA level was within 2-fold (Fig. 2), which is considerably less than the recent report of Chang et al. (2003), where a 240-fold variability was reported. This discrepancy might be attributable to the small sample size (four donors) in our study and/or to differences in the normalization method. Intriguingly, the hepatic expression of SV2 was almost comparable with that of SV0 among all the 4 donors. The relative expression level of each splice variant was 38 to 42% for SV2 and 6 to 10% for SV1, SV3, and SV4.

Hepatic mRNA levels of hCAR splice variants. Oligo(dT) primed cDNA was synthesized from human liver total RNA from four donors and subjected to SYBR Green real-time RT-PCR analysis using oligonucleotide primers specific for each splice variant. The results were normalized to the level of glyceraldehyde-3-phosphate-dehydrogenase expression for each sample. Results for each splice variant are expressed in terms of the relative level compared with the total amount of hCAR transcript for donor A.
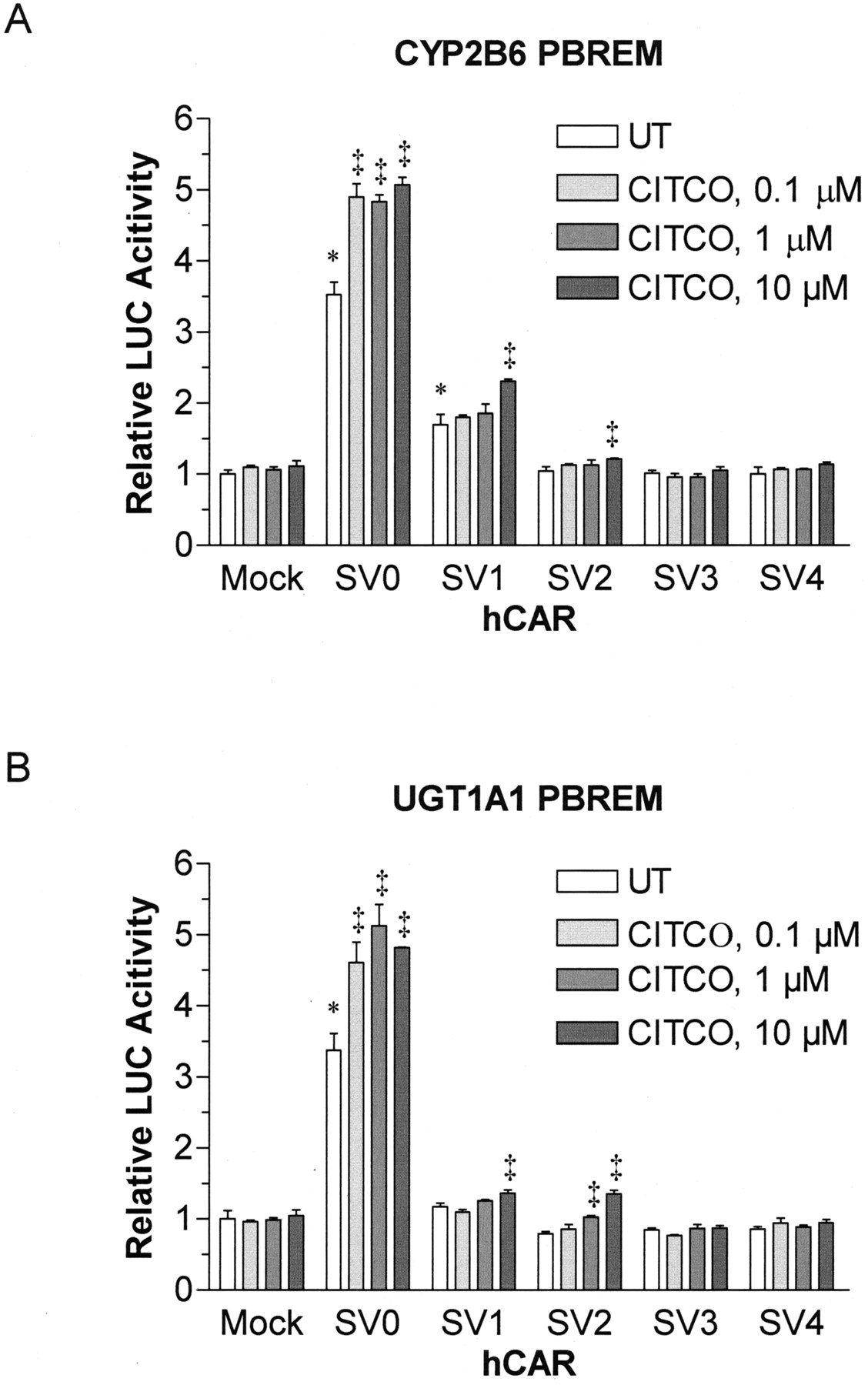
Functional Characterization of hCAR Splice Variants. The hCAR splice variants were functionally characterized in terms of ligand-dependent and ligand-independent transactivation of PBREM and intracellular localization. First, the reporter gene assays were performed using the reporter constructs carrying PBREM derived from the CYP2B6 and UGT1A1 gene promoters. hCAR, when expressed in immortalized cell lines such as HepG2 and HEK293 cells, has been shown to translocate spontaneously to the nucleus (Zelko et al., 2001) and transactivate the PBREM reporter genes in the absence of any exogenous ligands (Sueyoshi et al., 1999; Sugatani et al., 2001). In accordance with these previous studies, hCAR SV0 exhibited constitutive activities toward the PBREM reporters derived from both CYP2B6 and UGT1A1 (Fig. 3). In contrast, although SV1 transactivated the CYP2B6 PBREM (1.7-fold, p < 0.01), no activation was observed for UGT1A1 PBREM. SV2, SV3, and SV4 failed to transactivate either of the PBREM reporter genes. The reduced or abolished transcriptional activities of SV1 to SV4 could not be explained in terms of altered expression levels or stability, as judged by Western blotting of the N-terminal His6-tagged hCAR proteins (data not shown). We found that CITCO, a hCAR-specific ligand, enhanced the hCAR SV0-mediated transactivation of PBREM reporters (∼1.5-fold) at a concentration range between 0.1 and 10 μM (Fig. 3). A low but statistically significant activation by CITCO (>1 μM) was also observed for SV1 and SV2 that was reproducible among three independent PBREM reporter gene assays.

Transactivation of CYP2B6 and UGT1A1 PBREMs by hCAR splice variants. HepG2 cells in 24-well plates were transfected with CYP2B6 PBREM-TAL-Luc (A) or UGT1A1 PBREM-TAL-Luc (B) reporter plasmid (0.175 μg), pcDNA-DEST40 carrying one of the hCAR splice variants (0.175 μg) and phRL-Null vector (0.05 μg). Mock transfections were carried out using pcDNA3.1 without an insert. After 24 h, the cells were further treated with CITCO (0.1–10 μM) for 24 h. The control cells were treated with vehicle [0.4% (v/v) dimethyl sulfoxide] alone. The luciferase activities were measured using a dual luciferase assay system. The firefly luciferase activity was normalized with Renilla reniformis luciferase activity of the same sample, and the results were expressed as mean ± S.D. from three independent transfections. *, statistically different from the untreated mock-transfected cells at the level of p < 0.01 by one-way ANOVA and Dunnett's test. ‡, statistically different from the untreated cells transfected with the corresponding pEGFP-hCAR plasmid at the level of p < 0.01 by one-way ANOVA and Dunnett's test.
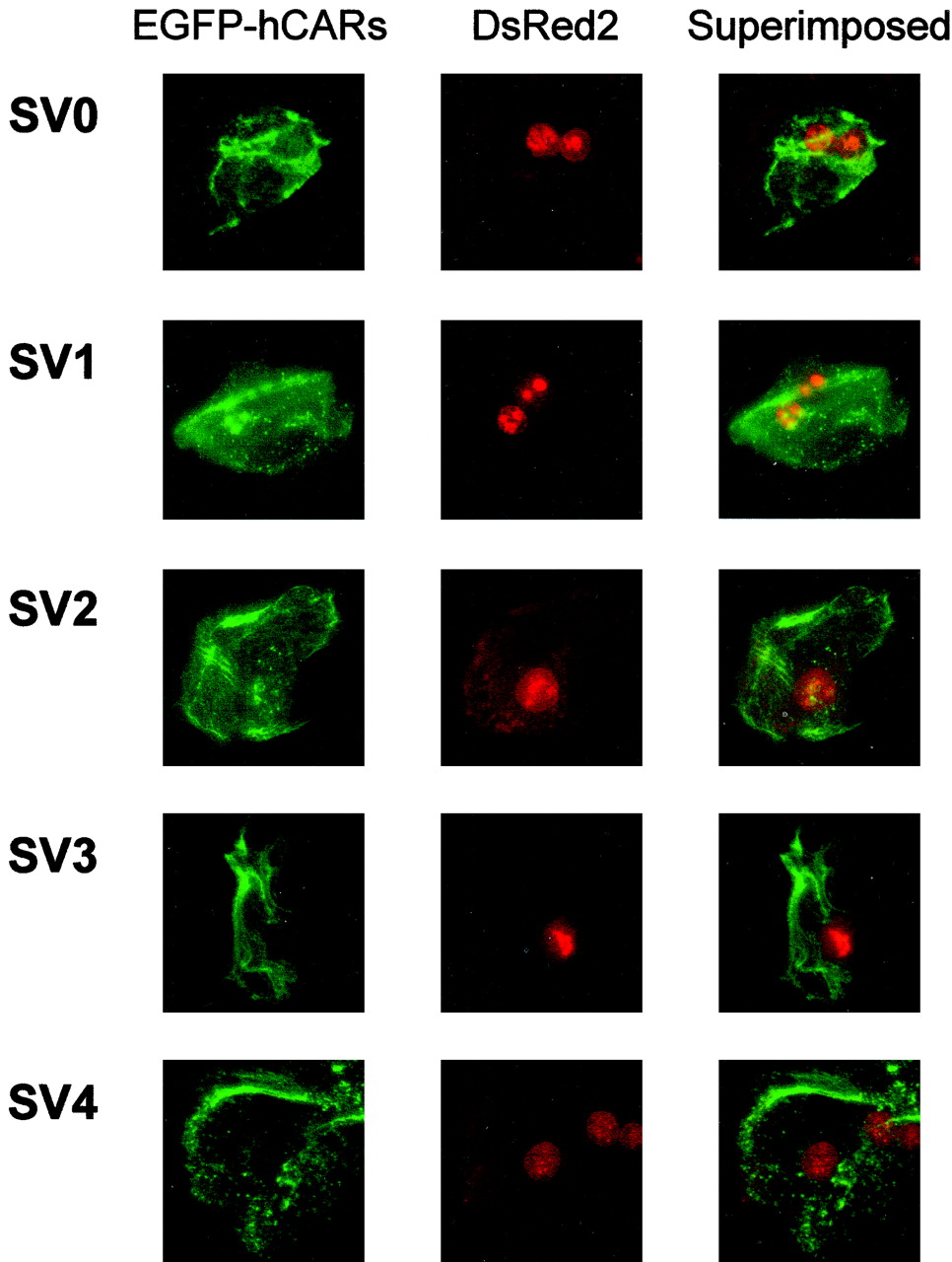
To investigate the intracellular localization of hCAR and its ligand-induced translocation from the cytoplasm to the nucleus, we performed a translocation assay in primary rat hepatocytes developed by Maglich et al. (2003). In the absence of ligand, the EGFP-hCAR splice variants were all present in both the cytoplasm and nucleus (Fig. 4). Consistent with a previous report (Maglich et al., 2003), the cells showed a somewhat crenulated pattern of fluorescence, suggesting that hCAR may be attached to a subcellular structure. As shown in Fig. 5, hCAR SV0 translocated to the nucleus in the presence of PB (1 mM) or CITCO (1 μM). Such translocation, however, was not observed for the other hCAR splice variants (SV1–SV4), even in the presence of 10 μM CITCO (Fig. 5).

Intracellular localization of EGFP-tagged hCAR splicing variants in primary rat hepatocytes. Rat hepatocytes were cotransfected with pEGFP-hCAR (2.8 μg; SV0–SV4) and pDsRed2-Nuc (0.4 μg) plasmids. After 24 h, fluorescence microscopic observation of the EGFP-hCAR fusion and DsRed2 proteins was performed as described under Materials and Methods.
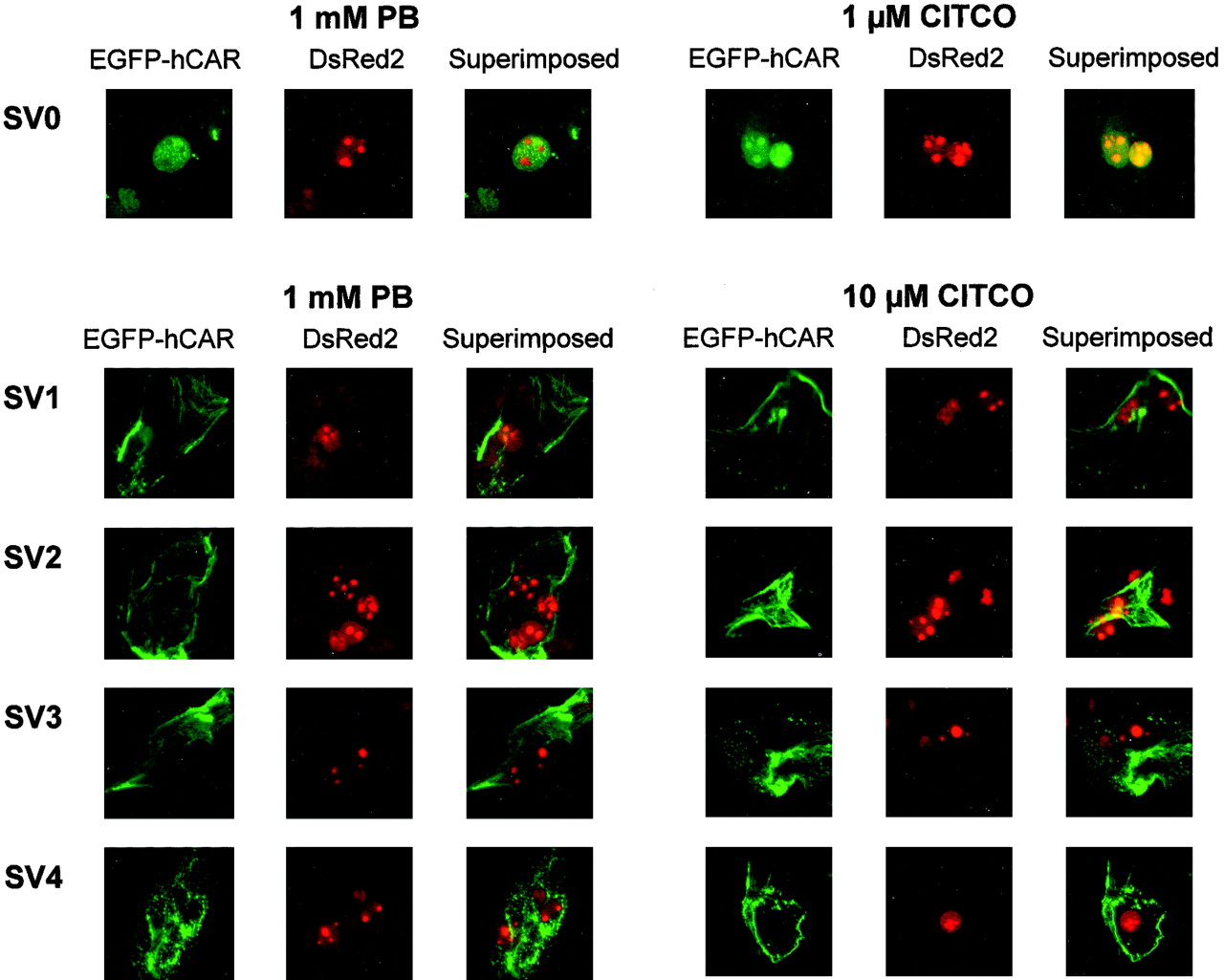
Ligand-induced nuclear translocation of EGFP-tagged hCAR splicing variants in primary rat hepatocytes. Twenty-four hours after transfection (pEGFP-hCAR and pDsRed2-Nuc), rat hepatocytes were treated with PB (1 mM) or CITCO (1 or 10 μM). After 4 h, fluorescence microscopic observation of the EGFP-hCAR fusion and DsRed2 proteins was performed as described under Materials and Methods.
Discussion
We identified four novel splice variants of hCAR that were rather abundantly expressed in the human liver. Although the physiological role of these splice variants is currently unclear, the structural significance of the regions in which the insertions or deletions occur suggests that the splice variants may exhibit different ligand specificity or coactivator requirements from hCAR SV0. The crystal structure of hCAR is not yet available, but mutational analyses and modeling studies have predicted the critical structural features of human and mouse CAR (Zelko et al., 2001; Dussault et al., 2002; Xiao et al., 2002). Figure 6 depicts the amino acid sequences and secondary structures of the LBD of hCAR and hPXR (Watkins et al., 2001, Xiao et al., 2002). As with other nuclear receptors, hCAR has the canonical fold comprising 10 α-helices and three β-strands, whereas hPXR has two extra β-strands (β1 and β1′). In addition, the hPXR sequences corresponding to the α6 helix of hCAR LBD have been shown to form a flexible surface-exposed loop (residues 309–321), which allows hPXR to accommodate extremely large ligands such as rifampicin (Watkins et al., 2001). In contrast to hPXR, hCAR lacks structural flexibility around the ligand-binding cavity (Xiao et al., 2002). The four-residue “VSPT” insertion of SV1 and SV3 is located in the α6 helix of hCAR LBD. In a model 3D-structure of the hCAR LBD (Xiao et al., 2002), helix α6 is located at the bottom of the ligand-binding cavity of hCAR, where four residues (Asp228, Gly229, Val232, and Phe234) line the ligand-binding cavity of hCAR. SV1 and SV3 have the insertion “VSPT”, containing two polar residues (S and T) between Arg231 and Val232, suggesting that these splice variants have a larger ligand-binding cavity than SV0. Intriguingly, an insertion of similar residues, “VSPA”, is found in the reported CAR amino acid sequence of rhesus monkey (GenBank accession number AAM76230). Thus, CAR may have evolved the ability to respond to a diverse set of ligands by using the alternative splicing site, although the three-dimensional structure model could not necessarily predict the precise ligand specificity of hCAR. To date, only a few compounds have been shown to interact with hCAR: 5β-pregnane-3,20-dione as an agonist, and clotrimazole as a potent deactivator (Moore et al., 2000). More recently, Maglich et al. (2003) have identified CITCO as a hCAR-specific agonist. Therefore, CITCO was examined as a potential ligand for the hCAR splice variants. In accordance with the previous report (Maglich et al., 2003), CITCO enhanced the SV0-mediated transactivation of PBREM reporters (∼1.5-fold) at a concentration range between 0.1 and 10 μM (Fig. 3). At concentrations above 1 μM, CITCO also reproducibly increased SV1- and SV2-mediated transactivation of the PBREM reporters. These results indicate that SV1 and SV2 retain, at least partially, the ability to interact with the hCAR-specific ligand CITCO, although additional factor(s) may be required for the full-activation of PBREM reporters observed for hCAR SV0.

Alignment of the amino acid sequences of hCAR and hPXR LBDs. The secondary structures, α-helices and β-strands, are labeled on the top for hCAR and on the bottom for hPXR (Watkins et al., 2001, Xiao et al., 2002). Arrows indicate the insertion sites of hCAR splicing variants (the inserted residues are “VSPT” for SV1 and SV3 and “APYLT” for SV2 and SV3).
Another five-residue insertion “APYLT” of SV2 and SV3 is located in the loop between helices α8 and α9 (Fig. 6). Although the functional significance of this loop is currently unknown, the amino acid sequence of the insertion site, “SPDRGV”, is identical among NR1I subfamily receptors including human, rat, and mouse. In the three-dimensional structure of hPXR LBD (Watkins et al., 2001), the loop α8–α9 lies adjacent in space to another highly conserved region in helix α4, which includes a nuclear receptor signature motif (Wurtz et al., 1996). This signature motif is known to play a critical role in the interaction with coregulators. Therefore the insertion of “APYLT” in the α8-α9 loop may affect the interaction of hCAR with coactivators such as PPARγ coactivator-1α (PGC-1α) (Shiraki et al., 2003). In mice, glucocorticoid receptor-interacting protein 1 (GRIP1), a p160 coactivator, has been reported to function in the nuclear translocation of CAR (Min et al., 2002). To clarify whether insertion in the loop α8–α9 (SV2 and SV3) could influence the nuclear translocation of hCAR splice variants, we examined their intracellular localization and ligand-induced translocation from the cytoplasm to the nucleus in primary rat hepatocytes (Fig. 4 and 5). Without a ligand, all the EGFP-hCAR splice variants were present in both the cytoplasm and nucleus. In the presence of PB or CITCO, hCAR SV0 translocated to the nucleus, whereas no alteration in the intracellular distribution was observed for the other splice variants. These results indicate that hCAR splice variants (SV1 to SV4) lack the responsiveness toward PB and CITCO, although relatively high concentrations of CITCO did enhance SV1- and SV2-mediated transactivation of the PBREM reporters.
In conclusion, we have discovered four novel splice variants of hCAR that are abundantly expressed in the human liver. The splice variants, except for SV1, were inactive in the reporter gene assays employing the PBREMs from CYP2B6 and UGT1A1. SV1 transactivated the CYP2B6 PBREM but not the UGT1A1 PBREM reporter. A nuclear translocation assay in rat hepatocytes revealed that all the splice variants lack the responsiveness toward PB and CITCO in terms of the ligand-dependent nuclear translocation. Further characterization of the variants will help elucidate the physiological significance of hCAR splice variants in the regulation of gene expression.
Acknowledgments
We thank Yuko Makino for technical assistance and Chie Knudsen and Reika Kou for generous support.
Footnotes
-
This work was supported in part by the Program for Promotion of Fundamental Studies in Health Sciences (MPJ-6) of the Organization for Pharmaceutical Safety and Research of Japan, and the Grant-in Aid-for the Development of Innovative Technology (13309) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
-
H.J. and T.T.-K. contributed equally to this work.
-
ABBREVIATIONS: hCAR, human constitutive androstane (or active) receptor; hPXR, human pregnane X receptor; DR, direct repeat; PBREM, phenobarbital-responsive enhancer module; LBD, ligand-binding domain; HNF, hepatocyte nuclear factor; PPAR, peroxisome proliferator-activated receptor; CITCO, 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; PB, phenobarbital sodium salt; bp, base pair(s); PCR, polymerase chain reaction; EGFP, enhanced green fluorescent protein; RT, reverse transcription; ANOVA, analysis of variance; UTR, untranslated region; Luc, luciferase.
- Received June 6, 2003.
- Accepted November 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}