


Abstract
Valproic acid (VPA) is a widely used antiepileptic agent that is undergoing clinical evaluation for anticancer therapy. We assessed the effects of VPA on angiogenesis in vitro and in vivo. In human umbilical vein endothelial cells, therapeutically relevant concentrations of VPA (0.25 to 1 mM) inhibited proliferation, migration, and tube formation. VPA 1 mM inhibited endothelial cell proliferation by 51 ± 5%, migration by 86 ± 11%, and tube formation by 82 ± 3%. These changes were preceded by the hyperacetylation of histone H4, indicating the inhibition of histone deacetylase (HDAC), and a decreased expression of the endothelial nitric-oxide synthase (eNOS). The inhibition of endothelial cell tube formation by VPA was prevented by addition of the nitric oxide donor (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate (DETA NONOate). The anticonvulsive active VPA derivative 2-ethyl-4-methylpentanoic acid, which does not inhibit HDAC, did not affect endothelial cell proliferation, tube formation, or eNOS expression. VPA was also found to inhibit angiogenesis in vivo in the chicken chorioallantoic membrane assay and in a Matrigel plug assay in mice. Embryos from VPA-treated mice showed disturbed vessel formation. These results indicate that therapeutic plasma levels of VPA inhibit angiogenesis by a mechanism involving a decrease in eNOS expression preceded by HDAC inhibition.
The antiepileptic drug valproic acid (VPA) is increasingly used and investigated for different additional pathological conditions, such as bipolar disorders, different forms of headache, schizophrenia, migraine, and different pathologies of the brain (Löscher, 2002). Moreover, VPA is currently under clinical investigation as an anticancer drug for the treatment of gliomas in children (Blaheta et al., 2002). Therefore, the collective of patients that might receive VPA is increasing. Antitumor activity is a characteristic that VPA shares with other short-chain fatty acids, such as sodium butyrate (NaBu), phenylbutyrate, or phenylacetate (Samid et al., 1992; Cinatl et al., 1993; Santini et al., 2001). A common mechanism contributing to the antitumor effects of short-chain fatty acids is their ability to inhibit histone deacetylase (HDAC) activity (Santini et al., 2001) as well as angiogenesis (Pili et al., 2001).
In addition to direct effects that result in the destruction of tumor cells or induction of differentiation (for review, see Marks et al., 2001), HDAC inhibitors are reported to inhibit hypoxia- and vascular endothelial growth factor (VEGF)-induced angiogenesis (Kim et al., 2001; Deroanne et al., 2002). More recently, the antiangiogenic effects of the HDAC inhibitors Trichostatin A, NaBu, and MS-275 were linked to a decrease in the generation of nitric oxide (NO) by endothelial cells and a marked reduction in the expression of the endothelial nitric-oxide synthase (eNOS) (Rössig et al., 2002).
Because VPA relieves HDAC-dependent transcriptional repression (Phiel et al., 2001) and up-regulates the antiangiogenic proteins thrombospondin-1 and activin A (Cinatl et al., 2002), we sought to determine whether VPA affects angiogenesis in vitro and in vivo. We report here that therapeutically relevant concentrations of VPA act as an effective inhibitor of angiogenesis as well as vasculogenesis. This effect, at least in vitro, is related to a decrease in the expression of eNOS; endothelial cell tube formation in VPA-treated endothelial cells can be restored by incubating VPA-treated endothelial cells with an NO donor.
Materials and Methods
Cells. Human umbilical vein endothelial cells (HUVEC) were isolated as described previously (Popp et al., 1996). Unless specified otherwise below, cells were seeded onto culture flasks coated with Matrigel (diluted 1:80 in culture medium described below; BD Biosciences, Heidelberg, Germany) and grown in Iscove's modified Dulbecco's medium (Sigma, Taufkirchen, Germany) supplemented with 10% fetal calf serum, 10% pooled human serum (Blood Bank of The German Red Cross, Frankfurt am Main, Germany), 100 IU/ml penicillin, 100 μg/ml streptomycin, and 5 ng/ml basic fibroblast growth factor (bFGF).
Immunocytochemical Staining. Immunocytochemical staining was performed as described previously (Cinatl et al., 2002). Briefly, HUVEC were incubated with either solvent (saline) or VPA (1 mM; Sigma, Taufkirchen, Germany) for 12 h, fixed with methanol/acetone (1/1), and incubated with monoclonal antibodies recognizing the acetylated form of histone H4 (Chemicon International, Temecula, CA). Immunoperoxidase staining was performed using biotin-labeled secondary antibodies. Cells stained with an isotype-matched irrelevant primary antibody served as a control for unspecific staining.
Immunoblotting. Cells were lysed in SDS sample buffer and separated by SDS-PAGE, as described previously (Fleming et al., 1998). Proteins were detected using specific antibodies against eNOS (Cell Signaling, Beverly, MA), β-actin (Sigma), or acetylated histone H4 (Upstate Biotechnology, Lake Placid, NY), and were visualized by enhanced chemiluminescence using a commercially available kit (Amersham Biosciences Europe, Freiburg, Germany).
To reprobe Western blots with alternative primary antibodies, the nitrocellulose membranes were incubated at 50°C for 30 min in a buffer containing 67.5 mM Tris/HCl, pH 6.8, 100 mM β-mercaptoethanol, and 2% SDS. After extensive washing, the filters were incubated in blocking buffer containing bovine serum albumin (3%), and subsequently with the primary antibody.
Proliferation Assay. Cell proliferation was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) dye reduction assay (Mosmann, 1983). HUVEC (100 μl, 104 cells/ml) were seeded onto 96-well microtiter plates and incubated with culture medium supplemented with 10% fetal calf serum and 10% pooled human serum containing serial dilutions of VPA. After 5 days of incubation, MTT (1 mg/ml) was added; after an additional 4 h, cells were lysed in a buffer containing 20% (w/v) SDS and 50% N,N-dimethylformamide adjusted to pH 4.5. Absorbance at 570 nm was determined for each well using a 96-well multiscanner. After subtracting background absorbance, results are expressed as cell number compared with control cells that were maintained in the presence of solvent. The inhibitory concentration of 50% (IC50) was calculated as the concentration of drug yielding 50% of dye reduction compared with untreated control.
Viability Assay. Viable and dead cells were discriminated via staining with 0.4% trypan blue dye. After adding trypan blue to cell suspension, blue-stained (dead) cells were counted in a Bürker chamber, and the percentage of dead (blue-stained) cells was calculated.
Migration Assay. Cell migration was investigated using HUVEC (passages 2–4) seeded at a density of 0.5 × 106 cells/ml onto the upper surface of polycarbonate filters (pore size, 8 μm; BD Biosciences) and cultured in Medium 199 (Biozol, Munich, Germany) containing 0.1% bovine serum albumin, 100 ng/ml gentamicin, and 2 mM HEPES buffer. The filters were placed into wells containing culture medium supplemented with bFGF (50 ng/ml). After 4-h incubation at 37°C, the reaction was stopped, and cells in the lower filter, including HUVEC that were attached to the inverse side of the filter, were counted under the microscope. The latter were obtained by swabbing off the cells on the upper side with a cotton wool tip, and by fixation (1% glutaraldehyde) and staining (Mayer's hematoxylin) the cells attached to the lower surface of the membranes. Five random fields (each 0.25 mm2) were counted at 200× magnification.
In Vitro Matrigel Angiogenesis Assay. The assay was performed as described previously (Ponce, 2001). Briefly, 96-well plates were coated with ice-cold Matrigel (50 μl/well), which was allowed to polymerize at room temperature for about 30 min. Thereafter, 100 μl of a suspension of HUVEC (5 × 104 cells/ml) that had been treated with saline, VPA, the NO-donor DETA NONOate, or a combination of VPA and DETA NONOate for up to 5 days were seeded onto the Matrigel and cultured overnight in Iscove's modified Dulbecco's medium, supplemented with 100 IU/ml penicillin, 100 μg/ml streptomycin, 1% (v/v) fetal calf serum, and 5 ng/ml bFGF but in the absence of VPA and DETA NONOate. Tube formation was assessed after 12 h and quantified by determining the mean number of branching points in three randomly chosen fields.
Chick Chorioallantoic Membrane Assay. All experiments with chick embryos were carried out in ovo as described previously (Michaelis et al., 2003). A window (7–10 mm in diameter) was cut into the egg shell of 3-day-old embryos, resealed with transparent film, and incubated for a further 5 days. For the CAM assay, solvent or VPA was mixed with a 1% methylcellulose solution. Aliquots (10 μl) of the resulting 0.5% methylcellulose solution were pipetted onto bacteriological-grade Petri dishes, air-dried for 1 h, and the resulting discs were placed onto the 8-day-old CAMs. Two to three discs were placed on each CAM about 10 mm apart. Control discs contained the appropriate solvent. Evaluation of the CAMs was performed 4 days after the application of the disc. To better visualize the vascular system of the CAM, 20% Luconyl Black (BASF, Ludwigshafen, Germany) in phosphate-buffered saline was injected into a vitelline vein using glass capillaries. Photographs were taken using a Nikon SMZ1000 stereomicroscope (Nikon, Tokyo, Japan).
In Vivo-Matrigel Plug Assay. C57/BL6 mice (AnLab Ltd, Prague, Czech Republic) were treated with VPA (60 to 240 mmol/kg, i.p.) for 5 days. After an additional 2 days without treatment, 0.5 ml of Matrigel (standard Matrigel, no growth factor-reduced Matrigel) supplemented with 75 ng/ml bFGF was introduced s.c. into the flank. VPA treatment was continued for a further 5 days, after which the animals were sacrificed, and the Matrigel plug and some surrounding tissue were removed. The tissue was fixed overnight in phosphate-buffered saline containing 10% formalin and 0.25% glutaraldehyde, embedded into paraffin, and stained with Masson's trichrome as described previously (Malinda, 2001). This procedure stains the Matrigel blue and endothelial cells/vessels red.
Determination of Influence of VPA on Vessel Formation in Mouse Embryos. Pregnant WinNMRI mice (Harlan-Winkelmann, Borchen, Germany) were treated three times (days 10, 11, and 12 of gestation) with solvent (saline) or with VPA (208 mmol/kg, s.c.). Twenty-four hours after the last injection, the animals were sacrificed, the embryos were removed and fixed using Bouin's solution (by Dubosq-Brasil), and then embedded in paraffin mixture Paraplast plus (Sherwood Medical Labs, St. Louis, MO). Tissue sections of 4 μm were deparaffinized and rehydrated sequentially in xylene, isopropanol, and ethanol (Romeis, 1989). The rehydrated sections were treated with hematoxylin and eosin to stain acidophilic structures red and basophilic structures blue. Photographs were taken using an inversed microscope and a digital camera (Coolpix 950; Nikon). All animal studies were conducted according to the National Institutes of Health's Guidelines for Care and Use of Experimental Animals.
Statistics. Values presented are the mean ± S.E.M. IC50 values for VPA was calculated as the concentration of drug resulting in a 50% of reduction in viable cells compared with untreated control. The IC50 values were determined using CalcuSyn for Windows. Comparisons between two groups were performed using Student's t test; three or more groups were compared by analysis of variance followed by the Newman-Keuls test. P values less than 0.05 were considered significant.
Results
Effect of Valproic Acid on Endothelial Cell Growth, Viability, and Migration. Endothelial cell proliferation and migration are initial steps in the angiogenic process, and to determine whether they are affected by VPA, bFGF-induced endothelial cell proliferation was assessed using the MTT assay and bFGF-induced cell migration was assessed in a filter system.
Incubation of endothelial cells with VPA for 5 days concentration-dependently decreased cell proliferation (Fig. 1a) with an apparent IC50 of 0.36 ± 0.09 mM relative to the maximal achievable effect (Emax) determined at 4 mM VPA (Emax = 59 ± 6% proliferation inhibition). VPA 1 mM, a concentration within the range of patients' plasma levels (Brodie and Dichter, 1996), inhibited cell growth by 51 ± 5%. The effects of VPA on cell number were relatively delayed; no significant effect was detected after 1 or 3 days of VPA-treatment.

Effect of valproic acid on endothelial cell proliferation and migration. Human umbilical vein endothelial cells were incubated with VPA (0.125 to 4 mM) for 5 days. a, influence of VPA on endothelial cell proliferation determined using MTT assay. b, influence of VPA (0.25 to 2 mM) on 20 ng/ml bFGF-induced endothelial cell migration in a Transwell filter (8-μm pores). Results are presented as the mean ± S.E.M. of data obtained in three independent experiments. *, P < 0.05; **, P < 0.01.
Because the MTT assay cannot discriminate between cytotoxic and cytostatic effects, results were confirmed by cell counting using a Bürker chamber and trypan blue staining. Cell proliferation was clearly inhibited by VPA. After 5 days of treatment with 1 mM VPA, cell number was reduced by 46 ± 11% (P = 0.003). Less than 5% of the cells stained positively for trypan blue after treatment with solvent or VPA (up to 4 mM for up to 7 days), indicating that VPA was not cytotoxic and that the effects observed can be attributed to the inhibition of cell proliferation.
To assess the ability of VPA to affect cell migration, we monitored the effects of VPA pretreatment on the 50 ng/ml bFGF-induced migration of endothelial cells through a polycarbonate filter. Cells pretreated with saline migrated readily through the 8-μm pores of the filter and 45.0 ± 10.5 cells (per 0.25 mm2) migrated to the underside of the filter within 4 h of seeding. The pretreatment of cells with VPA for up to 4 days failed to significantly affect endothelial cell migration (data not shown). However, treatment of endothelial cells with VPA (0.25 to 1 mM) for 5 days significantly inhibited the bFGF-induced migration (Fig. 1b). The effect was concentration-dependent, with a maximal effect observed with 1 mM VPA resulting in migration inhibition of 86 ± 11% (Fig. 1b). Higher concentrations failed to increase inhibition of HUVEC migration.
Effect of Valproic Acid on Histone Deacetylase Activity in Endothelial Cells. VPA, like other short-chain fatty acids, is reported to inhibit HDAC in several cell types (Göttlicher et al., 2001; Phiel et al., 2001). To determine whether VPA elicited a similar effect in endothelial cells, we assessed the levels of acetylated histone H4 in the absence and presence of VPA. Weak histone H4 acetylation was apparent in saline-treated endothelial cells (Fig. 2), whereas the addition of VPA (1 mM) markedly enhanced acetylated histone H4 levels within 6 h. This effect was maintained and was marginally enhanced in cells incubated with VPA for 18 h (Fig. 2b).

Effect of valproic acid on histone acetylation in endothelial cells. a, human umbilical vein endothelial cells were treated with either solvent (CTL) or 1 mM VPA for 12 h, fixed, incubated with a monoclonal antibody against the acetylated form of histone H4, and stained by immunoperoxidase staining. As a control for unspecific staining, cells were also labeled with an isotype-matched irrelevant primary antibody (IT-CTL). b, representative Western blot and densitometric analysis showing effect of 1 mM VPA on histone H4 acetylation in human endothelial cells at different time points. To demonstrate equal loading of each lane, the membranes were reblotted with a β-actin antibody. Results are presented as the mean ± S.E.M. of data obtained in three independent experiments. *, P < 0.05.
Effect of Valproic Acid on Endothelial Cell Tube Formation and eNOS Expression. Angiogenesis involves the migration of endothelial cells and their organization into a network of tube-like structures. In an in vitro assay of tube formation saline-pretreated endothelial cells formed tubes within 12 h of being seeded onto Matrigel. Between 28 and 35 branching points were counted per field in the control. This response was slightly attenuated in endothelial cells pretreated with the lowest concentration of VPA tested (0.25 mM) but was significantly inhibited in cells pretreated with 0.5 to 1 mM VPA (Fig. 3a). VPA 1 mM caused inhibition of tube formation by 82 ± 3%. The cells used in the assay had been incubated with and without VPA for 1, 3, or 5 days; the shorter incubation times failed to inhibit tube formation. Similar effects of VPA were observed when VEGF rather than bFGF was used (Fig. 3b).

Effect of valproic acid on tube formation in endothelial cells. Bar graphs showing the concentration-dependent effect of solvent (CTL) and VPA (0.25 to 1 mM) on tube formation by human umbilical vein endothelial cells in a Matrigel assay after stimulation with 5 ng/ml bFGF (a) or 10 ng/ml VEGF (b). Results are presented as the mean ± S.E.M. of data obtained in three independent experiments. *, P < 0.05; **, P < 0.01; ***, P ≤ 0.001.
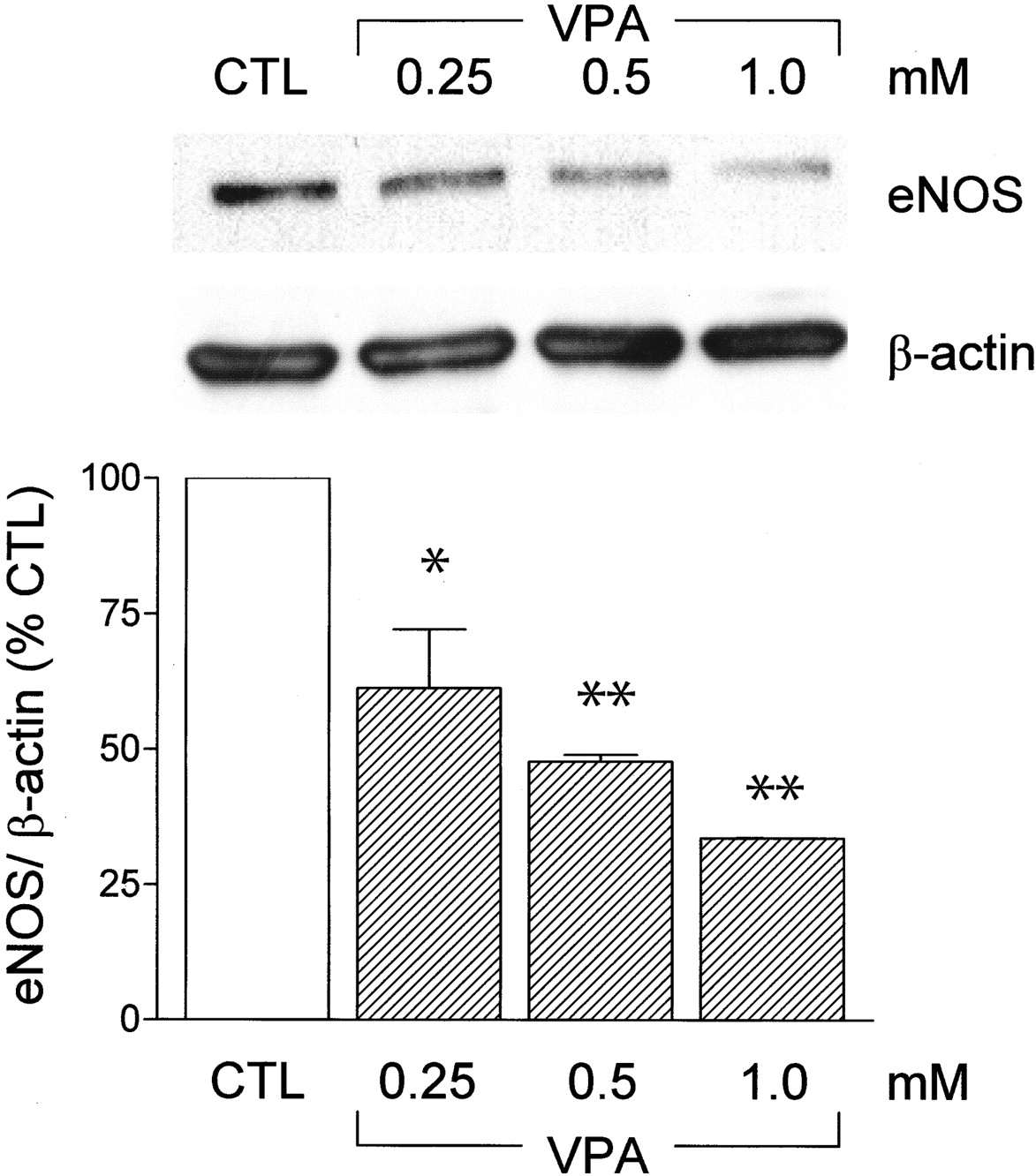
Because the generation of NO by endothelial cells is thought to be an important modulator of angiogenesis and the HDAC inhibitor trichostatin A decreased both eNOS protein and mRNA levels (Rössig et al., 2002), we assessed the effects of VPA on eNOS expression. In confluent cultures of human endothelial cells, VPA decreased eNOS protein expression in a concentration- and time-dependent manner.
A short delay was required for the effects of VPA to become evident, and no decrease in eNOS protein was detected in cells treated with VPA (0.25 to 1 mM) for 2 days or less. In contrast, pretreatment of endothelial cells with VPA for 3 days resulted in a concentration-dependent decrease in eNOS levels (Fig. 4). More prolonged treatment (up to 6 days) did not result in any further decrease in eNOS expression.

Effect of valproic acid on eNOS expression in endothelial cells. Representative Western blot and densitometric analysis showing the effects of VPA (0.25, 0.5, or 1 mM; 72 h) on eNOS expression in human umbilical vein endothelial cells. To demonstrate equal loading of each lane, the membranes were reblotted with a β-actin antibody. Results are presented as the mean ± S.E.M. of data obtained in three independent experiments. *, P < 0.05; **, P < 0.01.
To determine whether a causal relationship exists between the VPA-induced decrease in eNOS expression and inhibition of tube formation, we assessed the effects of an NO donor on tube formation in cells pretreated with VPA. As mentioned above, VPA (1 mM) pretreatment markedly attenuated endothelial cell tube formation. However, cells pretreated with a combination of VPA and the NO donor DETA NONOate (10 μM, added each 24 h over the 5-day incubation period) demonstrated a normal angiogenic response comparable with that of cells pretreated with saline or the NO donor alone (Fig. 5). Under the experimental conditions employed, either alone or in combination with VPA, the NO donor was not associated with cytotoxicity.

Effect of exogenous NO on the inhibition of endothelial cell tube formation by valproic acid. Representative photographs and bar graph showing the effect of exogenous application of the NO donor, DETA NONOate (NO, 10 μM) on the inhibition of tube formation by VPA. Endothelial cells were pretreated with either solvent (CTL) or VPA (1 mM) for 5 days before seeding onto Matrigel. Experiments were performed in the absence and presence of DETA NONOate (10 μM). Results are presented as the mean ± S.E.M. of data obtained in three independent experiments. ***, P ≤ 0.001.
To clarify the role of eNOS in our system, HUVEC were incubated with the NOS-inhibitor Nω-nitro-l-arginine (l-NNA) and the effect of eNOS inhibition in our system was investigated. l-NNA inhibited tube formation in a manner comparable with that of VPA. VPA did not totally suppress eNOS-expression. In concordance with this, l-NNA further increased the inhibitory effect of VPA on tube formation (Fig. 6).

Bar graph showing influence of 1 mM VPA, the NOS inhibitor l-NNA (300 nM), and a combination of both after 5-day treatment on tube formation of human umbilical vein endothelial cells compared with control. Tube formation was quantified by counting branching points. HUVEC were stimulated using 5 ng/ml bFGF. Experiments were performed in triplicate. *, P < 0.05; **, P < 0.01; ***, P≤0.001.
NO activates soluble guanylyl cyclase and thus elicits an increase in intracellular levels of cGMP, which then further mediates proangiogenic effects in endothelial cells (Schlossmann et al., 2003). To determine whether cGMP analogs are able to prevent antiangiogenic effects of VPA, HUVEC were incubated with of VPA (1 mM) in either the absence or presence of 8-bromo-cGMP (8-Br-cGMP; 1 mM) for 5 days. At day 5, tube formation assay was performed. Tube formation in VPA-treated cells was 35.1 ± 6.0% compared with control. 8-Br-cGMP led to tube formation of 88.7 ± 2.9% and the combination of VPA and 8-Br-cGMP led to 90.7 ± 11.8% tube formation compared with untreated control. Statistical analysis revealed differences only between VPA and the three other groups, but no statistical significant differences between control, 8-Br-cGMP, and combination of VPA with 8-Br-cGMP. P values were 0.044 for VPA compared with control, 0.007 for VPA compared with 8-Br-cGMP, and 0.026 for VPA compared with VPA and 8-Br-cGMP. These data show that VPA-induced inhibition of tube formation could be reverted by addition of cGMP analogs, suggesting that in our model, antiangiogenic effects of VPA are caused by down-regulation of eNOS followed by down-regulation of cGMP.
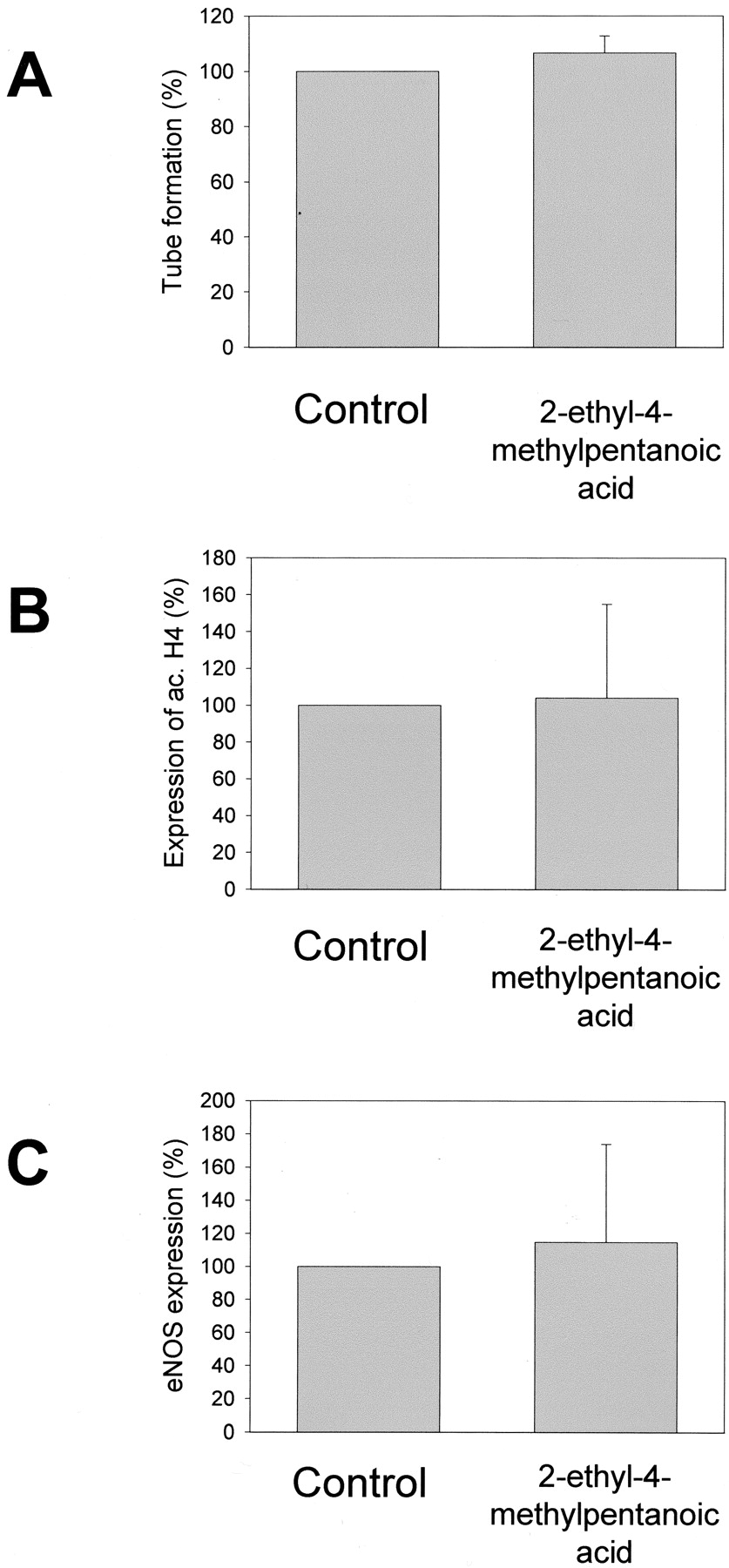
To find out whether short-chain fatty acids that do not inhibit HDAC might be antiangiogenic effective, the influence of 2-ethyl-4-methylpentanoic acid, an anticonvulsive active VPA derivative, was investigated. 2-Ethyl-4-methylpentanoic acid did not influence accumulation of acetylated histone H4, eNOS expression, or in vitro-tube formation (Fig. 7).

Influence of 2-ethyl-4-methylpentanoic acid on tube formation, histone acetylation, and eNOS expression in human umbilical vein endothelial cells. A, bar graph showing influence of 1 mM 2-ethyl-4-methylpentanoic acid after 5-day treatment on tube formation of human umbilical vein endothelial cells compared with control. Tube formation was quantified by counting branching points. HUVEC were stimulated using 5 ng/ml bFGF. B, bar graph showing influence 1 mM of 2-ethyl-4-methylpentanoic acid after 18-h treatment on accumulation of acetylated histone H4 in human umbilical vein endothelial cells compared with control. C, bar graph showing influence of 1 mM 2-ethyl-4-methylpentanoic acid after 3-day treatment on eNOS expression in human umbilical vein endothelial cells compared with control. All experiments were performed in triplicate. No statistically significant differences were detected.
Effect of VPA on Angiogenesis in Vivo. To evaluate the antiangiogenic effect of VPA in vivo, a Matrigel plug assay was used. Matrigel plugs supplemented with bFGF were introduced into mice that had been pretreated with either solvent (saline) or VPA (60, 120, and 240 mmol/kg) for 5 days. After an additional 10 days, the Matrigel plugs isolated from saline-treated animals were clearly vascularized (Fig. 8). Although endothelial cells were detected in the plugs isolated from VPA-treated animals, few distinct and perfused vessels were formed. Although the effects were concentration-dependent, a significant inhibition of angiogenesis was observed in animals treated with the lowest dose of VPA.

Effect of VPA on angiogenesis in an in vivo Matrigel plug assay. Representative sections from Matrigel plugs stained with Masson's trichrome showing the effect of VPA (60–240 mmol/kg) on the formation of blood vessels in a bFGF-supplemented Matrigel plug. The number of perfused vessels in each plug was counted, and the bar graph shows the effect of VPA treatment on vessel number. Results are presented as the mean ± S.E.M. of plugs from 5 mice, ***, P ≤ 0.001 versus CTL.
To assess vessel development in the CAM assay, methylcellulose discs containing either solvent (saline) or VPA (1 mM) were applied to the CAM on day 8 of embryo development. After an additional 4 days, the CAM underlying the saline-containing disc had matured normally whereas the CAM underlying the VPA-containing disc was poorly vascularized (Fig. 9). Antiangiogenic effects were evident based on reduced vessel ingrowth, the development of irregular and brittle vessels, and a markedly reduced perfusion compared with control CAMs.

Effect of valproic acid on vessel formation in the chick chorioallantoic membrane assay. Methylcellulose discs containing either solvent (CTL) or 1 mM VPA were placed onto the CAM at day 8. The photographs shown (final magnification, 30×) were taken after a further 4 days and are representative of the results obtained in an additional seven eggs per group.
To determine whether VPA was also able to affect angiogenesis in an embryo, we assessed the development of blood vessels in the liver of mouse embryos (gestation day 13). The period between days 10 and 25 of gestation is characterized by strong angiogenesis and vasculogenesis in the liver (Gouysse et al., 2002). Embryos removed from saline-treated mice appeared developmentally normal, and the livers were clearly vascularized and well perfused. In contrast, the livers of embryos from VPA-treated mice were markedly smaller than those of the control embryos, and blood vessel density was drastically reduced, as is evident from the lack of red staining in the preparation (Fig. 10).

Effect of valproic acid on vessel formation in mice embryos. Hematoxylin & eosin-stained sections of embryonal liver tissue (gestation day 13) from mouse embryos after treatment of the mother with either solvent (CTL) or VPA (208 mmol/kg) on days 10, 11, and 12 of gestation. The photographs are representative of an additional five embryos per group. The insets indicate the distinct differences in liver morphology between the treatment groups. White arrows indicate perfused vessels.
Discussion
The results of the present investigation clearly demonstrate that VPA is an effective inhibitor of angiogenesis in vitro and in vivo. In cultured endothelial cells, in any case, this effect is associated with the inhibition of endothelial cell proliferation, migration, and capillary tube formation. The antiangiogenic effects were observed in vitro using concentrations between 0.25 and 1 mM (i.e., concentrations that are frequently reached in the plasma of VPA-treated patients) (Brodie and Dichter, 1996). Because VPA inhibited HDAC and decreased eNOS expression in human endothelial cells and the supplementation of VPA-treated cells with an exogenous NO donor prevented the VPA-induced inhibition of angiogenesis, the antiangiogenic effect of this short-chain fatty acid seems to be linked to interference with NO-mediated signaling. Pro-angiogenic effects of NO are usually further mediated by soluble guanylyl cyclases that catalyze cGMP (Schlossmann et al., 2003). The addition of the cGMP analog 8-Br-cGMP prevented antiangiogenic effects of VPA, suggesting that decreased cGMP formation caused by decreased NO production played a role in angiogenesis inhibition by VPA. The anticonvulsive active VPA derivative 2-ethyl-4-methylpentanoic acid, which did not inhibit HDAC, did not inhibit eNOS expression and in vitro tube formation.
HDAC inhibitors have recently emerged as a new class of anticancer agents. VPA has recently been identified as an HDAC inhibitor in different cell types (Göttlicher et al., 2001; Phiel et al., 2001). Most recently, VPA was shown to induce proteasomal degradation of HDAC2 in addition to selectively inhibiting the catalytic activity of class I HDAC (Krämer et al., 2003). HDAC inhibitors are reported to induce cell death through an apoptotic process; however, little is really known about the molecular events mediating this effect. Moreover, different cell types react differently to VPA: in some cell types, VPA is cytoprotective; in others, it induces apoptosis (Blaheta et al., 2002). The differential sensitivity to HDAC inhibitors has recently been attributed to the ability of this substance to stimulate nuclear factor-κB–dependent transcription and cell survival (Mayo et al., 2003). In the present investigation, we observed that VPA induces histone hyperacetylation but found no evidence to suggest that VPA induces endothelial cell apoptosis. Thus, the effects observed could be attributed to the inhibition of cell proliferation. Indeed, we have previously reported that VPA inhibits the proliferation of cultured human neuroblastoma cells in vitro and in vivo (Cinatl et al., 1996, 1997, 2002). Moreover, our observations are in agreement with results from studies showing that VPA inhibits cell cycle in the mid-G1 phase (Blaheta et al., 2002).
Although VPA elicited a relatively rapid increase in histone acetylation the VPA-induced decrease in eNOS expression as well as the inhibitory effects on cell migration, proliferation, and angiogenesis were delayed for 3 to 5 days. This time course is in accordance with that for differentiation initiation in different cell types (Cinatl et al., 1996, 2002; Blaheta et al., 2002) for example. Taking into account the differences in replication rate of different cells as well as different sensitivities of different proliferation assays, our results seem to be in line with those of other investigators.
The generation of NO by eNOS plays a central role in the regulation of cell proliferation and angiogenesis, and angiogenesis is impaired in eNOS-/- animals (Lee et al., 1999; Fukumura et al., 2001; Zhao et al., 2002). The relationship between NO and VEGF is relatively complex; not only does NO influence the expression of VEGF (Tuder et al., 1995) but also VEGF stimulates the short-term release of NO (Dimmeler et al., 1999; Fulton et al., 1999) as well as a more prolonged increase in eNOS expression (Papapetropoulos et al., 1997; Bouloumié et al., 1999). The activity of eNOS is regulated by a number of different mechanisms, including its serine phosphorylation by the protein kinase Akt (Dimmeler et al., 1999; Fulton et al., 1999). A recent report demonstrated not only that eNOS is activated by Akt but also that NO itself leads to Akt activation. NO promoted endothelial cell migration and neovascularization via cGMP-dependent activation of phosphatidylinositol 3-kinase, which then in turn led to Akt phosphorylation and activity mediating NO-induced angiogenesis (Kawasaki et al., 2003). However, we failed to detect any effects of VPA on the phosphorylation of Akt or eNOS in human umbilical vein endothelial cells (our unpublished observations). Moreover, the fact that a pronounced delay in the effects of VPA was observed and 5 days of treatment were generally necessary for the antimigratory effects and the inhibition of tube formation to become apparent suggested that VPA may exert its effects by regulating eNOS expression. eNOS levels are regulated by a number of hemodynamic and hormonal stimuli and the HDAC inhibitors trichostatin A, NaBu, and MS-275, were recently reported to decrease eNOS mRNA stability, most probably by prompting the transcription of a RNA-destabilizing protein that binds to the 5′-untranslated region of the eNOS RNA (Rössig et al., 2002). In the present study, we observed that eNOS expression levels are also decreased by VPA and that the resulting decrease in NO production plays an important role in the antiangiogenic action of VPA because the angiogenic response was completely normal in endothelial cells cotreated with VPA and an NO donor or a cGMP analog. Although the mechanism underlying this effect remains to be determined, the results obtained highlight the importance of NO signaling in the regulation of angiogenesis.
The study of Rössig et al. (2002) also offers an explanation for the time course of eNOS protein down-regulation. The data of this report suggest that the inhibition of HDAC leads to the induction of an eNOS mRNA destabilizing protein, which causes a decrease of eNOS mRNA and finally leads to the decline in eNOS protein.
In contrast to the in vitro situation, in which significant antiangiogenic effects were observed after 5 days of treatment, in vivo results in the CAM as well as in the liver of mouse embryos showed antiangiogenic effects after 4 days of treatment. Although caution is necessary when extrapolating in vitro data to explain in vivo findings, previous investigations have suggested that HDAC inhibitors influence the production of pro- and antiangiogenic factors by nonendothelial cells and lead to inhibition of matrix metalloproteinase activation (Kim et al., 2001; Cinatl et al., 2002; Liu et al., 2003). Therefore, additional effects that are independent of endothelial cell signaling are likely to act together with mechanisms identified within this report.
Although VPA has low toxicity, the compound has been associated with two rare but severe toxic effects: fatal hepatotoxicity (incidence about 1:500 to 1:20,000) and teratogenicity (about 1–2% of human fetuses are affected). Fatal hepatotoxicity is age-related, and the patient groups at highest risk are those under 2 years of age and those on general anticonvulsant polytherapy. The incidence of prenatal hepatotoxicity has not been investigated. Teratogenic effects of VPA include neural tube defects and multiple malformations (Nau et al., 1991). Recent studies on structure-activity relationships have revealed a correlation between the potential of VPA to activate the peroxisome proliferator activated receptor δ and its ability to induce the differentiation of F9-teratocarcinoma cells and the neural tube defects (exencephaly in mice) (Lampen et al., 2001; Werling et al., 2001). Our finding that the treatment of pregnant mice with VPA leads to a marked reduction in embryonic vessel formation in liver tissue indicates a further link to teratogenic and hepatotoxic effects of VPA, especially to multiple malformations that might be caused by angiogenesis inhibition.
Taken together, the results of the present investigation show that VPA is an effective antiangiogenic agent. This might have general impact for the use of VPA. This newly discovered biological action might lead to new indications as well as contraindications for VPA and help to explain the different observed effects of the drug. The relationship between antiangiogenic effects and hepatoxicity and/or teratogenicity of VPA remains to be investigated. Clinical use of VPA as antiangiogenic drug offers a number of significant advantages. In contrast to other antiangiogenic drugs, which are still undergoing clinical evaluation, VPA is clinically well established, and its toxicological and pharmacological profiles have been well studied. Antiangiogenic substances are of enormous interest, because many common diseases, such as cancer, psoriasis, arthritis, blindness, obesity, asthma, atherosclerosis, and infectious diseases are characterized or caused by excessive angiogenesis (Carmeliet, 2003). Compared with other short-chain fatty acids that also inhibit HDAC activity, such as NaBu, phenylacetate, or phenylbutyrate, VPA possesses pharmacokinetics that are more optimal for clinical use, and its plasma half-life (7–19 h in humans) is substantially longer, which means that therapeutic levels in plasma can easily be reached (Brodie and Dichter, 1996).
Footnotes
-
This work was supported by the friendly society “Hilfe für krebskranke Kinder Frankfurt e.V.” and its foundation “Frankfurter Stiftung für krebskranke Kinder”. The work was in part supported by the foundation “Arthur und Margarete Ebert-Stiftung” and by the Deutsche Forschungsgemeinschaft (SFB 553, B1 and B5).
-
ABBREVIATIONS: VPA, valproic acid; NaBu, sodium butyrate; HDAC, histone deacetylase; MS-275, N-(2-aminophenyl)-4-[N-(pyridin-3-ylme-thoxycarbonyl)aminomethyl]benzamide; CAM, chick chorioallantoic membrane; NO, nitric oxide; eNOS, endothelial nitric-oxide synthase; HUVEC, human umbilical vein endothelial cells; bFGF, basic fibroblast growth factor; MTT, 3-/4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; DETA NONOate, (Z)-1-[2-(2-aminoethyl)-N-(2-ammonioethyl)amino]diazen-1-ium-1,2-diolate; VEGF, vascular endothelial growth factor; l-NNA, Nω-nitro-l-arginine; 8-Br-cGMP, 8-bromo-cGMP.
- Received June 16, 2003.
- Accepted November 19, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}