


Abstract
Diversity of neuronal nicotinic acetylcholine receptor binding was measured using [3H]epibatidine after deletion of α7, β2, or β4 subunits. [3H]Epibatidine binding is distinctly biphasic. Densities of higher (Kd ≈ 0.02 nM) and lower (Kd ≈ 5 nM) affinity sites in whole brains of wild-type mice are very similar. Relative sensitivity to inhibition by cytisine or α-bungarotoxin was used to evaluate pharmacological subsets of the higher- and loweraffinity sites, respectively. Deletion of each subunit had distinct effects on the binding sites. Deletion of α7 did not affect higher-affinity sites but reduced the numbers of lower-affinity sites. This reduction was confined to the [3H]epibatidine binding sites sensitive to inhibition by α-bungarotoxin. Deletion of the β2 subunit had the largest effect. Higher-affinity sites sensitive to inhibition by cytisine were eliminated, and cytisine-resistant sites were reduced. Deletion of the β2 subunit also significantly reduced the number of lower-affinity sites insensitive to α-bungarotoxin. β4 Gene deletion partially reduced cytisine-resistant and α-bungarotoxin-resistant sites with lower and higher affinity for [3H]epibatidine, respectively. Gene deletion in four brain regions (thalamus, hippocampus, superior colliculus, and inferior colliculus) elicited changes generally similar to whole brain. However, relative expression of the binding sites differed among the regions. [3H]Cytisine and 125I-α-bungarotoxin binding sites were eliminated by β2 and α7 gene deletion, respectively. These studies establish that the lower-affinity sites represent a structurally diverse set of sites that require expression of either α7, β2, or β4 subunits and extend and confirm previous classifications of the higher-affinity [3H]epibatidine binding sites.
Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels that are expressed in skeletal muscle, autonomic ganglia, and the central nervous system (Lindstrom, 2000). These receptors influence simple and complex behaviors (Picciotto, 2003) and short- and long-term responses to nicotine and similar drugs (Dani and DeBiasi, 2001). nAChRs have been implicated in the causes of Alzheimer's and Parkinson's diseases and in psychopathologies such as schizophrenia, Tourette's syndrome, and anxiety (Leonard et al., 2001; Bourin et al., 2003; Quik, 2004). Because different nAChRs are likely to regulate various behaviors and diseases, identification and measurement of nAChR subtypes expressed in brain is important.
Mammalian neuronal nAChRs are pentameric assemblies of homologous subunits, 11 of which have been identified (α2-α7, α9-10, β2-β4) (Lindstrom, 2000). Although the potential for nAChR diversity is substantial, rules of receptor assembly restrict the number of subtypes actually expressed in the brain. Of these nAChR subunits, only α7 subunits assemble to form functional homomeric receptors. Heteromeric receptors must include β2 and/or β4 subunits assembled with α2, α3, α4, and/or α6 subunits (Millar, 2003). Diversity is also limited by the specific cellular expression of mRNA encoding the subunits. In rodent brain, α4, α7, and β2 subunit mRNAs are widely expressed (Wada et al., 1989; Marks et al., 1992, 1996), whereas α2, α3, α5, α6, β3, and β4 subunit mRNAs have limited distributions (Wada et al., 1989; Marks et al., 1992; Whiteaker et al., 2000a; Champtiaux et al., 2002). Even with these restrictions, overlapping expression of compatible subunits makes identifying the compositions of the nAChRs actually expressed in the brain challenging.
Radioligand binding has been very useful for the identification of neuronal nAChR subtypes. Nicotinic agonists [nicotine, cytisine, and methylcarbachol (Anderson and Arneric, 1994)] relatively selectively label α4β2-nAChR (Whiting and Lindstrom, 1988; Flores et al., 1992; Picciotto et al., 1995; Ross et al., 2000). The elapid neurotoxin α-bungarotoxin (αBgt) labels α7-nAChR subtypes selectively (Chen and Patrick, 1997; Orr-Urtreger et al., 1997). The selectivity of these ligands, although useful for the identification of specific subtypes, limits their utility for the measurement of different nAChR subtypes.
The demonstration that epibatidine is an extraordinarily potent nicotinic agonist (Badio and Daly, 1994) prompted the development of binding assays using radiolabeled epibatidine to measure additional nAChR subtypes in the brain (Houghtling et al., 1995; Marks et al., 1998; Zoli et al., 1998; Perry et al., 2002). Immunochemical (Whiting and Lindstrom, 1988; Flores et al., 1992; Gotti et al., 2005; Marritt et al., 2005) or gene deletion (Picciotto et al., 1995; Orr-Urtreger et al., 1997; Zoli et al., 1998; Xu et al., 1999; Ross et al., 2000; Whiteaker et al., 2002) methods confirm the existence of multiple epibatidine binding subtypes. The further demonstration that epibatidine binds with high affinity to several different nAChR subtypes heterologously expressed in Xenopus laevis oocytes (Parker et al., 1998; Kuryatov et al., 2000) or human embryonic kidney cells (Xiao and Kellar, 2004) confirms the ability of this ligand to label diverse nAChRs.
Analysis of specific, saturable [3H]epibatidine binding to whole mouse brain membranes revealed two sites differing markedly in affinity (Kd ≈ 0.02 and 5 nM) but with similar site densities (Marks et al., 1999; Whiteaker et al., 2000b). Most studies have focused on the higher-affinity [3H]epibatidine binding sites. For example, pharmacological studies identified higher-affinity [3H]epibatidine binding sites differentially sensitive to inhibition by cytisine and A85380 (Marks et al., 1998; Whiteaker et al., 2000a; Perry et al., 2002). In contrast, the lower-affinity sites have not been well characterized. It has not even been clearly demonstrated whether these sites are actually composed of nAChR subunits, although they are reduced by deletion of the β2 subunit (Marks et al., 1999).
The studies described here used pharmacological and null mutant analyses to evaluate these lower- and higher-affinity sites. Given what is known about subunit assembly and expression, β2*-nAChR, β4*-nAChR, or α7-nAChR should account for all nicotinic receptors in brain (Millar, 2003). Therefore, the effects of β2, β4, and α7 gene deletion on both higher- and lower-affinity epibatidine binding sites were investigated.
Materials and Methods
Mice. Mice engineered by homologous recombination to express null mutations for the α7 (Orr-Urtreger et al., 1997) and β4 (Xu et al., 1999) nAChR genes were originally obtained from Baylor University School of Medicine (Houston, TX), whereas mice expressing the β2 nAChR null mutation (Picciotto et al., 1995) were obtained from Yale University (New Haven, CT). Animals were subsequently maintained in the specific pathogen-free facility at the University of Colorado (Boulder, CO). Mice expressing each mutation had been backcrossed with C57BL/6J mice for 8 to 10 generations (β2),5to8 generations (α7), or 2 to 5 generations (β4) at the time of the experiments. Crossing heterozygotes generated experimental subjects. Mice were weaned at 25 days of age, and like-sexed litter mates were housed together (two to five mice per cage). Mice were allowed free access to food (Harlan Teklad, Madison, WI) and water. The vivarium, in which they were housed, was maintained at 22 ± 1°C. Lights were on from 7 AM to 7 PM.
Genotypes were determined using DNA extracted from tail clippings obtained around postnatal day 40 as described previously (Orr-Urtreger et al., 1997 for α7; Xu et al., 1999 for β4; Picciotto et al., 1995 for β2; see Salminen et al., 2004 for details). Animals were 60 to 100 days old at the time of the experiments. The University of Colorado Animal Care and Utilization Committee approved animal care and experimental procedures.
Tissue Preparation. Each mouse was killed by cervical dislocation. The brain was rapidly removed and placed on an ice-cold platform. For some experiments, the whole brain was used, whereas for others, four brain regions (hippocampus, thalamus, superior colliculus, and inferior colliculus) were dissected. Samples were placed in 10 volumes (w/v) of ice-cold hypotonic buffer (14 mM NaCl, 0.15 mM KCl, 0.2 mM CaCl2, 0.1 mM MgSO4, and 2.5 mM HEPES, pH 7.5) and homogenized using a motor-driven pestle. Homogenized samples were centrifuged at 12,000g for 20 min. The pellet was resuspended in hypotonic buffer and again centrifuged. The resuspension/centrifugation cycle was repeated two more times. The resulting pellet was stored frozen under fresh hypotonic buffer until assay.
On the assay day, the sample was thawed, resuspended in the overlying buffer, and centrifuged at 12,000g. The resulting pellet was resuspended in water for use in the [3H]epibatidine binding assays.
Assay of Whole-Brain Samples. Tissue prepared from the whole brains of mice of each genotype [wild type (+/+), heterozygote (+/-), and null mutant (-/-)] was assayed for [3H]epibatidine binding (PerkinElmer Life and Analytical Sciences, Boston, MA; several lots with specific activity of 33.8-66.6 Ci/mmol). All binding experiments were performed in a buffer of the following composition: 144 mM NaCl, 1.5 mM KCl, 2 mM CaCl2, 1 mM MgSO4, and 25 mM HEPES, pH 7.5. Experiments that evaluated the effects of αBgt also included 0.05% bovine serum albumin (Type V; Sigma Chemical Co., St. Louis, MO) to protect the αBgt. All incubations were conducted for 3 h at room temperature (22°C). After the incubation, particulate protein was collected onto glass fiber filters that had been soaked in 0.5% polyethylenimine (top filter: type GB; Micro Filtration Systems, Dublin, CA; bottom filter: Type A/E, Gelman Instrument Co., Ann Arbor, MI) under vacuum using an Inotech Cell Harvester (Inotech Biosystems, Rockville, MD). Samples were subsequently washed six times with cold (4°C) buffer. After filtration and wash, filters were transferred to 5-ml scintillation vials, to which 1 ml of Budget Solve scintillation fluid (RPI, Mt. Prospect, IL) was added. Radioactivity was determined using a Tricarb 1600 liquid scintillation analyzer (PerkinElmer Life and Analytical Sciences). Counting efficiency was 45%.
[3H]Epibatidine saturation binding was determined using ligand concentrations from 0.01 to 32 nM. Incubation volume for the eight lower concentrations (0.01-1.28 nM) was 500 μl, whereas the incubation volume for the eight higher concentrations (0.25-32 nM) was 65 μl. All samples were arranged in a standard 96-well format. Incubations in the lower concentration range were conducted in 1.2-ml polypropylene tubes, whereas those in the higher concentration range were conducted in polystyrene plates. Concentrations were chosen such that the three higher concentrations from the lower concentration range (0.32, 0.64, and 1.28 nM) were similar to the three lower concentrations from the higher concentration range (0.25, 0.5, and 1.0 nM) to ensure that binding under the two conditions was comparable. Results for the overlapping samples were averaged, yielding a saturation curve with 13 concentrations of [3H]epibatidine. Aliquots of each ligand concentration were counted to determine initial concentration. Free ligand concentrations were estimated by adjusting the initial ligand concentrations for the amount of bound ligand, thereby correcting for ligand depletion.
Inhibition of [3H]epibatidine binding by cytisine was measured using a ligand concentration of approximately 300 pM, whereas inhibition by d-tubocurarine and αBgt was determined using approximately 10 nM [3H]epibatidine. Actual concentrations in each experiment were determined by counting aliquots of the appropriate [3H]epibatidine solution.
An incubation volume of 500 μl was used with 0.3 nM [3H]epibatidine. Ten concentrations of cytisine (0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, 100 nM, 300 nM, 1 μM, 3 μM, 10 μM, and 30 μM) were used. Blanks were established by measuring binding in the presence of 100 μM nicotine.
An incubation volume of 65 μl was used with 10 nM [3H]epibatidine. Ten concentrations of d-tubocurarine (300 nM, 1 μM, 3 μM, 10 μM, 30 μM, 100 μM, 300 μM, 1 mM, 3 mM, and 10 mM) and eight concentrations of αBgt (0.03 nM, 0.1 nM, 0.3 nM, 1 nM, 3 nM, 10 nM, 30 nM, and 100 nM) were used. Samples containing αBgt were incubated with tissue homogenates for 1 h before the addition of [3H]epibatidine to allow association of the toxin before the addition of ligand. Blanks were established using 1 mM nicotine.
Assay of [3H]Epibatidine Binding to Brain Regions. Hippocampus, thalamus, superior colliculus, and inferior colliculus were dissected, homogenized, and particulate fractions were prepared as described above for whole brain. Differential inhibition of [3H]epibatidine (0.3 nM) binding by 50 and 150 nM cytisine was measured to estimate the cytisine-sensitive and cytisine-resistant components of high-affinity [3H]epibatidine binding. In addition, the effect of 100 nM αBgt and 300 μM d-tubocurarine on binding measured with 10 nM [3H]epibatidine was determined to estimate αBgt-sensitive and αBgt-resistant lower-affinity [3H]epibatidine binding. These inhibitor concentrations were selected after experiments that measured inhibition of [3H]epibatidine binding in whole brain. Tissue samples were incubated with αBgt and αBgt plus d-tubocurarine for 1 h before the addition of [3H]epibatidine to allow toxin to associate with its binding sites. Incubation volumes and times, sample filtration and wash, and liquid scintillation counting were as described above.
[3H]Cytisine and 125I-αBgt Binding. Binding of [3H]cytisine, final concentration 6.8 nM (specific activity, 33 Ci/mmol; PerkinElmer Life and Analytical Sciences) and 125I-αBgt, final concentration 1 nM (specific activity, 200 Ci/mmol; GE Healthcare, Little Chalfont, Buckinghamshire, UK) was determined essentially as described previously (Marks et al., 1999). The final incubation volume for both assays was 65 μl, and both assays were conducted at room temperature (22°C). The buffer composition was the same as that used for [3H]epibatidine binding, and 0.05% bovine serum albumin was included in the buffer when 125I-αBgt binding was measured. A 30-min incubation was used for [3H]cytisine, and a 4-h incubation was used for 125I-αBgt. Filtration and wash were the same as that used for [3H]epibatidine. [3H] was measured at 45% efficiency using a PerkinElmer scintillation counter as described above. 125I was measured at 80% efficiency using a Cobra γ Counter (PerkinElmer Life and Analytical Sciences).
Data Calculation and Analyses. All curve fits were conducted using the nonlinear least-squares algorithm in Sigma Plot 2001 (SPSS Inc., Chicago, IL). [3H]Epibatidine saturation curves were analyzed using a two-site model: B = [Bm1*Epi/(Kd1 + Epi)] + [Bm2*Epi/(Kd2 + Epi)], where B is the specific [3H]epibatidine bound at each concentration of [3H]epibatidine (Epi), whereas Bm1 and Bm2 are the maximal high- and low-affinity sites with apparent binding affinities of Kd1 and Kd2, respectively. Inhibition of [3H]epibatidine binding by either cytisine or d-tubocurarine was analyzed using a two-site model: B = [B1/(1 + I/K1)] + [B2/(1 + I/K2)], where B is the [3H]epibatidine bound at any concentration of inhibitor, I; B1 and B2 are the density of binding sites sensitive to inhibition with apparent inhibition constants, K1 and K2, respectively. Inhibition of [3H]epibatidine binding by αBgt was analyzed using the following model: B = B1/(1 + I/Ki) + B2, where B1 is binding inhibited by αBgt with an apparent inhibition constant of Ki and B2 is the binding insensitive to inhibition by αBgt.
Cytisine-sensitive and cytisine-resistant components of the higher-affinity [3H]epibatidine binding sites were calculated from data obtained in the absence of cytisine and in the presence of 50 and 150 nM cytisine using the two-site inhibition equation with the Ki values for the two cytisine sites (0.5 and 30 nM) fixed, and the apparent IC50 values at these sites were calculated using the Cheng-Prussoff equation (IC50 = Ki*(1 + L/KD), where L is the concentration of [3H]epibatidine, and Kd is the average high-affinity binding constant calculated from saturation binding experiments for the concentration of [3H]epibatidine used in a specific experiment (Marks et al., 1998; Perry et al., 2002). Lower-affinity, αBgt-sensitive sites were calculated as the difference in binding between samples containing no αBgt and those containing 100 nM αBgt. αBgt-resistant sites were calculated as the difference in binding between samples containing 100 nM αBgt and those containing 100 nM αBgt plus 300 μM d-tubocurarine.
SPSS PC program was used for statistical comparisons. To compare the effects of gene (α7, β2, and β4) and genotype (+/+, +/-, and-/-) on binding parameters in whole brain, a two-way analysis of variance (ANOVA) was used. Thereafter, binding data were analyzed using one-way ANOVA followed by Duncan's post hoc test to evaluate the effect of genotype (gene deletion) on the binding parameters within each nAChR gene (α7, β2, or β4). To compare the effects of gene (α7, β2, and β4), genotype (+/+, +/-, and -/-), and brain region (four regions), all binding data were analyzed using a three-way ANOVA (gene by genotype by brain region). Thereafter, the effects of genotype and brain region were analyzed by two-way ANOVA for each nAChR gene. Finally, the effects of deletion of a given gene in each brain region were analyzed by one-way ANOVA followed by Duncan's post hoc test.
Results
Pharmacologically Identifiable [3H]Epibatidine Binding
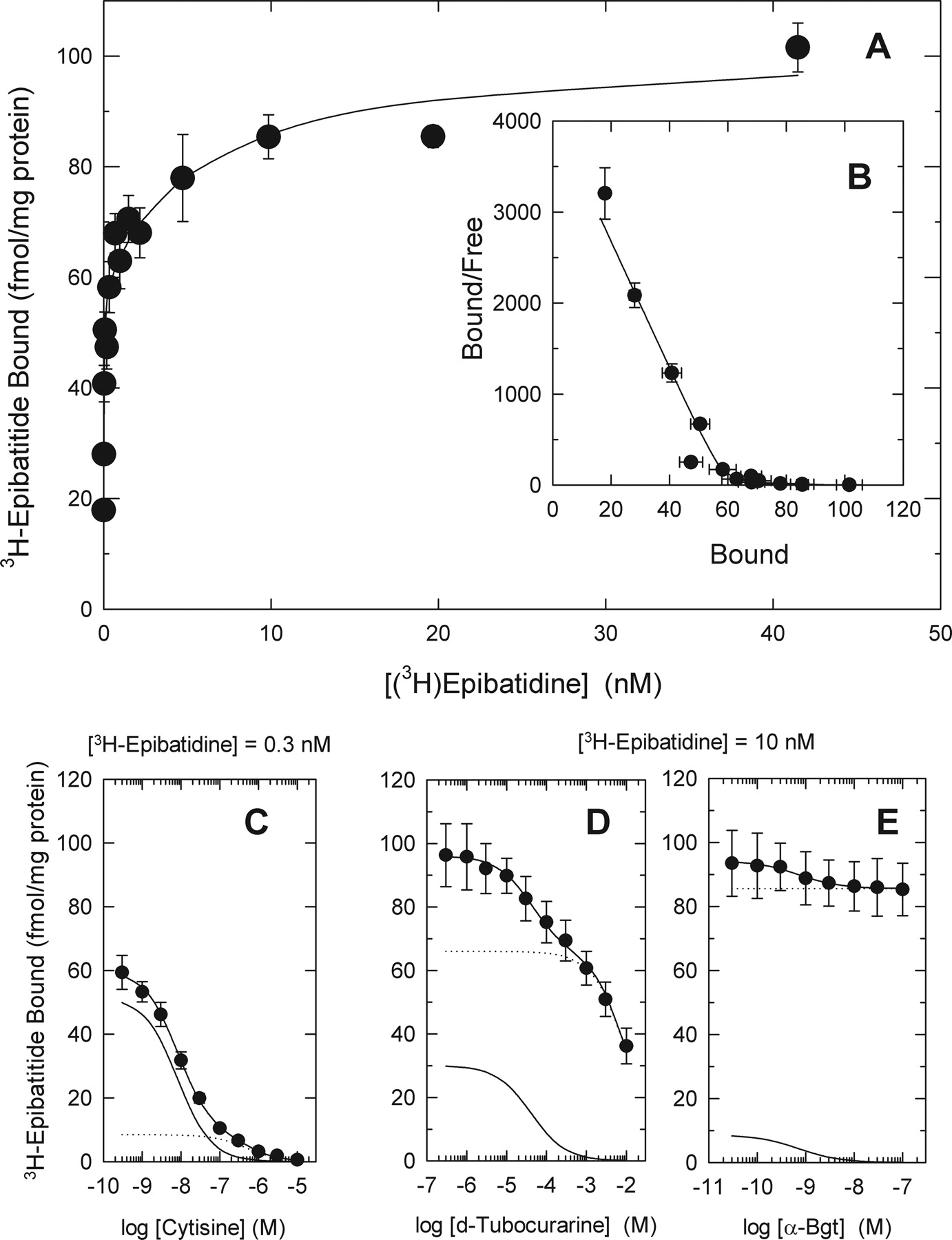
The results presented in Fig. 1 illustrate the analysis of [3H]epibatidine binding to whole mouse (C57BL/6) brain and demonstrate the heterogeneity of these sites.
The concentration-dependence of [3H]epibatidine binding in whole brain is illustrated in Fig. 1A. When a wide concentration range (0.005-40 nM) of ligand is used, [3H]epibatidine binding is distinctly biphasic as demonstrated by the Scatchard plot shown in B. Apparent Kd values of 0.014 ± 0.001 and 7.2 ± 2.2 nM for the higher- and lower-affinity sites, respectively, were estimated by nonlinear curve-fitting. Maximal binding for the higher- and lower-affinity sites were comparable (57.2 ± 2.3 and 47.2 ± 2.2 fmol/mg protein, respectively).
Differential sensitivity of the [3H]epibatidine binding to inhibition by the nicotinic agonist, cytisine, and the antagonists, d-tubocurarine and αBgt, illustrates the pharmacological heterogeneity of the higher- and lower-affinity sites. Inhibition of [3H]epibatidine (0.3 nM) binding by cytisine is shown in Fig. 1C. The inhibition profile deviates from that expected for a single site and can be resolved into two components differentially sensitive to inhibition by cytisine. Assuming similar Kd values (0.014 nM) for these two higher-affinity components of [3H]epibatidine binding, Ki values of 0.5 and 30 nM were calculated from the IC50 values of 7.9 ± 0.5 and 440 ± 85 nM measured for the cytisine-sensitive and -resistant sites, respectively. Sites with higher affinity for cytisine are more numerous (51.1 ± 3.1 fmol/mg protein) than the lower-affinity sites (9.3 ± 1.2 fmol/mg protein) and represent approximately 85% of the higher-affinity [3H]epibatidine binding sites. αBgt does not inhibit binding measured at this concentration of [3H]epibatidine.
Both higher- and lower-affinity [3H]epibatidine binding sites are measured at high ligand concentrations. Given the observation that d-tubocurarine has very similar affinity for the higher- and lower-affinity [3H]epibatidine binding sites, differential inhibition by d-tubocurarine is useful to estimate the number of lower-affinity sites (Marks et al., 1999) and is illustrated in Fig. 1D. Inhibition of [3H]epibatidine (10 nM) by d-tubocurarine is biphasic. Under these conditions, for which binding to the lower-affinity site was not maximal, site density for binding to the component with higher-affinity for d-tubocurarine was 30.1 fmol/mg protein, and site density for the component with lower-affinity for d-tubocurarine was 66.0 fmol/mg protein. The IC50 values calculated for the higher- and lower-affinity phases were 41 and 11,000 μM, respectively. However, given the large differences in Kd values for the lower (7.2 nM) and higher (0.014 nM) affinity [3H]epibatidine binding sites, Ki values for d-tubocurarine calculated for these two phases were similar (16 and 17 μM, respectively).
At high concentrations of [3H]epibatidine, αBgt inhibits some binding as illustrated in Fig. 1E. In whole brain under these conditions, αBgt-sensitive (IC50, 0.72 nM) [3H]epibatidine binding represents a small fraction of the total binding sites in whole brain (8.8 of a total of 94.3 fmol/mg protein, approximately 9%).
Subsets of [3H]Epibatidine Binding Sites Defined by Nicotinic Receptor Gene Deletion, Assays with Whole Brain Preparations
The experiments described in this section used mice in which the α7 subunit (Orr-Urtreger et al., 1997), the β2 subunit (Picciotto et al., 1995), or the β4 subunit (Xu et al., 1999) have been deleted. Given that all known nAChR expressed in mammalian brain are either homomeric α7-nAChR or require either the β2 subunit (β2*-nAChR) or the β4 subunit (β4*-nAChR) for assembly of functional receptors (Millar, 2003), studies with null mutants of these three subunits should define the full complement of nAChR subtypes.
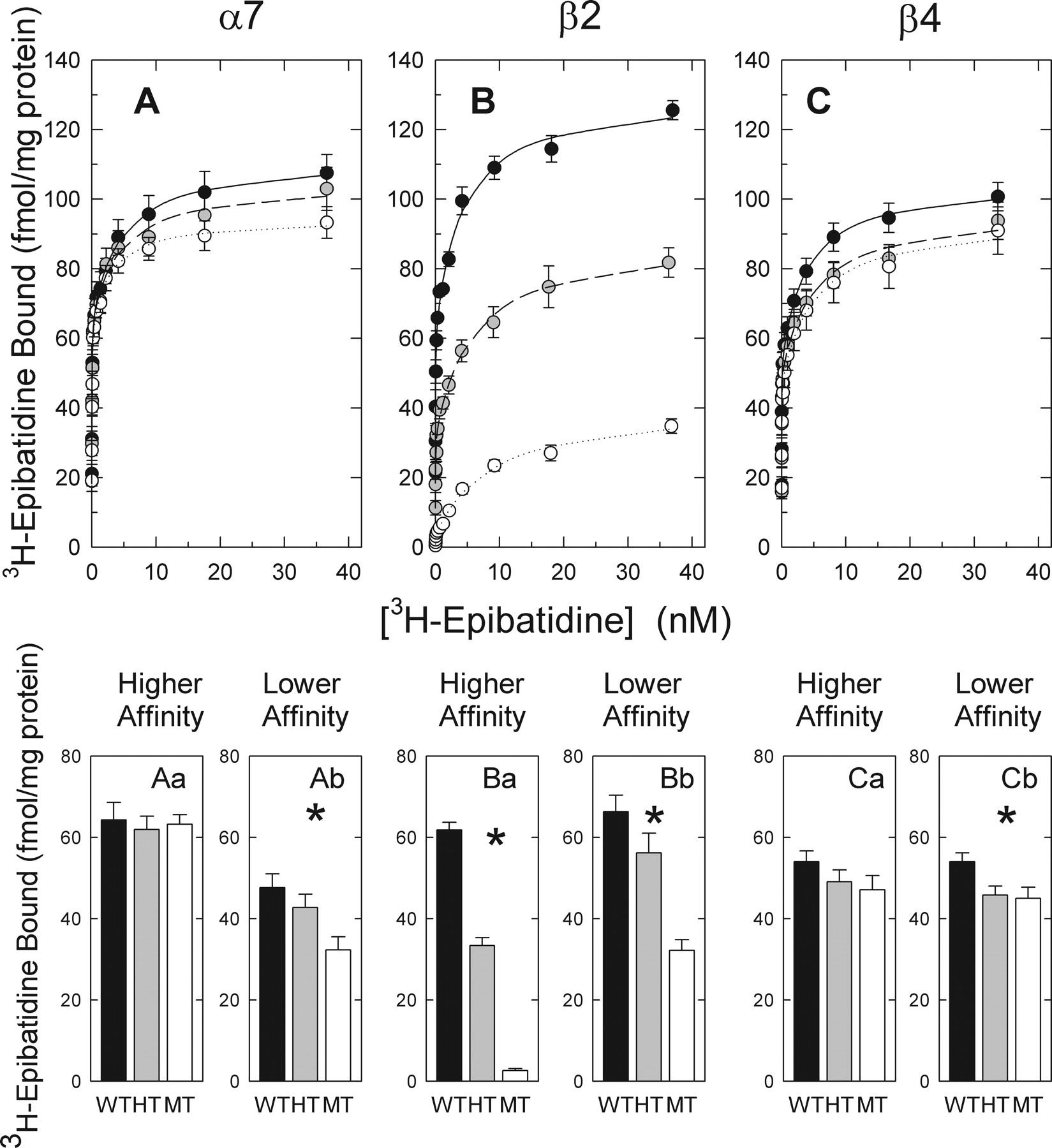
[3H]Epibatidine saturation curves constructed using particulate fractions isolated from whole brains of α7+/+, α7+/-, α7-/-, β2+/+, β2+/-, β2-/-, β4+/+, β4+/-, and β4-/-mice are presented in Fig. 2. Each curve was resolved into a higher- and lower-affinity component to evaluate the effects of targeted gene deletion on these components of [3H]epibatidine binding. [3H]Epibatidine binding to whole brain fractions of wild-type mice of each genotype was similar. However, differential effects of deletion of the α7, β2, or β4 nAChR genes on the higher- and lower-affinity [3H]epibatidine binding sites were observed.

Saturation and inhibition curves for [3H]epibatidine binding in whole brain fractions of C57BL/66 mice. Concentration dependence of [3H]epibatidine binding (A) and inhibition by cytisine (C), d-tubocurarine (D), or αBgt (E) was measured with particulate fractions prepared from whole brains of C57/BL6 mice. The Scatchard plot for the binding data are shown in B. Each point represents the mean ± S.E.M. for four mice. All curves through the points represent the best fit of the data to two-site models. For the inhibition curves, theoretical higher-affinity sites are represented by the solid lines, whereas theoretical lower-affinity sites are represented by the broken lines.
Effects of α7 gene deletion on [3H]epibatidine binding are shown in Fig. 2A. Deletion of the α7 gene had no significant effect on either the number (Bmax: α7+/+, 64.7 ± 1.6 fmol/mg protein; α7+/-, 62.4 ± 1.7 fmol/mg protein; α7-/-, 61.3 ± 1.6; Fig. 2Aa) or apparent affinity (Kd: α7+/+, 0.017 ± 0.002 nM; α7+/-, 0.016 ± 0.002 nM; α7-/-, 0.018 ± 0.002 nM) of higher-affinity [3H]epibatidine binding sites. In contrast, a statistically significant gene dose-dependent reduction in lower-affinity [3H]epibatidine binding was observed after deletion of the α7 gene (Bmax: α7+/+, 45.8 ± 2.2 fmol/mg protein; α7+/-, 40.3 ± 2.0 fmol/mg protein; α7-/-, 34.0 ± 1.7 fmol/mg protein, approximately a 25% decrease; Fig. 2Ab). An increase in the apparent affinity (Kd: α7+/+, 4.3 ± 0.8 nM; α7+/-, 3.6 ± 0.8 nM; α7-/-, 2.7 ± 0.6 nM) for [3H]epibatidine at the lower-affinity site was observed, suggesting that the α7-nAChR sites that have been deleted have a slightly lower affinity for [3H]epibatidine than those remaining.
Effects of β2 gene deletion on [3H]epibatidine binding are shown in Fig. 2B. A large gene dose-dependent reduction of the higher-affinity [3H]epibatidine binding sites occurred after deletion of the β2 nAChR gene (Bmax: β2+/+, 61.3 ± 2.3 fmol/mg protein; β2+/-, 32.9 ± 1.2 fmol/mg protein; β2-/-, 3.1 ± 0.6 fmol/mg protein, approximately a 95% decrease; Fig. 2Ba). The number of sites remaining in the β2-null mutants is small but detectable. The Kd value for [3 H]epibatidine at the higher-affinity site was unaffected by deletion of the β2 gene, indicating that the affinity for [3H]epibatidine of the deleted β2*-nAChR sites is similar to that of the residual sites. A gene dose-dependent reduction in lower-affinity [3H]epibatidine binding was also observed after deletion of the β2 gene. The reduction in these lower-affinity sites was less extensive than the reduction of the higher-affinity component (Bmax: β2+/+, 64.8 ± 3.0 fmol/mg protein; β2+/-, 54.9 ± 2.2 fmol/mg protein; β2-/-, 35.4 ± 1.2 fmol/mg protein, approximately 55% decrease; Fig. 2Bb). In addition, a modest β2 gene dose-dependent reduction in affinity was observed (Kd: β2+/+, 4.0 ± 0.8 nM; β2+/-, 6.1 ± 0.9 nM; β2-/-, 8.3 ± 1.0 nM) suggesting that the β2*-nAChR that have been deleted have higher affinity for [3H]epibatidine than the remaining sites.

[3H]Epibatidine binding in whole brain fractions of mice differing in α7, β2, or β4 genotype. Particulate fractions prepared from whole brains of mice differing in expression of the three nAChR subunits were incubated with the indicated concentrations of [3H]epibatidine. Each point represents the mean ± S.E.M. of 8 to 14 separate experiments. Data for wild-type mice are indicated by •, for heterozygotes by  , and for mutants by ○. A two-site binding model was used to fit the data and is represented by the lines through the points. Effects of deletion of α7, β2, and β4 are shown in A, B, and C, respectively. Two-way ANOVA [gene (α7, β2, and β4) by genotype (+/+, +/-, and -/-)] revealed main effects of gene [F(2,85) = 60.43] and genotype [F(2,85) = 31.20] and a gene-by-genotype interaction [F(4,850) = 23.77] for the higher-affinity component and main effects of gene [F(2,85) = 7.35] and genotype [F(2,85) = 21.79] and a gene-by-genotype interaction [F(4,85) = 5.15] for the lower-affinity component (P < 0.001 for every F value). The effect of genotype on Bmax values for the α7, β2, and β4 genes were subsequently analyzed by one-way ANOVA. Deletion of the α7 gene had no significant effect on the higher-affinity component [F(2,24) = 1.20, P > 0.05] (Aa), but the lower-affinity component was reduced by deletion of α7 [F(2,24) = 4.17, P < 0.05] (Ab). Deletion of the β2 gene significantly reduced the Bmax values for both the higher [F(2,23) = 318.32, P < 0.001] (Ba) and lower [F(2,23) = 18.16, P < 0.001] (Bb) affinity [3H]epibatidine binding sites. Bmax values for the higher-affinity site [F(2,39) = 1.34, P > 0.05] (Ca) were not significantly changed by deletion of the β4 subunit, but Bmax values for the lower-affinity site [F(2,39) = 4.37, P < 0.05] (Cb) were reduced after deletion of β4.
, and for mutants by ○. A two-site binding model was used to fit the data and is represented by the lines through the points. Effects of deletion of α7, β2, and β4 are shown in A, B, and C, respectively. Two-way ANOVA [gene (α7, β2, and β4) by genotype (+/+, +/-, and -/-)] revealed main effects of gene [F(2,85) = 60.43] and genotype [F(2,85) = 31.20] and a gene-by-genotype interaction [F(4,850) = 23.77] for the higher-affinity component and main effects of gene [F(2,85) = 7.35] and genotype [F(2,85) = 21.79] and a gene-by-genotype interaction [F(4,85) = 5.15] for the lower-affinity component (P < 0.001 for every F value). The effect of genotype on Bmax values for the α7, β2, and β4 genes were subsequently analyzed by one-way ANOVA. Deletion of the α7 gene had no significant effect on the higher-affinity component [F(2,24) = 1.20, P > 0.05] (Aa), but the lower-affinity component was reduced by deletion of α7 [F(2,24) = 4.17, P < 0.05] (Ab). Deletion of the β2 gene significantly reduced the Bmax values for both the higher [F(2,23) = 318.32, P < 0.001] (Ba) and lower [F(2,23) = 18.16, P < 0.001] (Bb) affinity [3H]epibatidine binding sites. Bmax values for the higher-affinity site [F(2,39) = 1.34, P > 0.05] (Ca) were not significantly changed by deletion of the β4 subunit, but Bmax values for the lower-affinity site [F(2,39) = 4.37, P < 0.05] (Cb) were reduced after deletion of β4.
Effects of β4 gene deletion on [3H]epibatidine binding are shown in Fig. 2C. The small apparent reduction in the numbers of higher-affinity [3H]epibatidine binding sites was not statistically significant (Bmax: β4+/+, 53.9 ± 0.8 fmol/mg protein; β4+/-, 49.6 ± 1.3 fmol/mg protein; β4-/-, 47.5 ± 1.2 fmol/mg protein, approximately a 12% decrease; Fig. 2Ca). However, the reduction in the density of lower-affinity binding sites was statistically significant (Bmax: β4+/+, 54.6 ± 2.2 fmol/mg protein; β4+/-, 45.8 ± 2.2 fmol/mg protein; β4-/-, 45.0 ± 2.7 fmol/mg protein, approximately a 17% decrease; Fig. 2Cb). No significant effects on the apparent affinity for [3H]epibatidine after deletion of the β4 subunit were observed.
Effect of α7, β2, or β4 nAChR Gene Deletion on Subsets of [3H]Epibatidine Binding Sites in Whole Brain
Both the higher- and lower-affinity [3H]epibatidine binding sites are pharmacologically heterogeneous (Marks et al., 1998; Zoli et al., 1998; Perry et al., 2002). Differential sensitivity to inhibition by cytisine defines two major subsets of higher-affinity [3H]epibatidine binding sites (termed cytisine-sensitive and cytisine-resistant), whereas differential sensitivity to inhibition by αBgt defines two major subsets of lower-affinity [3H]epibatidine binding sites (termed αBgt-sensitive and αBgt-resistant). The effects of deletion of α7, β2, and β4 nAChR genes on these four pharmacologically defined [3H]epibatidine binding sites in whole brain were evaluated.
The effects of nAChR gene deletion on subsets of higher-affinity [3H]epibatidine binding sites are illustrated in the upper half of Fig. 3.
Saturation binding shown in Fig. 2 indicated that deletion of α7 did not affect total higher-affinity [3H]epibatidine binding sites. This lack of effect was confirmed when the cytisine-sensitive and cytisine-resistant subsets of these higher-affinity sites were measured (Fig. 3, top left).
Saturation binding analysis demonstrated that deletion of the β2 subunit reduced total higher-affinity binding by approximately 95% (Fig. 2). Differential inhibition assays revealed that the cytisine-sensitive sites were completely eliminated by β2 gene deletion and reduced by approximately 50% in β2+/-mice (Fig. 3, top middle). β2 gene deletion also partially reduced cytisine-resistant, higher-affinity [3H]epibatidine binding sites (38.4 ± 6.6%).
The apparent small reduction in total higher-affinity [3H]epibatidine binding after deletion of the β4 subunit was not statistically significant (Fig. 2Ca). Further analysis confirmed that deletion of β4 had no significant effect on cytisine-sensitive, higher-affinity [3H]epibatidine binding. However, the density of cytisine-resistant, higher-affinity [3H]epibatidine binding sites was significantly reduced by deletion of the β4 gene (55.2 ± 5.0%) (Fig. 3, top right).
Deletion of α7, β2, or β4 genes each reduced maximal binding of the lower-affinity [3H]epibatidine binding sites (Fig. 2). The effects of deletion of each of these genes on the αBgt-sensitive and αBgt-resistant subsets of these lower-affinity sites are summarized in the lower portion of Fig. 3.
The decrease in lower-affinity [3H]epibatidine binding observed after deletion of the α7 gene was confined to the αBgt-sensitive subset of these sites (Fig. 3, bottom left). Indeed, deletion of α7 completely eliminated αBgt-sensitive binding without affecting the αBgt-resistant component.
Deletion of the β2 gene also selectively decreased total lower-affinity binding sites. No significant effects of β2 gene deletion were noted for αBgt-sensitive sites. However, a partial, statistically significant gene-dose-dependent reduction in αBgt-resistant lower-affinity [3H]epibatidine binding of approximately 40% was observed (from 28.5 fmol/mg protein in the β2+/+ brain to 17 fmol/mg protein in β2-/-brain) (Fig. 3, bottom middle).
Deletion of the β4 gene had no effect on the number of αBgt-sensitive, lower-affinity [3H]epibatidine binding sites. As was observed for the β2 gene, a partial, statistically significant gene-dose-dependent reduction in the αBgt-resistant sites of approximately 27% was noted upon deletion of the β4 gene (from 20.5 to 15 fmol/mg protein) (bottom right of Fig. 3).

Effects of deletion of α7, β2, or β4 genes on subsets of [3H]epibatidine binding sites in whole brain. Histograms illustrate the effects of α7, β2, or β4 gene deletion on cytisine-sensitive and cytisine-resistant higher-affinity [3H]epibatidine binding sites and on αBgt-sensitive and αBgt-resistant lower-affinity [3H]epibatidine binding sites for wild-type (WT), heterozygotic (HT), and null mutant (MT) mice. Mean ± S.E.M. values are given for each sample. Cytisine-sensitive and cytisine-resistant [3H]epibatidine binding was measured using an average ligand concentration of 0.35 nM, whereas αBgt-sensitive and αBgt-resistant lower-affinity [3H]epibatidine binding was measured using an average ligand concentration of 8 nM. Those subsets for which significant effects of a specific gene deletion was detected by one-way ANOVA (P < 0.05) are marked with an asterisk (*).
Effects of Deletion of α7, β2, or β4 Genes on Nicotinic Binding in Four Brain Regions
The differential effects of α7, β2, or β4 gene deletion on [3H]epibatidine binding in whole brain indicate that the nAChR families dependent on the expression of these subunits comprise distinct subsets of both the higher- and lower-affinity [3H]epibatidine binding sites. These analyses also allow the calculation of the relative expression of nAChR subtypes requiring the α7, β2, or β4 subunits. However, given the relatively low expression of several of the pharmacologically identifiable subtypes in whole brain, examination of [3H]epibatidine binding subsets using whole brain is technically difficult. Several brain regions express larger proportions of the various nicotinic binding sites than those expressed in whole brain, making evaluation of the effects of gene deletion in these regions more precise than is possible with whole brain preparations. Therefore, four brain regions (hippocampus, thalamus, superior colliculus, and inferior colliculus), which previous studies indicate express pharmacologically identifiable subsets of binding sites (Marks et al., 1998; Whiteaker et al., 2000a), were dissected from the brains of mice differing in α7, β2, and β4 gene expression. Cytisine-sensitive and cytisine-resistant subsets of higher-affinity [3H]epibatidine binding sites and αBgt-sensitive and αBgt-resistant subsets of lower-affinity [3H]epibatidine binding sites were subsequently measured. In addition, to measure directly two well-characterized nAChR binding sites, the effects of nAChR gene deletion on [3H]cytisine and 125I-αBgt binding were also assessed. Results of the statistical analyses of the effects of nAChR gene deletion on each of these sites are summarized in Table 1.
ANOVA results for effects of nAChR gene, genotype, and brain region on binding sites for [3H]epibatidine, [3H]cytisine, and 125I-αBgt
F values for the main effects of gene (α7, β2, or β4), genotype (+/+,+/− or −/−) and brain region (thalamus, hippocampus, superior colliculus, or inferior colliculus) on each of the six binding sites are presented. All results were initially analyzed by three-way ANOVA and subsequently analyzed for each gene by two-way ANOVA. Results of subsequent one-way ANOVA are presented in the figure legends.
Subsets of Higher-Affinity Binding Sites After nAChR Gene Deletion, Assays in Four Brain Regions
[3H]Cytisine Binding Sites. [3H]Cytisine binding was measured to determine the effects of α7, β2, or β4 gene deletion on these well-characterized sites (believed to be primarily α4β2-nAChR). Statistical analysis of [3H]cytisine binding sites yielded highly significant effects of nAChR subunit gene, genotype, and brain region (Table 1). The significant two-way and three-way interaction terms indicated that specific gene deletion differentially affected these binding sites. These differential effects are illustrated at the top of Fig. 4.
Binding-site density differed markedly among the brain regions. Deletion of either the α7 or β4 gene had no effect on [3H]cytisine binding in any brain region (Fig. 4, A and C, respectively). In contrast, deletion of the β2 subunit virtually eliminated [3H]cytisine binding in every brain region (Fig. 4B). Binding in regions of β2+/-mice was intermediate between that of wild type and null mutants.
Cytisine-Sensitive, Higher-Affinity [3H]Epibatidine Binding Sites. Highly significant effects of nAChR subunit gene, genotype, and brain region were noted for the cytisine-sensitive high-affinity [3H]epibatidine binding sites. Furthermore, the significant two-way and three-way interaction terms indicated that specific gene deletion differentially affected these binding sites. These differential effects are illustrated in Fig. 4, D to F, and the statistics are shown in Table 1. Binding-site density differed markedly among the brain regions, but deletion of the α7 (D) or the β4 (F) subunit did not significantly affect cytisine-sensitive [3H]epibatidine binding in any brain region. In contrast, deletion of the β2 subunit virtually eliminated cytisine-sensitive, higher-affinity [3H]epibatidine binding in every brain region (Fig. 4E). Binding in β2+/-mice was intermediate between the wild-type and null mutant mice in each region.
As anticipated, the effects of gene deletion on the cytisine-sensitive, higher-affinity [3H]epibatidine binding sites were virtually identical with the effects on [3H]cytisine binding.
Cytisine-Resistant, Higher-Affinity [3H]Epibatidine Binding. Effects of nAChR gene deletion and brain region on cytisine-resistant, higher-affinity [3H]epibatidine binding sites were also observed. Significant main effects of gene, genotype, and brain region and significant two-way and three-way interactions were obtained (Table 1). However, the responses of these binding sites to the effects of gene deletion were more complex than the effects on [3H]cytisine and cytisine-sensitive, higher-affinity [3H]epibatidine binding sites.
Deletion of the α7 subunit had no significant effect on the cytisine-resistant, high-affinity [3H]epibatidine binding sites in any brain region (Fig. 4G).
Significant effects of genotype and significant genotype-bybrain region interactions were observed after deletion of either β2 or β4. The histograms illustrate the effects of deletion of the β2 (Fig. 4H) and β4 (Fig. 4I) subunits, respectively. These results indicate that both β2 and β4 subunits contribute to the cytisine-resistant, higher-affinity [3H]epibatidine binding sites, as was observed for whole brain. β2 and β4 gene deletion differentially affected these sites in the brain regions assayed.
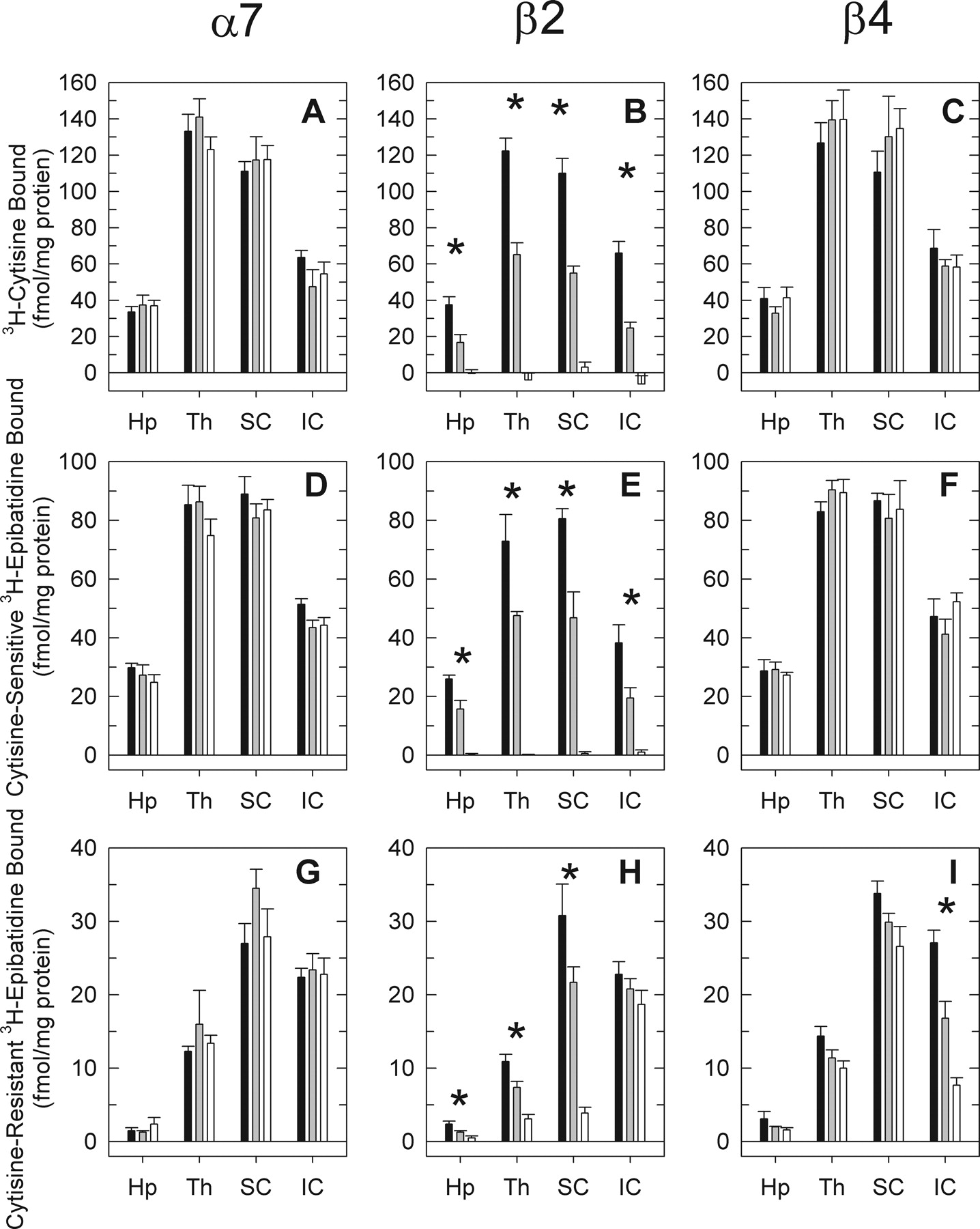
Effects of α7, β2, or β4 gene deletion on [3H]cytisine binding and on cytisine-sensitive and cytisine-resistant higher-affinity [3H]epibatidine binding in hippocampus, thalamus, superior colliculus, and inferior colliculus. Binding of [3H]cytisine (average concentration ≈ 6.8 nM) and [3H]epibatidine (average concentration ≈ 0.3 nM) was measured in four brain regions dissected from α7, β2, or β4 wild-type, hetero-zygote, or null mutant mice: hippocampus (Hp), thalamus (Th), superior colliculus (SC), and inferior colliculus (IC). Results for three-way and two-way ANOVA are presented in Table 1. Brain regions in which a significant effect of genotype was observed by one-way ANOVA are marked with an asterisk (*).
The pattern exhibited after deletion of the β2 subunit is illustrated in Fig. 4H. A significant gene dose-dependent reduction in cytisine-resistant, higher-affinity [3H]epibatidine binding sites was observed for three of the brain regions (superior colliculus, thalamus, and hippocampus). However, deletion of β2 did not completely eliminate these sites. Residual binding was noted in each of these three regions. The small reduction observed for inferior colliculus after deletion of β2 was not statistically significant.
In contrast, deletion of β4 elicited a significant reduction in cytisine-resistant, higher-affinity [3H]epibatidine binding sites in inferior colliculus. The modest effects on cytisine-resistant, higher-affinity [3H]epibatidine binding sites in hippocampus, thalamus, and superior colliculus, although not statistically significant, were similar in magnitude to the residual cytisine-resistant, higher-affinity [3H]epibatidine binding sites persisting in β2-/-mice.
Subsets of Lower-Affinity [3H]Epibatidine Binding Sites after nAChR Gene Deletion, Assays in Four Brain Regions
As noted previously and reiterated above, a subset of the sites with lower affinity for [3H]epibatidine in whole brain is sensitive to inhibition by αBgt. This subset of sites is eliminated by deletion of α7 but is unaffected by deletion of either β2 or β4. Examination of the effects of deletion of the α7, β2, or β4 genes on the residual lower-affinity, αBgt-resistant sites demonstrate for the first time that these sites are primarily, if not completely, nicotinic and are heterogeneous. To provide a more comprehensive analysis of the effects of nAChR gene deletion on the lower-affinity [3H]epibatidine binding sites, the effects of α7, β2, or β4 gene deletion were examined in four brain regions.
125I-αBgt Binding Sites.125I-αBgt binding was measured to examine whether the effects of nAChR gene deletion affected these well-characterized sites. Significant effects of brain regions, nAChR gene (α7, β2, or β4), and genotype and significant higher-order interactions were observed (Table 1).
Deletion of either β2 (Fig. 5B) or β4 (Fig. 5C) genes did not affect 125I-αBgt binding in any brain region. In contrast, and as expected, deletion of the α7 reduced the number of these sites by at least 95% in each brain region (Fig. 5A).
αBgt-Sensitive, Lower-Affinity [3H]Epibatidine Binding Sites. Significant differences among brain regions, nAChR gene (α7, β2, and β4), and genotype and significant interactions between gene and genotype and between gene and brain region were observed for the αBgt-sensitive, lower-affinity [3H]epibatidine binding sites (Table 1). No significant effects on these binding sites were observed for any brain region after deletion of either β2 (Fig. 5E) or β4 (Fig. 5F) genes. In contrast, αBgt-sensitive, lower-affinity [3H]epibatidine binding sites were abolished by deletion of the α7 gene in every brain region (Fig. 5D).
The effects of nAChR gene deletion on 125I-αBgt and on αBgt-sensitive, lower-affinity [3H]epibatidine binding sites were consistent. However, it should be noted that the average site density of the 125I-αBgt sites was 25% greater than the density of the αBgt-sensitive, lower-affinity [3H]epibatidine sites.

Effects of α7, β2, or β4 gene deletion on 125I-αBgt binding and on αBgt-sensitive and αBgt-resistant lower-affinity [3H]epibatidine binding in hippocampus, thalamus, superior colliculus, and inferior colliculus. Binding of 125I-αBgt (average concentration ≈ 1 nM) and αBgt-sensitive and αBgt-resistant lower-affinity [3H]epibatidine (average concentration ≈ 8 nM) was measured in four brain regions dissected from α7, β2, or β4 wild-type, heterozygote, or null mutant mice. The figure illustrates the effects of gene deletion in tissue prepared from hippocampus (Hp), thalamus (Th), superior colliculus (SC), and inferior colliculus (IC). Results for the three-way and two-way ANOVA of these data are given in Table 1. Brain regions in which a significant effect of genotype was observed by one-way ANOVA are marked with an asterisk (*).
αBgt-Resistant Lower-Affinity [3H]Epibatidine Binding Sites. The effects of deletion of either α7, β2, or β4 genes on αBgt-resistant lower-affinity [3H]epibatidine binding sites were more complex than the patterns observed for the αBgt-sensitive, lower-affinity [3H]epibatidine or 125I-αBgt binding sites, which were affected only by α7 gene deletion. Significant differences among brain regions, nAChR gene, and genotype and significant interactions between gene and genotype and between gene and brain region were indicated for the αBgt-resistant lower-affinity [3H]epibatidine binding sites by the three-way ANOVA (Table 1).
No significant effects on the αBgt-resistant lower-affinity [3H]epibatidine binding sites were revealed by two-way ANOVA after deletion of either the α7 or β4 subunit (Table 1).
In contrast, deletion of the β2 subunit resulted in highly significant reduction in the numbers of lower-affinity, αBgt-resistant [3H]epibatidine binding sites in thalamus (82%), superior colliculus (87%), and to a lesser extent in inferior colliculus (54%), whereas the apparent decrease in hippocampus (34%) was not statistically significant (Fig. 5H).
Although reductions in binding after deletion of the β4 subunit were not statistically significant in any brain region, apparent decreases were noted in thalamus (26%), hippocampus (27%), and inferior colliculus (40%), but not in superior colliculus. These apparent reductions suggest that it is possible that both β2*-nAChR and β4*-nAChR contribute to the lower-affinity, αBgt-resistant [3H]epibatidine binding sites in hippocampus, thalamus, and inferior colliculus, but that the relatively modest contribution of β4*-nAChR on these binding sites prevents definitive identification. The experiments outlined in the following section address this possibility.
Effect of Deletion of Both α7 and β2 Genes on [3H]Epibatidine Binding Sites
As presented above, the effects of deletion of either the α7 or β2 subunit on specific nicotinic binding sites were easily detectable, whereas the effects of deletion of β4 subunit were relatively small and were not always obvious. The sites requiring the β4 subunit were included in heterogeneous subsets of sites that were also affected by deletion of β2. To verify whether the small effects observed after deletion of the β4 subunit were measurable with an alternative method, sites remaining after deletion of both α7 and β2 subunits were measured.
The saturation curve for the binding of [3H]epibatidine to whole brain of (α7-/-)/(β2-/-) mice is shown in Fig. 6A. This saturation curve could be resolved into components with higher-affinity for [3H]epibatidine (Kd, 0.018 ± 0.006 nM) and lower-affinity (Kd,10 ± 7.7 nM), with Bmax values of 5.7 ± 0.1 and 14.3 ± 4.0 fmol/mg protein, respectively. The Scatchard plot shown as an inset to A further illustrates the heterogeneity. Reduction in higher- and lower-affinity binding sites after deletion of the β4 subunit was 5.8 ± 1.4 and 9.6 ± 3.5 fmol/mg protein, respectively (see Fig. 2, compare G with I), values comparable with those remaining in (α7-/-)/(β2-/-) brain.
Subsets of higher and lower-affinity [3H]epibatidine binding sites were subsequently measured in four brain regions of (α7-/-)/(β2-/-) double mutant mice to evaluate whether the residual binding sites in these regions were also similar to the sites eliminated by deletion of the β4 subunit. Binding in these four brain regions (hippocampus, thalamus, superior colliculus, and inferior colliculus) is shown in Fig. 6.

Whole-brain [3H]epibatidine saturation curve and regional [3H]epibatidine binding sites in (α7-/-)/(β2-/-) mice. [3H]Epibatidine binding was measured in particulate fractions prepared from whole brains of three (α7-/-)/(β2-/-) mice as illustrated on the left (A). Points represent mean ± S.E.M. of three mice. Curve is the fit of the data to a two-site model. The inset presents these data as a Scatchard plot. Effects of double gene deletion of cytisine sensitive (B) and cytisine-resistant (C) higher-affinity [3H]epibatidine binding and αBgt-sensitive (D) and αBgt-resistant (E) lower-affinity [3H]epibatidine binding in four brain regions measured in six (α7-/-)/(β2-/-) mice are illustrated on the right of the figure. Each point is the mean ± S.E.M. for six mice.
Cytisine-sensitive, higher-affinity [3H]epibatidine binding was undetectable (did not differ significantly from 0) in any of the regions assayed (Fig. 6B). This result is consistent with the observation that deletion of the β2 subunit virtually eliminated these sites (Figs. 3 and 4).
Nonzero levels of cytisine-resistant, higher-affinity [3H]epibatidine binding were detected in thalamus, superior colliculus, and particularly inferior colliculus, although in most of the brain regions assayed, residual binding sites were quite low (less than 5 fmol/mg protein) (Fig. 6C). However, cytisine-resistant, higher-affinity [3H]epibatidine binding sites remaining in inferior colliculus (18.1 ± 1.3 fmol/mg) of α7/β2 double mutants represented 32% of the site density measured for wild-type mice.
αBgt-sensitive, lower-affinity [3H]epibatidine binding sites were not detected in any brain region of α7/β2 double mutants (Fig. 6D) as would be expected for animals lacking α7-nAChR (Figs. 3 and 5).
Inferior colliculus was the only brain region of those assayed in which αBgt-resistant lower-affinity [3H]epibatidine binding sites were detectable in α7/β2 double mutants (Fig. 6E).
Discussion
A schematic summarizing [3H]epibatidine binding sites in mouse brain is illustrated in Fig. 7. Pharmacological and null mutational analyses revealed six major classes of [3H]epibatidine binding sites and defined a minimum number of sites based on the role of subunits essential for the assembly of known brain nAChRs. Inclusion of two lower-affinity [3H]epibatidine binding sites adds to the four classes described by Zoli et al. (1998). The results establish that virtually all [3H]epibatidine binding sites were eliminated by the deletion of α7, β2, and β4 nAChR subunits. Thus, both the higher- and lower-affinity sites are nicotinic. Furthermore, data on the relative expression of the six [3H]epibatidine binding sites in whole mouse brain have been obtained.
[3H]Epibatidine binds with high affinity to nAChR assembled from α2, α3, and α4 subunits and β2 or β4 subunits expressed heterologously in X. laevis oocytes (Parker et al., 1998) or stably transfected human embryonic kidney cells (Xiao and Kellar, 2004) and to α6 subunits coexpressed with β2 or β4in X. laevis oocytes (Kuryatov et al., 2000). Epibatidine also interacts with lower affinity (3-10 nM) with the 125I-αBgt binding site of human α7-nAChR (Gerzanich et al., 1995). Therefore, the response of [3H]epibatidine binding sites to nAChR gene deletion provides a reasonable estimate of the relative numbers of putative nAChR subtypes in the brain.
The proportions of higher- and lower-affinity [3H]epibatidine binding sites in whole brains of wild-type mice are similar (51 and 49% of the total, respectively). Reductions in binding for the higher-affinity sites of 1.7 ± 7.6% by deletion of α7, of 95.8 ± 0.9% by deletion of β2, and of 12.9 ± 7.8% by deletion of β4 were observed and accounted overall for 110.4 ± 10.9% of these sites. Reductions in binding for the lower-affinity sites of 27.9 ± 8.3% by deletion of α7, of 51.4 ± 5.0% by deletion of β2, and of 16.8 ± 6.1% by deletion of β4 accounted overall for 96.1 ± 11.4% of the sites. Thus, deletion of the α7, β2, and β4 genes virtually eliminated all higher- and lower-affinity [3H]epibatidine binding sites, establishing their nicotinic nature.
Analysis of the effects of gene deletion on [3H]epibatidine binding sites after division into pharmacological subtypes (higher-affinity sites into cytisine-sensitive and cytisine-resistant and lower-affinity sites into αBgt-sensitive and αBgt-resistant) further delineated the contribution of the three nAChR subunits to [3H]epibatidine binding sites.
Cytisine-sensitive, higher-affinity [3H]epibatidine binding and [3H]cytisine binding in each of the four brain regions assayed are entirely dependent on expression of the β2 nAChR subunit. Deletion of α7 or β4 had no effect. Thus, the patterns of nAChR gene deletion on cytisine-sensitive, higher-affinity [3H]epibatidine binding and [3H]cytisine binding were identical and simple and confirmed results reported previously for β2-null mutant mice (Picciotto et al., 1995; Whiteaker et al., 2000a). Cytisine-sensitive, higher-affinity [3H]epibatidine binding sites are the most numerous of the six subsets defined here, accounting for approximately 45% of the total in whole brain. Most of these sites are probably α4β2*-nAChR (Whiting and Lindstrom, 1988; Flores et al., 1992; Ross et al., 2000).

Major epibatidine binding subtypes identified using pharmacological and null mutant analysis. Putative nAChR subtypes identified by pharmacological methods are listed in the rectangular boxes. Further subdivision by examination of the effects of α7, β2, or β4 nAChR subunit gene deletion is shown by the gray ovals. Numbers above each subdivision are the average percentages of each binding subtype in whole wild-type mouse brain.
Higher-affinity cytisine-resistant [3H]epibatidine binding sites represent approximately 6% of total [3H]epibatidine binding in whole brain. Deletion of α7 had no effect on these sites. However, cytisine-resistant, higher-affinity [3H]epibatidine binding sites were reduced by the deletion of either β2 (38.4%) or β4 (55.2%). Thus, this subset of higher-affinity [3H]epibatidine binding sites is heterogeneous, and it requires the expression of either β2 or β4 nAChR subunits (the sum effect of β2 and β4 deletion was a 93.6 ± 8.3% reduction). The cytisine-resistant, higher-affinity binding sites in whole brain requiring the β2 subunit accounted for approximately 2.5%, whereas those requiring the β4* subunit accounted for approximately 3.5% of total [3H]epibatidine binding.
Numbers of cytisine-resistant, higher-affinity [3H]epibatidine binding sites differed markedly among the brain regions, as did the effects of β2 and β4 gene deletion on these sites. Both superior colliculus and inferior colliculus express relatively high levels of cytisine-resistant binding sites, but these sites respond quite differently to nAChR gene deletion. Sites in superior colliculus are predominantly β2*-nAChR, whereas those in inferior colliculus are predominantly β4*-nAChR. This observation is consistent with results of other studies that assessed potential subunit compositions of this diverse population of nAChR. For example, deletion of the α3-nAChR subunit significantly decreased A85380-resistant 125I-epibatidine binding in inferior colliculus (>80%) but had little effect on binding in superior colliculus (Whiteaker et al., 2002), indicating the presence α3β4*-nAChR in the inferior colliculus. Consistent with this assignment was the observation that the pharmacological properties of residual nAChR in the inferior colliculus of β2-/- mice were those expected for an α3β4-nAChR (Marks et al., 2002). In contrast, immunoprecipitation and null mutant analyses of the [3H]epibatidine binding sites have demonstrated that β4*-nAChR are relatively rare in superior colliculus and that the β2*-nAChR in this brain region are complex and include α3, α4, α6, β3, and α5 subunits (Gotti et al., 2005). Furthermore, the composition of nAChRs in retinal afferents differs from that in other neurons in the superior colliculus. The complexity of retinal nAChR has been confirmed independently (Marritt et al., 2005). Thus, the cytisine-resistant, higher-affinity [3H]epibatidine binding sites are a very complex mixture of nAChR requiring either β2 or β4 subunits for assembly.
In contrast to the considerable progress made in the characterization of the higher-affinity [3H]epibatidine binding sites, much less has been known about the lower-affinity sites even though these sites account for approximately one half of the total [3H]epibatidine binding. The current study demonstrates that deletion of the α7, β2, or β4 nAChR subunits defines unique subsets of the lower-affinity [3H]epibatidine binding sites and provides an initial characterization of these sites.
125I-αBgt binding is entirely dependent on expression of the α7 nAChR subunit, consistent with the effects reported previously for α7-/- mice (Orr-Urtreger et al., 1997). αBgt-sensitive, lower-affinity [3H]epibatidine binding is also completely α7-dependent. Although this subset of binding sites is not frequently investigated, an αBgt-sensitive, lower-affinity [3H]epibatidine binding site has also been identified in cells transfected with the α7 subunit (Peng et al., 2005). αBgt-sensitive sites were unaffected by the deletion of β2 or β4 subunits. Thus, αBgt-sensitive [3H]epibatidine binding can be used, as can 125I-αBgt binding, to measure α7-nAChR. However, results obtained by direct measurement of these sites with 125I-αBgt are more reliable than those obtained by measuring αBgt inhibition of [3H]epibatidine binding. Furthermore, site density measured by 125I-αBgt binding is higher than that measured by αBgt inhibition of [3H]epibatidine binding (Fig. 5).
Surprisingly, αBgt-sensitive, lower-affinity [3H]epibatidine binding sites account for only one third of the total lower-affinity sites in whole brain, although site density measured in this manner is lower than that measured with 125I-αBgt directly and may somewhat underestimate this subset of binding sites. The relatively high levels of the αBgt-resistant sites indicate that these are important [3H]epibatidine binding sites that merit further investigation. Deletion of α7 did not affect αBgt-resistant lower-affinity [3H]epibatidine binding, but deletion of either β2 or β4 significantly reduced expression of the αBgt-resistant sites. The differential responses to deletion of the β2 or β4 gene demonstrate that this subset of sites is composed of structurally diverse nAChR subtypes. In many brain regions, these sites require the expression of β2; in some brain regions, these sites require the expression of β4; whereas in other brain regions, both β2*-nAChR and β4*-nAChR are present. No information is presently available about the α-subunit composition of these αBgt-resistant sites. Furthermore, it is unknown whether these lower-affinity sites detect a different affinity state of known receptors, identify incompletely assembled receptors or measure mature, possibly functional, but lower-affinity receptors. α4β2-nAChRs with different stoichiometry and different sensitivity to activation by nicotinic agonists are assembled in heterologous expression systems (Zwart and Vijverberg, 1998; Zhou et al., 2003). If nAChRs with different α/β stoichiometries also have different affinities for ligand binding, higher- and lower-affinity sites could reflect these differentially assembled receptors. Indeed, [3H]epibatidine binding in cells transfected with α4 and β2 nAChR subunits is biphasic (Shafaee et al., 1999).
Deletion of one of the major structural subunits α7, β2, or β4 did not substantially change the expression of residual [3H]epibatidine binding sites. This is further supported by the observation that the number of sites remaining in α7/β2 double-null mutants was comparable with that measured after deletion of the β4 subunit. These results indicate that direct compensation for deletion of an nAChR subunit essential for assembly through the induction of a unique receptor subtype or increased expression of another existing subtype does not occur. In contrast, it should be noted that changes in relative expression of nAChR after deletion of auxiliary α5 or β3 subunits have been observed (Salminen et al., 2004).
In summary, the results reported here reveal two additional and novel classes of lower-affinity [3H]epibatidine binding sites in addition to the one that also interacts with αBgt. The results also confirm and expand the general classification of nAChR subtypes that bind [3H]epibatidine with high affinity (Zoli et al., 1998). Given the prevalence of these lower-affinity sites, which are unequivocally nicotinic, additional analyses including pharmacological, immunochemical, and genetic experiments are warranted.
Acknowledgments
We greatly appreciate the expert assistance of Natalie Meinerz, Julie Kachinski, and Theresa DelVecchio in genotyping the mutant mice.
Footnotes
-
This study was supported by National Institute on Drug Abuse research grant DA03194 and animal resources grant DA15663.
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; A85380, 3-((2S)-azetidinylmethoxy)pyridine; αBgt, α-bungarotoxin; ANOVA, analysis of variance; Epi, epibatidine.
- Received April 6, 2006.
- Accepted May 23, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}