


Abstract
5-(Trifluoromethyl)-6-(1-methyl-azepan-4-yl)methyl-1H-quinolin-2-one (TMAQ) is a novel nicotinic acetylcholine receptor (nAChR) agonist with strong selectivity for β4-containing receptors. TMAQ also exhibits remarkable species selectivity, being a potent agonist of nAChRs containing the human β4 subunit but having no detectable agonist activity on nAChRs containing the rat β4 subunit. With the aim of identifying subunit domains and individual amino acids, which contribute to the species selectivity of TMAQ, a series of chimeric and mutated β4 subunits has been constructed. Recombinant receptors containing wild-type, chimeric, or mutated β4 subunits have been examined by radioligand binding, intracellular calcium assays, and electrophysiological recording. Two adjacent amino acids located within the extracellular loop D domain of the β4 subunit (amino acids 55 and 56) have been identified as playing a critical role in determining the agonist potency of TMAQ. Mutagenesis of these two residues within the rat β4 subunit to the corresponding amino acids in the human β4 subunit (S55N and I56V mutations) confers sensitivity to TMAQ. The converse mutations in the human β4 subunit (N55S and V56I) largely abolish sensitivity to TMAQ. In contrast, these mutations have little or no effect on sensitivity to the nonselective nicotinic agonist epibatidine. Despite acting as a potent agonist of human β4-containing nAChRs, TMAQ acts as an antagonist of rat β4-containing receptors. Our experimental data, together with homology models of the rat and human α3β4 nAChRs, suggest that amino acids 55 and 56 may be involved in the coupling of agonist binding and channel gating.
Nicotinic acetylcholine receptors (nAChRs) are a family of pentameric ligand-gated ion channels that display considerable subunit diversity (Le Novère et al., 2002; Millar, 2003, 2006). In addition to nAChRs expressed at the neuromuscular junction (which are assembled from five different subunits: α1, β1, γ, δ, and ϵ), a heterogeneous population of neuronal nAChRs is expressed within the central and peripheral nervous systems. Twelve neuronal nAChR subunits (α2-α10 and β2-β4) have been identified in vertebrates, all of which (with the exception of α8) are expressed in mammals. The influence of subunit composition upon the pharmacological diversity of nAChRs is incompletely understood but is being investigated by a variety of experimental approaches. There is strong evidence, for example, that receptors containing α4 and β2 subunits are important subtypes within the brain, whereas receptors containing α3 and β4 subunits are important postsynaptic receptors in autonomic ganglia (McGehee and Role, 1995).
Neuronal nAChRs have been implicated in several neurological disorders and are being seen increasingly as important target sites for therapeutic drug discovery (Lloyd and Williams, 2000; Jensen et al., 2005). As a consequence, considerable efforts have been aimed at identifying subtypeselective nAChR ligands. As part of a program aimed at the development of subtype-selective nAChR agonists, a series of compounds has been identified that exhibits selectivity for nAChRs containing the β4 subunit. Here we describe one such compound, 5-(trifluoromethyl)-6-(1-methyl-azepan-4-yl)-methyl-1H-quinolin-2-one (TMAQ) (Fig. 1), a novel nicotinic agonist that exhibits selectivity for β4-containing nAChRs. It is interesting that TMAQ also exhibits strong species selectivity. TMAQ is a potent agonist of receptors containing the human β4 subunit but not of receptors containing the rat β4 subunit. This is somewhat surprising, given the relative similarity in the primary amino acid sequence of the human and rat β4 subunits.

Structure of TMAQ and epibatidine.
In the present study, we identified amino acids responsible for conferring species selectivity of TMAQ by the construction and characterization of a series of chimeric and mutated β4 subunits. Recombinant nAChRs have been examined by radioligand binding, intracellular calcium assays, and by electrophysiological recording. Experimental data are interpreted with reference to homology models of the human and rat α3β4 nAChR extracellular domain, which are based on the atomic resolution structure of the acetylcholine binding protein (AChBP) from Lymnaea stagnalis (Brejc et al., 2001; Celie et al., 2004).
Materials and Methods
Chemical Synthesis. TMAQ was synthesized by Lilly Research Laboratories. In brief, (4-nitro-2-trifluoromethylbenzyl)-phosphonic acid diethyl ester and 1-tert-butoxycarbonylazepan-4-one were coupled under standard Wadsworth Emmons conditions. The product was subjected to Pd/C-catalyzed hydrogenation to give 4-(4-amino-2-trifluoromethyl-benzyl)-azepane-1-carboxylic acid tert-butyl ester. Quinolone ring formation and concomitant removal of the N-boc group was achieved with 3-ethoxyacryloyl chloride/concentrated sulfuric acid followed by a reductive methylation of the azepan nitrogen with aqueous formaldehyde/sodium triacetoxyborohydride. Purification by flash chromatography on silica gel followed by chiral chromatography on a Chiralcel OD column (collecting to first enantiomer to elute) gave TMAQ as a white solid. Purity was determined to be >99% by liquid chromatographic mass spectrometry and proton NMR. 1H NMR (400 MHz, CDCl3) δ 1.32-1.92 (7H, m), 2.34 (3H, s), 2.43-2.68 (4H, m), 2.83 (2H, d), 6.81-8.23 (4H, m), 12.92 (1H, br s); flow injection analysis-mass spectrometry MH+= 339.
Subunit cDNAs, Subunit Chimeras, and Site-Directed Mutagenesis. Human nAChR α3 and β4 subunit cDNAs (Elliott et al., 1996) were obtained from Merck Research Laboratories (La Jolla, CA). Rat nAChR α3 and β4 subunit cDNAs (Boulter et al., 1986; Duvoisin et al., 1989) were provided by Jim Patrick (Baylor College of Medicine, Houston, TX). All nAChR subunit cDNAs were subcloned into plasmid expression vector pcDNA3 (Invitrogen, Paisley, UK). A unique BstEII restriction site, present in both the rat and human β4 subunit cDNAs, located between the second and third transmembrane domains, was used to construct chimeric cDNAs in which the N- and C-terminal regions were exchanged (h/rβ4 and r/hβ4). Site-directed mutagenesis of nAChR subunit cDNAs was performed using the QuikChange mutagenesis kit (Stratagene, Amsterdam, the Netherlands). All chimeric and mutated subunits were verified by nucleotide sequencing using the Big Dye Terminator Cycle Sequencing kit and ABI Prism 3100-Avant automated sequencer according to the manufacturer's instructions (Applied Biosystems, Warrington, UK).
Mammalian Cell Lines Stably Expressing nAChRs. Human embryonic kidney (HEK) 293 cell lines stably expressing human recombinant neuronal nAChR subtypes α2β4, α3β4, α4β4, and α4β2, andaGH4C1 cell line stably expressing the human α7 nAChR were obtained from Merck Research Laboratories. Conditions used to culture these stable cell lines have been described previously (Broad et al., 2002). A human embryonic rhabdomyosarcoma cell line (RD) expressing the muscle-type nAChR was obtained from the American Type Culture Collection (Manassas, VA).
Transient Expression of nAChRs in Mammalian Cells. Human kidney tsA201 cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen) containing 10% fetal calf serum (Sigma, Poole, UK), penicillin (100 U/ml), and streptomycin (100 mg/ml) (Invitrogen). Cells were maintained in a humidified incubator containing 5% CO2 at 37°C. Cells were transfected using the Effectene reagent (QIAGEN, Crawley, UK) according to the manufacturer's instructions. After overnight incubation in Effectene, cells were incubated at 37°C for 24 h before being assayed for radioligand binding or intracellular calcium recording.
Radioligand Binding. [3H]Epibatidine (55.5 Ci/mmol) was purchased from PerkinElmer Life and Analytical Sciences (Beaconsfield, UK). Radioligand binding to transiently transfected tsA201 cells was performed essentially as described previously (Baker et al., 2004). Cell membranes (typically 80-150 μg of protein) were incubated with radioligand for 150 min at 4°C in a total volume of 300 μl in the presence of protease inhibitors leupeptin (2 μg/ml) and pepstatin (1 μg/ml). Nonspecific binding was determined in the presence of nicotine (1 mM) and carbamylcholine (1 mM). To avoid ligand depletion, binding was performed in a larger volume (2 ml) for concentrations of [3H]epibatidine lower than 0.3 nM. Radioligand binding was assayed by filtration onto Whatman GF/B filters (presoaked in 0.5% polyethylenimine), followed by rapid washing with ice-cold 10 mM phosphate buffer using a Brandel cell harvester. Bound radioligand was quantified by scintillation counting. For competition binding experiments with TMAQ, a fixed concentration (typically 0.3 nM) of [3H]epibatidine was used. IC50 values were converted to Ki values using the equation Ki = IC50/1 + ([L]/Kd), in which L is the free concentration of [3H]epibatidine used in the assay and Kd is the dissociation constant for binding of [3H]epibatidine. Curves for equilibrium binding were fitted with the Hill equation using Prism version 4 (GraphPad Software, San Diego, CA).
Intracellular Calcium Assays. Transfected tsA201 cells were replated onto poly(l-lysine)-coated black-walled 96-well plates (Marathon Laboratories, London, UK) approximately 18 to 20 h after transfection. After ∼24 h, cell medium was removed, and the cells were incubated in 50 to 100 μl of 1 μM Fluo-4 acetoxymethyl ester (Invitrogen) in Hanks' balanced salt solution with 0.02% Pluronic F-127 (Invitrogen) for 45 to 60 min at room temperature. Cells were rinsed once with 160 μl of assay buffer (Hanks' balanced salt solution supplemented with 18.8 mM CaCl2, 8.8 mM sucrose, and 6.3 mM HEPES), and the cells were assayed using a fluorometric imaging plate reader (FLIPR) (Molecular Devices, Winnersh, UK), as described previously (Lansdell et al., 2005). Drug dilutions in assay buffer were prepared in a separate 96-well plate. Parameters for drug addition to the cell plate were preprogrammed, and delivery was automated through a 96-tip head pipettor. Experimental conditions for FLIPR experiments conducted with stably transfected HEK 293 and GH4C1 cells and with cultured human rhabdomyosarcoma RD cells were as described previously (Broad et al., 2002; Craig et al., 2004). Dose-response curves were constructed by measuring peak responses with TMAQ and normalizing to maximal peak response with acetylcholine or epibatidine. Curve-fitting was performed with GraphPad Prism version 4 (GraphPad Software).

Agonist concentration-response curves for TMAQ determined by FLIPR-based intracellular calcium assay. Initial characterization of the nAChR subtype selectivity of TMAQ was performed using a 96-well FLIPR-based intracellular calcium assay with cultured cells expressing either native or recombinant nAChRs. Data were obtained from stably transfected HEK cell lines expressing the following human recombinant neuronal nAChR subtypes: α2β4 (▪), α3β4 (•), α4β4 (▴), and α4β2 (□). Additional data were obtained from a stably transfected GH4C1 cell line expressing the human α7 receptor (○) and from a human embryo rhabdomyosarcoma cell line (RD) expressing the native muscle-type nAChRs (▵). Results obtained with TMAQ are normalized to the maximum response obtained with 1 mM acetylcholine in the presence of 3 μM atropine for each subunit combination. Data points are means of six to eight independent experiments.
Xenopus laevis Oocyte Expression and Electrophysiology. Adult female X. laevis frogs were obtained from Xenopus Express (Vernassal, France). X. laevis oocytes (stages V-VI) were removed from schedule 1 sacrificed toads and defolliculated by treatment with 6 mg/ml collagenase type I (Sigma) in calcium-free Barth's solution for 4 h at room temperature. Plasmids containing the human α3, human β4, and rat β4 subunit coding sequences were suspended in distilled water and injected into the nuclei of the oocytes immediately after the collagenase treatment using a Drummond variable volume microinjector. Subunit cDNAs were coinjected in a 1:1 ratio, and approximately 2 ng of cDNA was injected in a total injection volume of 18.4 nl/oocyte. After injection, oocytes were incubated at 18°C in a modified Barth's solution containing 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.3 mM Ca(NO3)2, 0.41 CaCl2, 0.82 mM MgSO4, 15 mM HEPES, and 50 mg/l neomycin, pH 7.6 with NaOH (osmolarity, 235 mOsM). Experiments were performed on oocytes after 3 to 5 days of incubation. Oocytes were placed in a recording chamber (internal diameter, 3 mm), which was continuously perfused with a saline solution (115 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, and 10 mM HEPES, pH 7.3 with NaOH, 235 mOsm) at a rate of ∼10 ml/min. Dilutions of drugs in external saline were prepared immediately before the experiments and applied by switching between control and drug-containing saline using a BPS-8 solution exchange system (ALA Scientific Inc., Westbury, NY). Agonist applications were alternated by 5 min of superfusion with agonist-free saline to allow the receptors to recover from desensitization. Oocytes were impaled by two microelectrodes filled with 3 M KCl (0.5-2.5 MΩ) and voltage-clamped using a Geneclamp 500B amplifier (Molecular Devices). The external saline was clamped at ground potential by means of a virtual ground circuit using an Ag/AgCl reference electrode and a Pt/Ir current-passing electrode. The membrane potential was held at -60 mV. The current required to keep the oocyte membrane at the holding potential was measured. Membrane currents were low-pass-filtered (four-pole low-pass Bessel filter, -3dB at 100 Hz), digitized (300 Hz), and stored on disk for offline computer analysis. Data are expressed as mean ± S.E.M. All experiments were performed at room temperature. For antagonist concentration-response experiments an EC20 concentration of ACh (30 μM) was applied for 45 s. After 15 s of ACh application, ACh was coapplied with antagonist for 15 s, after which another 15-s period followed of ACh application in the absence of antagonist. Current amplitudes at the end of the coapplication of ACh with antagonist were measured and normalized to the current amplitudes of the ACh control response just before coapplication. Concentration-response curves were fitted to the data obtained in separate experiments, and mean ± S.E.M. of estimated parameters were calculated for n oocytes. Standard inhibition curves were fitted according to the equation i/imax = 1/{1 + ([antagonist]/IC50)nH. Curve-fitting was performed using Prism version 4 (GraphPad Software).
Computer Modeling. A homology model of the human α3β4 nAChR extracellular domain was constructed using the crystal structure of the Lymnaea stagnalis AChBP with bound nicotine (Protein Data Bank code 1UW6; protomer units A&E) as a starting template (Celie et al., 2004). Amino acid sequence alignments were generated using the CLUSTAL X program (Thompson et al., 1997). Nicotinic receptor α3 and β4 subunit models were generated with the MODELER program (Sali and Blundell, 1993), and individual α3 and β4 subunit models were then refitted back onto the AChBP structure to generate an α3β4 subunit dimer model in which α3 corresponds to the principal subunit and β4 to the complementary subunit.
Results
The ability of TMAQ to activate diverse nAChR subtypes was examined in a series of mammalian cell lines expressing native or recombinant receptors. Cells were plated in 96-well tissue culture plates, and agonist-induced changes in intracellular calcium were recorded with a FLIPR. TMAQ exhibited potent agonist activity on several human recombinant β4-containing nAChRs (α2β4, α3β4, and α4β4). In contrast, little or no agonist activity was observed on human recombinant α4β2 or α7 nAChRs or on native human muscle-type nAChRs expressed in the rhabdomyosarcoma RD cell line (Fig. 2).
The ability of TMAQ to act as an agonist of human α3β4 nAChRs was confirmed by FLIPR-based assays with transiently transfected human embryonic kidney cells (Fig. 3A). Despite clear evidence that TMAQ acts as a potent agonist of human α3β4 nAChRs (EC50 = 0.2 ± 0.1 μM; n = 4), TMAQ displayed no detectable agonist activity in cells transfected with rat α3β4 nAChRs (Fig. 3B). In contrast, epibatidine acted as a potent agonist of both human and rat α3β4 nAChRs (Fig. 3, A and B). To examine the contribution of α3 and β4 subunits to TMAQ sensitivity, experiments were performed with human-rat hybrid nAChRs. Responses to TMAQ were detected in cells expressing the rat α3 subunit with human β4 (EC50 = 0.3 ± 0.03 μM; n = 3), but not in cells transfected with human α3 subunit with rat β4 (Fig. 3C). This provided evidence that the species-selectivity of TMAQ was a consequence of differences in the human and rat β4 subunits.
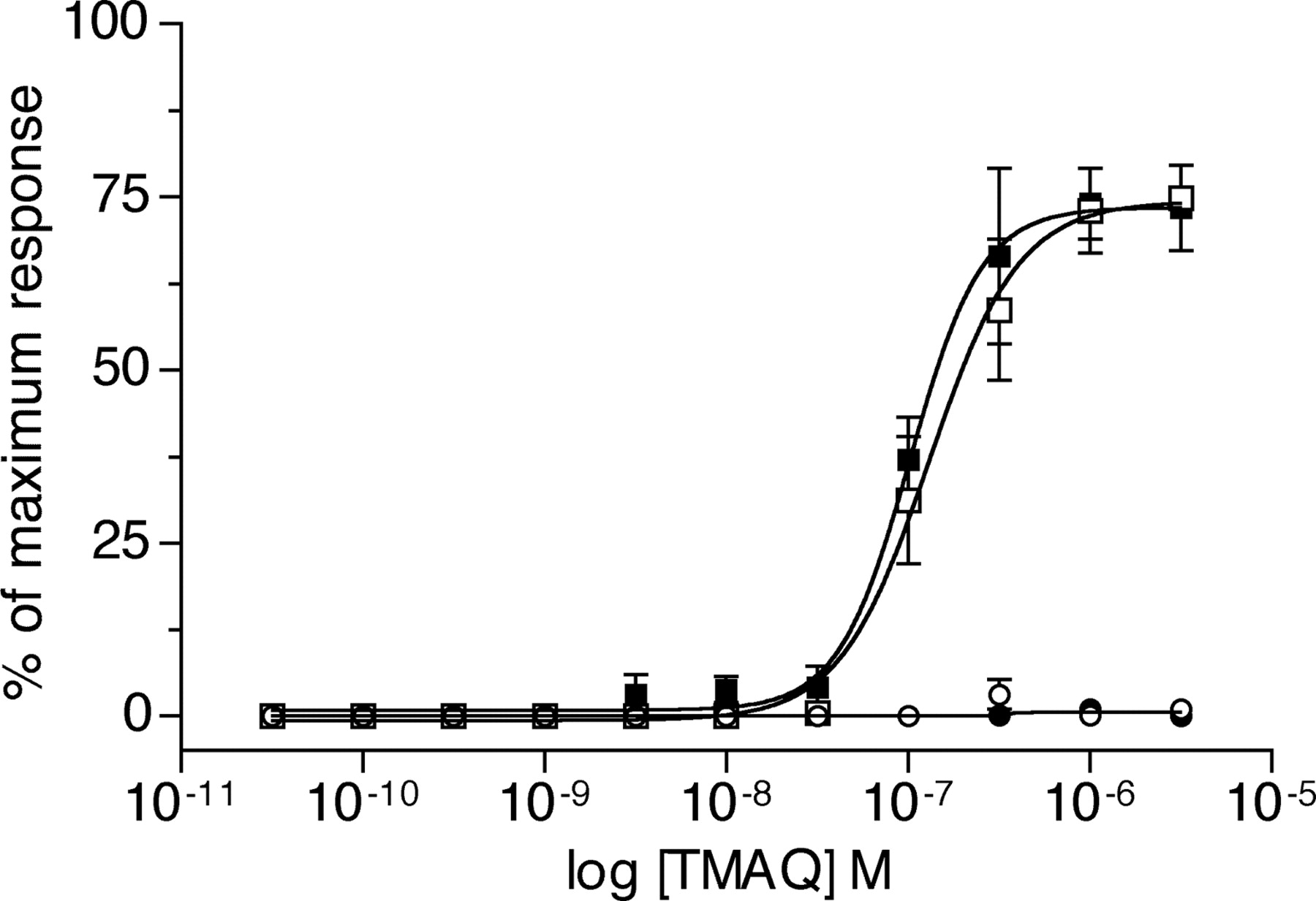
With the aim of identifying subunit domains conferring sensitivity to TMAQ, two subunit chimeras (h/rβ4 and r/hβ4) were constructed using a unique BstEII site located within the M3 transmembrane domain of both subunits. Alignments of the human and rat β4 subunit amino acid sequences revealed that their mature proteins differ by 82 amino acids. Of these amino acid differences, 19 are located in the region N-terminal to the BstEII site that was used to construct the chimeras (Fig. 4), and 63 are C-terminal to this site. The ability of TMAQ to activate nAChRs containing the human/rat and rat/human β4 subunit chimeras (h/rβ4 and r/hβ4) was examined by FLIPR-based intracellular calcium assays. Cells were cotransfected with the human α3 subunit in combination with wild-type or chimeric β4 subunits. In all cases, responses to TMAQ were compared with maximal responses obtained with epibatidine, an agonist that activates both human β4 (hβ4) and rat β4 (rβ4) containing nAChRs. As shown in Fig. 5, TMAQ induced dose-dependent responses in cells transfected with either the hβ4 subunit or the h/rβ4 subunit chimera. The EC50 value (0.1 ± 0.1 μM; n = 4) and maximum response observed with nAChRs containing the h/rβ4 subunit chimera were not significantly different from that observed with hα3 + hβ4 nAChRs. In contrast, no responses were detected to TMAQ, even at high agonist concentrations, in cells transfected with either the rβ4 subunit or with the r/hβ4 subunit chimera (Fig. 5).

Agonist potency of epibatidine and TMAQ upon nAChR subtypes determined by FLIPR-based intracellular calcium assay. Representative responses obtained with 1 μM epibatidine (a maximally effective concentration, •) and 3 μM TMAQ (a maximally effective concentration, ○) in cells transfected with human α3β4 (A) and rat α3β4 (B). C, concentration-response curves showing responses to TMAQ normalized to the maximum response observed with epibatidine. Data were obtained with transiently transfected human embryonic kidney (tsA201) cells expressing the human α3β4 nAChRs (▪), rat α3β4 nAChRs (•), or to hybrid receptors containing hα3 + rβ4 (○) or rα3 + hβ4 (□). Data are means (± S.E.M.) derived from four independent experiments, each of which was performed in quadruplicate.

Alignment of amino acid sequences in the human and rat β4 subunits. The amino acid sequence of each subunit is shown from the first amino acid in the predicted mature protein up to a position within the M3 transmembrane domain corresponding to the BstEII site that was used in the construction of chimeric subunits. Horizontal lines above the sequences indicate the positions of transmembrane domains M1 to M3 and of loops D to F. The position of the amino acids mutated in this study (at positions 55 and 56) are indicated by asterisks below the amino acid sequences.
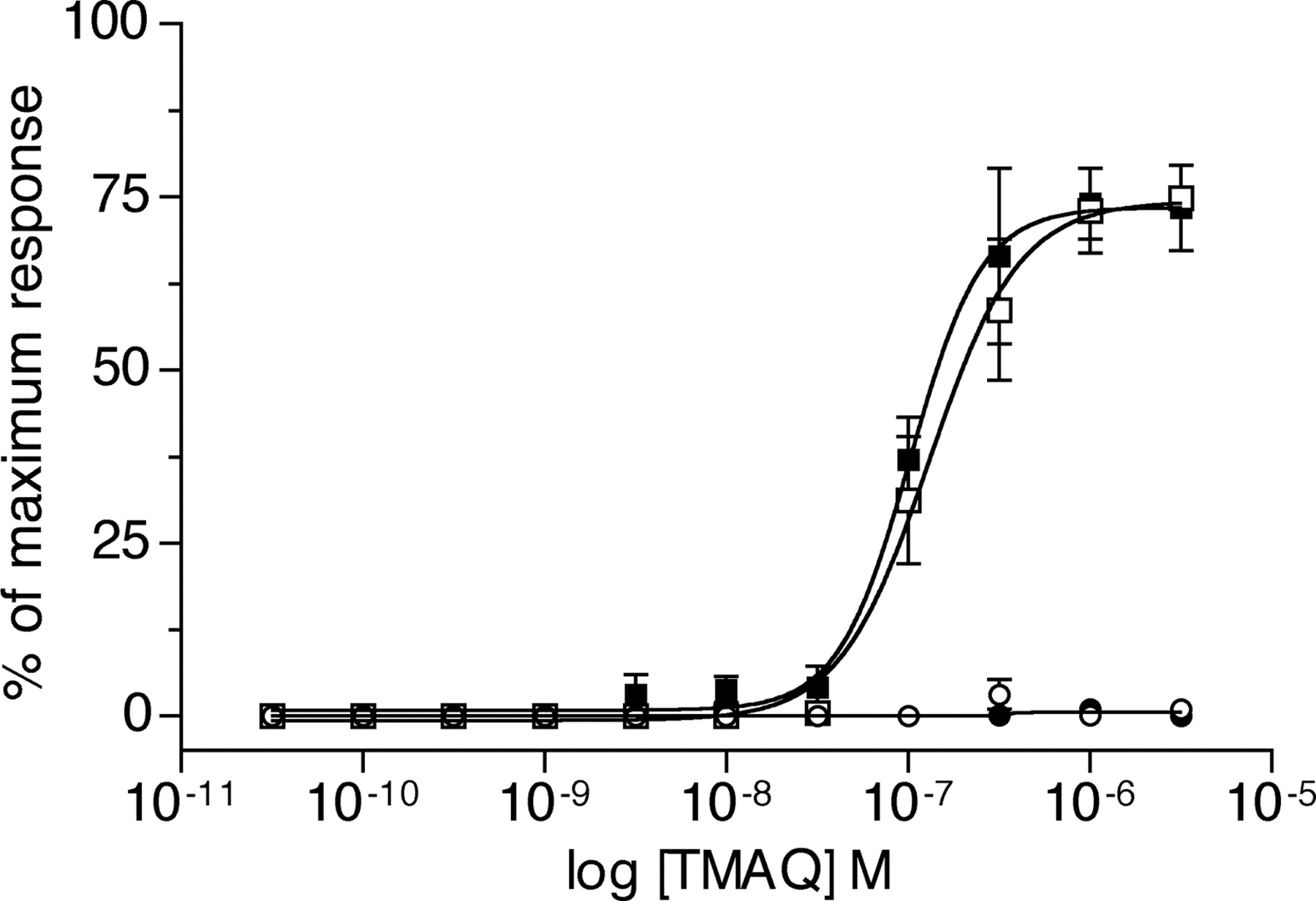
Agonist concentration-response curves of TMAQ upon nAChR subtypes containing chimeric β4 subunits determined by FLIPR-based intracellular calcium assay. Data were obtained with transiently transfected tsA201 cells expressing the human α3 subunit coexpressed with human β4 (▪), rat β4 (•), chimeric h/rβ4 (□), or chimeric r/hβ4 (○) subunits. Results obtained with TMAQ are normalized to the maximum response obtained with 1 μM epibatidine for each subunit combination. Data are means (± S.E.M.) derived from four independent experiments, each of which was performed in quadruplicate.
The previous findings indicate that species-selectivity of TMAQ is conferred by residues present within the N-terminal region of the β4 subunit. Of the 19 amino acid differences in the N-terminal region of the β4 subunits, two amino acids (at positions 55 and 56), which are located within the loop D domain of the β4 subunit, were examined by site-directed mutagenesis. As shown in Fig. 4, the human β4 subunit contains asparagine and valine at these positions, whereas the rat β4 subunit contains serine and isoleucine. Initially both amino acids were mutated individually in the rat β4 subunit to their corresponding amino acids in the human β4 subunit (to create mutated subunits rβ4S55N and rβ4I56V).

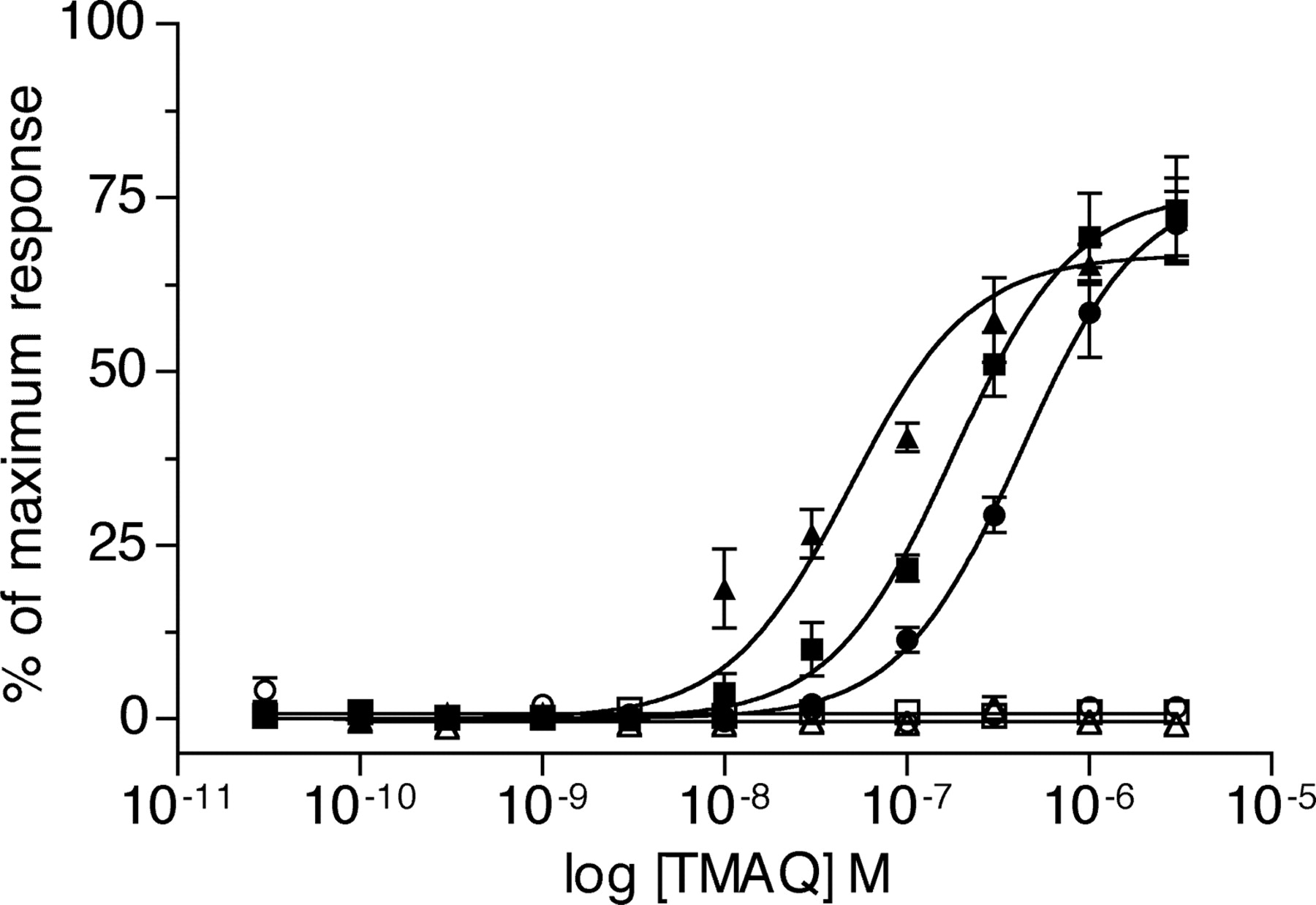
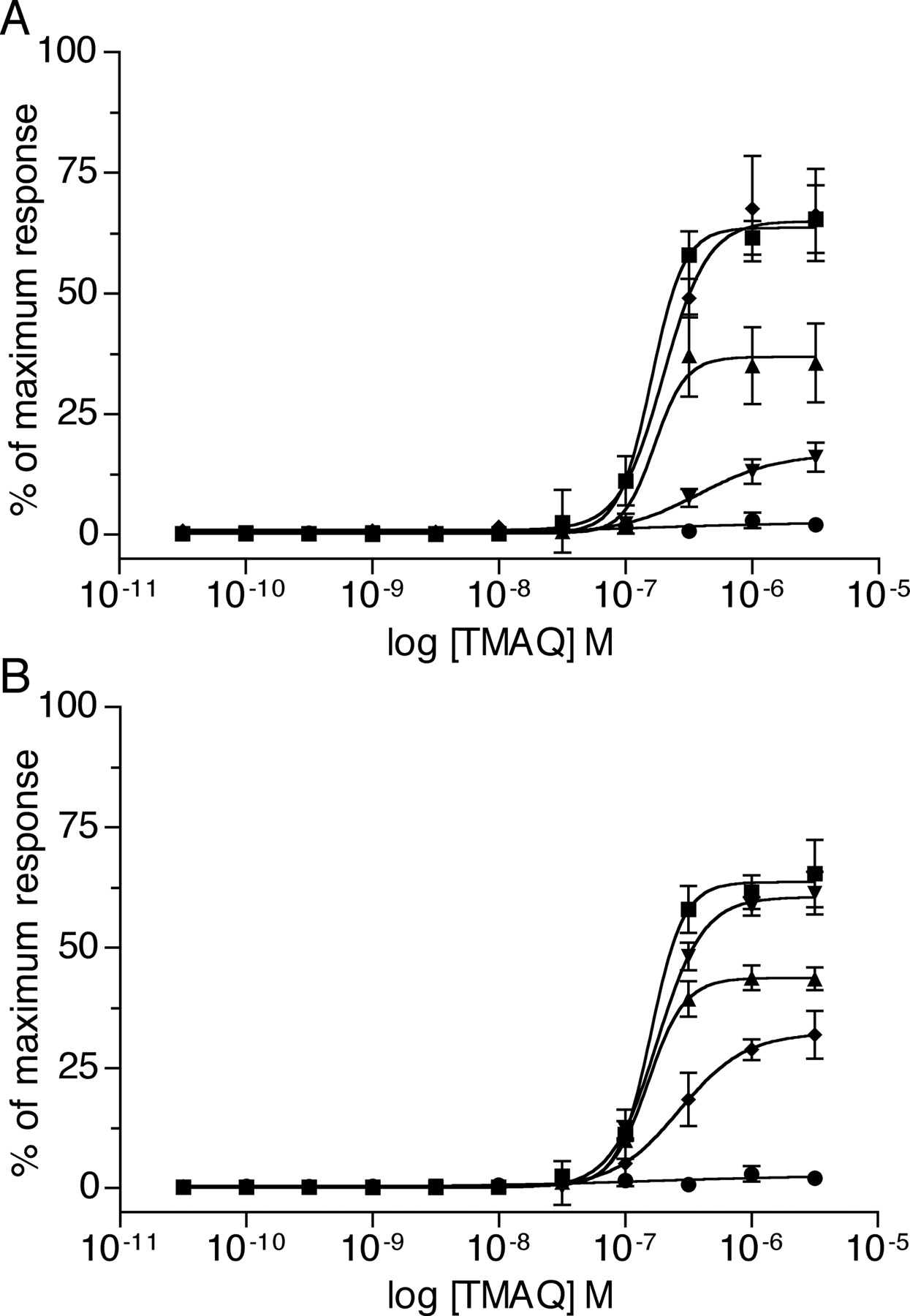
Agonist concentration-response curves of TMAQ upon nAChR subtypes containing mutated β4 subunits determined by FLIPR-based intracellular calcium assay. In all cases, the human α3 subunit has been coexpressed in transiently transfected tsA201 cells with either human β4 (▪), rat β4 (•), or with mutated β4 subunits. A, data obtained with mutated rat β4 subunits: rβ4S55N (▴), rβ4I56V (▾), or rβ4S55N,I56V (♦). B, data obtained with mutated human β4 subunits: hβ4N55S (▴), hβ4V56I (▾), or hβ4N55S,V56I (♦). Results obtained with TMAQ are normalized to the maximum response obtained with epibatidine for each subunit combination. Data are means (± S.E.M.) derived from three to four independent experiments, each of which was performed in quadruplicate.

Characterization of recombinant nAChRs by radioligand binding. A, saturation binding with [3H]epibatidine to tsA201 cells expressing hα3 + hβ4 nAChRs (squares) and hα3 + rβ4 nAChRs (circles). Both specific binding (filled symbols) and nonspecific binding (open symbols) is shown. B, binding of TMAQ examined by competition radioligand binding. Data are from nAChRs containing the human α3 subunit coexpressed with hβ4 (▪), rβ4 (•), hβ4N55S,V56I (□), or rβ4S55N,I56V (○). Results are presented as a percentage of maximum binding with [3H]epibatidine. Data are from a representative experiment performed in triplicate, typical of three independent experiments.
The mutated rat β4 subunits (coexpressed with human α3) were examined by means of FLIPR-based intracellular calcium assays. As illustrated in Fig. 6A, when the human α3 subunit was coexpressed with either of the singly mutated rat β4 subunits (rβ4S55N or rβ4I56V), nAChRs were generated that were sensitive to TMAQ. In both cases, however, a lower maximal response was seen for nAChRs containing the mutated rβ4 subunits than for nAChRs containing the wild-type human β4 subunit (Fig. 6A). The maximum responses observed with rβ4S55N and rβ4I56V were 58 ± 9% (n = 4) and 26 ± 3% (n = 3) of the maximum response detected with the wild-type human β4 subunit. Further mutagenesis was performed to create a rat β4 subunit containing both of these mutations (rβ4S55N,I56V). The EC50 value and maximum response observed with TMAQ upon receptors containing the human α3 subunit coexpressed with rβ4S55N,I56V was indistinguishable from that observed with the wild-type human α3β4 nAChRs (Fig. 6A). In contrast, mutation of two adjacent amino acids within the loop E domain of the rat β4 subunit (to create rβ4I118V,Q119L) had no effect on maximal agonist responses with TMAQ when examined by FLIPR-based intracellular calcium assays (data not shown).
To confirm the influence of the two loop D amino acids upon the species-selectivity of TMAQ, a series of mutations was created within the human β4 subunit. The two amino acids within the human β4 subunit were mutated individually and in combination to create mutated subunits hβ4N55S, hβ4V56I, and hβ4N55S,V56I. As previously, the mutated subunits were coexpressed with the human α3 subunit and examined by a FLIPR-based intracellular calcium assay (Fig. 6B). Receptors containing the hβ4V56I mutation did not exhibit a significant reduction in maximal agonist response observed with TMAQ, but receptors containing the hβ4N55S mutation showed a maximum response to TMAQ, which was 69 ± 2% (n = 4) of that with the human β4 subunit. An even more dramatic reduction in maximal response with TMAQ was observed in receptors containing hβ4N55S,V56I (maximum responses with TMAQ were reduced to 51 ± 5%; n = 3 of that observed with the human β4 subunit). Thus, although sensitivity to TMAQ was not completely abolished by mutation of the human β4 subunit (Fig. 6B), these findings, taken together with the dramatic results observed with the mutated rat β4 subunit (Fig. 6A), help to demonstrate the importance of these two adjacent amino acid in conferring species-selectivity of TMAQ.
Competition radioligand binding was performed to examine the affinity of TMAQ binding to nAChRs containing wildtype and mutated β4 subunits. Saturation binding studies were first performed to determine the affinity of [3H]epibatidine for nAChRs containing the human α3 subunit coexpressed with either human β4orrat β4 (Fig. 7A). Epibatidine bound with a similar high affinity to both nAChRs. The calculated Kd values were not significantly different for hα3 + hβ4 nAChRs (0.34 ± 0.02 nM; n = 3) and hα3 + rβ4 nAChRs (0.33 ± 0.03 nM; n = 3). Competition binding studies were then performed to determine the affinity of TMAQ binding to nAChRs containing the human α3 subunit coexpressed with wild-type and mutant β4 subunits (Fig. 7B). TMAQ bound with high affinity to hα3 + hβ4 nAChRs (Ki = 1.2 ± 0.1 nM; n = 3) but with significantly lower affinity (P = 0.028) to hα3 + rβ4 nAChRs (Ki = 17 ± 5 nM; n = 3). TMAQ binding to nAChRs containing rβ4S55N,I56V (Ki = 2.2 ± 0.6 nM; n = 3) was not significantly different to the high-affinity binding seen to nAChRs containing the human β4 subunit. TMAQ binding to nAChRs containing hβ4N55S,V56I (Ki = 11 ± 3 nM; n = 3) was not significantly different from the lower-affinity binding to nAChRs containing the rat β4 subunit. We can conclude that amino acids 55 and 56 are responsible for a modest but significant (∼5- to 14-fold) difference in the affinity of binding of TMAQ to nAChRs containing the human and rat β4 subunit. Thus, although TMAQ exhibits little or no agonist activity of rβ4-containing nAChRs, it retains the ability to bind to rβ4-containing nAChRs with high affinity. In contrast, competition binding experiments revealed that TMAQ bound to recombinant human α4β2 and α7 nAChRs with much lower affinity (1.3 ± 0.1 and >30 μM, respectively; n = 3; data not shown).
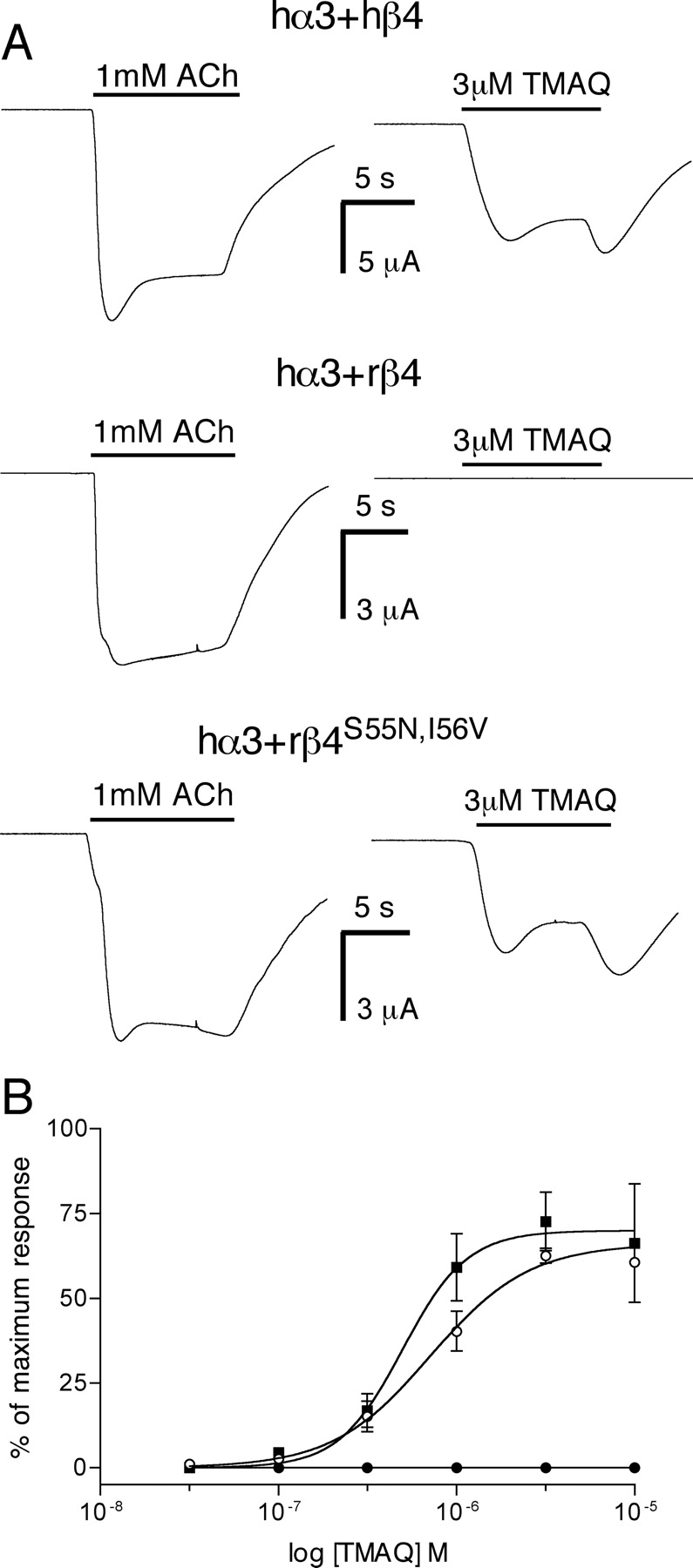
Characterization of recombinant nAChRs by two-electrode voltage clamp recording in X. laevis oocytes. A, both ACh and TMAQ generated agonist responses in oocytes expressing hα3 + hβ4 (top) and in hα3 + rβ4S55N,I56V (bottom). In contrast, TMAQ did not generate a measurable agonist response in oocytes expressing hα3 + rβ4 (middle). B, doseresponse experiments revealed that TMAQ was a potent agonist of hα3 + hβ4 (▪) and in hα3rβ4S55N,I56V nAChRs (○) but showed no significant agonist activity on hα3 + rβ4 nAChRs (•) at concentrations up to 10 μM. Data points are means (± S.E.M.) from three independent experiments.
Further experiments were performed with recombinant α3β4 nAChRs expressed in X. laevis oocytes (Fig. 8). Acetylcholine induced large inward currents in oocytes expressing hα3 + hβ4, hα3 + rβ4, and hα3 + rβ4S55N,I56V nAChRs, whereas agonist responses to TMAQ were detected only in oocytes expressing hα3 + hβ4 and hα3 + rβ4S55N,I56V (Fig. 8A), findings that are in agreement with the data obtained by intracellular calcium recordings. Dose-response experiments were performed (Fig. 8B), which revealed that TMAQ had no significant agonist activity on hα3 + rβ4 nAChRs (at concentrations up to 10 μM) but was a potent agonist of hα3 + hβ4 nAChRs (EC50 = 0.5 ± 0.1 μM; n = 3). TMAQ was also a potent agonist on hα3 + hβ4N55S,V56I nAChRs, with an EC50 of 0.3 ± 0.1 μM (n = 3), which is not significantly different from that observed on hα3 + hβ4 nAChRs (Fig. 8B).
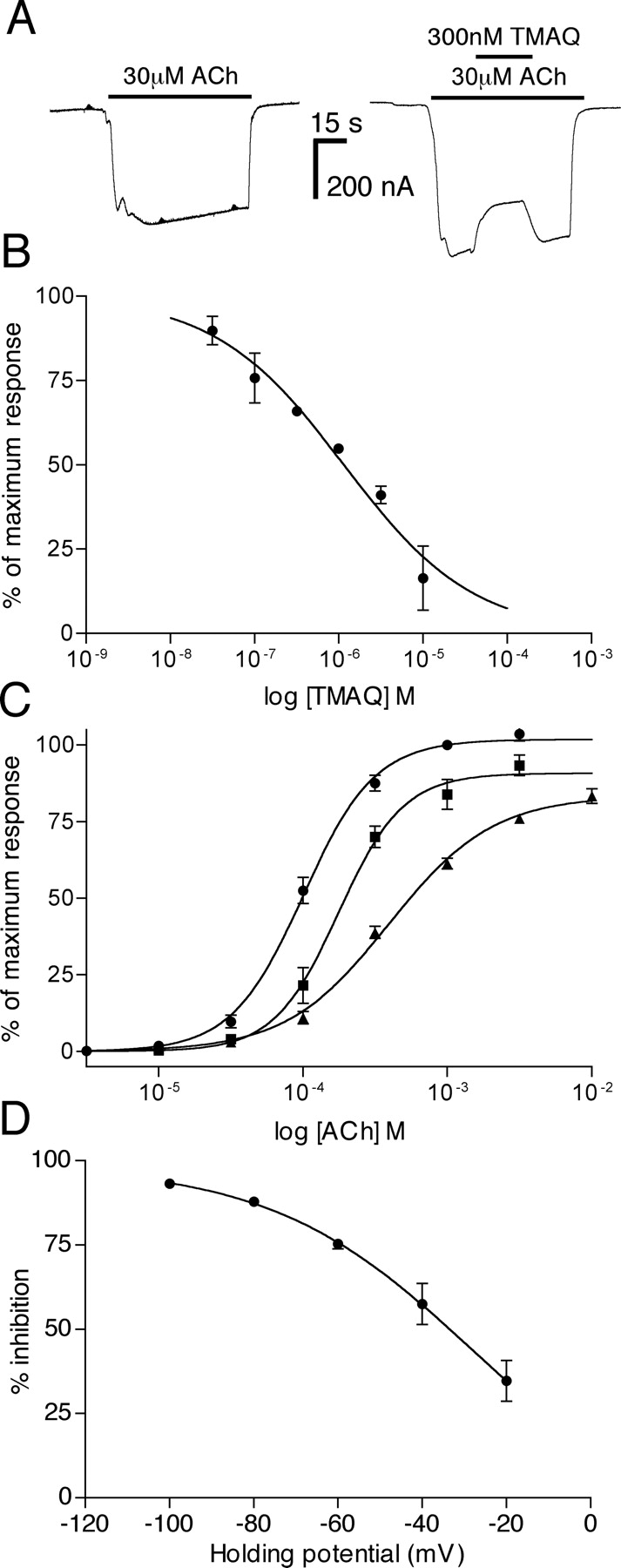
It is interesting that when TMAQ was coapplied with ACh to oocytes expressing hα3 + rβ4, evidence of dose-dependent, reversible antagonism was observed (Fig. 9, A and B). The antagonist activity of TMAQ on hα3 + rβ4 nAChRs was examined further by construction of ACh dose-response curves in the absence and presence of TMAQ (Fig. 9C). TMAQ caused a significant rightward shift of the ACh doseresponse curve (P = 0.014, for both 1 and 10 μM TMAQ). The EC50 for ACh determined in the absence of TMAQ was 107 ± 8 μM (n = 3) but was 177 ± 15 μM (n = 3) in the presence of 1 μM TMAQ and 407 ± 71 μM (n = 3) in the presence of 10 μM TMAQ. TMAQ caused a reduction in the maximal ACh response (91 ± 4% of maximum with 1 μM TMAQ and 84 ± 3% of maximum with 10 μM TMAQ). The voltage-dependence of TMAQ's antagonist activity was examined by determining the % inhibition caused by 1 μM TMAQ upon responses to 100 μM ACh at a range of holding potentials (from -20 to -100 mV; Fig. 9D). Taken together, these results suggest that the antagonist activity of TMAQ on hα3 + rβ4 nAChRs is at least partly a consequence of noncompetitive antagonism caused by channel block.
With the aim of investigating further the role of amino acids 55 and 56 upon nAChR function, a homology model of the extracellular domain of the human α3β4 nAChR was generated (Fig. 10) based on the atomic-resolution structure of the L. stagnalis AChBP with bound nicotine (Celie et al., 2004). Examination of this homology model, together with a model described previously of the rat α3β4 nAChRs (Costa et al., 2003), indicates that amino acids 55 and 56 of the β4 subunit do not lie within the interaction shell of the agonist binding site (Fig. 10).
Based on the nAChR α3β4 homology models, amino acid 55 (Ser55 in rβ4 and Asn55 in hβ4) is predicted to be external to the nAChR subunit core (Fig. 10) and is located at the interface of the β4 and α3 subunits. The main subunit interactions in this region are between loop domains linking β-sheet structures (the β4-β5 loop on the α3 subunit and the β1-β2 loop on the β4 subunit). In the human α3β4 model, the Asn55 amine side chain is 4 Å from the backbone amide carbonyl, whereas in the rat α3β4 model, the Ser55 hydroxyl side chain is 5.3 Å from this position. This suggests that a hydrogen bond interaction would be possible for Asn55 but, if present, would be much weaker for Ser55. Hence, the mutation rβ4S55N may help to stabilize subunit-subunit interactions and may account for the increased maximal response of TMAQ (Fig. 6). It should be noted, however, that there is a single amino acid insertion in the nAChR α3 sequence (YNNAV) compared with the AChBP sequence (YN-AI) within the β4-β5 loop region of the principal subunit. As a consequence, accurate modeling of this region is difficult.

Antagonist action of TMAQ on hα3 + rβ4 nAChRs. A, in oocytes expressing hα3 + rβ4 nAChRs, TMAQ exhibited antagonist activity when coapplied with ACh. B, in oocytes expressing hα3 + rβ4 nAChRs, TMAQ exhibited dose-dependent antagonist activity (IC50 = 1.2 μM; nH = 0.6) when coapplied with ACh (30 μM). C, dose-response curves to ACh were performed in the absence of TMAQ (•) and presence of TMAQ (either 1 μM TMAQ, ▪, or 10 μM TMAQ, ▴). D, the influence of the membrane holding potential upon the antagonist activity of TMAQ on hα3 + rβ4 nAChRs was examined and is presented as the percentage of inhibition of a response to 100 μM ACh by 1 μM TMAQ. Data points in B through D are means (± S.E.M.) from three independent experiments.
Based on the nAChR α3β4 homology models, amino acid 56 (Ile56 in rβ4 and Val56 in hβ4) is predicted to be internal to the nAChR subunit core (Fig. 10). The surrounding residues are hydrophobic in nature and would be predicted to stabilize the subunit tertiary structure. The increase in the maximal response for TMAQ observed with the rβ4I56V mutant (Fig. 6A), and the decrease in maximal response for the hβ4V56I mutant (Fig. 6B) highlights how the replacement of one branched hydrophobic side chain for another similar side chain results in only a relatively small effect on TMAQ efficacy. It is, perhaps, logical that an increase in side-chain size (in hβ4V56I) might result in a reduction in agonist efficacy (Fig. 6B) as a result of perturbation of the hydrophobic subunit core, but it is interesting that a decrease in side-chain size (in rβ4I56V) causes an increase in agonist efficacy for TMAQ (Fig. 6A).
It is possible that the effect of the mutations within the α3β4 nAChR is to create a binding site in the locality of the mutations that is suitable for TMAQ to stabilize an open conformation of the receptor. Analysis of the homology model of the rat α3β4 nAChR (Costa et al., 2003) has identified possible alternative regions for ligand binding, which are consistent with photoaffinity-labeling experiments. However, without similar direct biophysical evidence, a binding site for TMAQ near amino acids 55 and 56 would be purely speculative. It seems more probable that TMAQ binds in a similar manner to the binding of nicotine to the AChBP (Celie et al., 2004), a conclusion that is consistent with our evidence for competitive binding of TMAQ and [3H]epibatidine (Fig. 7B).
Discussion
Information derived from a variety of experimental approaches has led to a model in which the nAChR ligand binding site is located at the interface of two subunits (Corringer et al., 2000). In a heteromeric nAChR such as α3β4, the “principal” component is considered to be that contributed by the α3 subunit and the “complementary” component by β4. It has been proposed that amino acids from six discrete subunit domains (loops A-F) contribute to the nicotinic ligand binding site (Corringer et al., 2000). Three of these (loops A-C) are contributed by the principal subunit and three (loops D-F) by the complementary subunit.
At first, nAChR α subunits were considered to be “agonist-binding” subunits, whereas non-α subunits have been referred to as either “non-agonist-binding” or “structural” subunits (Deneris et al., 1988; Schoepfer et al., 1988). There is extensive experimental evidence, however, to indicate that non-α subunits such as β4 are able to contribute to the pharmacological diversity of nAChRs (Luetje and Patrick, 1991; Parker et al., 1998). Studies using chimeric subunits and site-directed mutagenesis have demonstrated the importance of amino acids located within loops D, E, and F in determining selectivity for nicotinic agonists and antagonists (Figl et al., 1992; Czajkowski et al., 1993; Prince and Sine, 1996; Bren and Sine, 1997). Previous studies with chimeric and mutant β2 and β4 subunits have demonstrated that the loop D region between residues 54 and 63 is a major determinant of sensitivity to agonists and competitive antagonists (Harvey and Luetje, 1996; Parker et al., 2001). Within this region, residue 59 (tyrosine in β2 and lysine in β4) has been identified as being the critical residue in determining sensitivity to both agonists and antagonists.
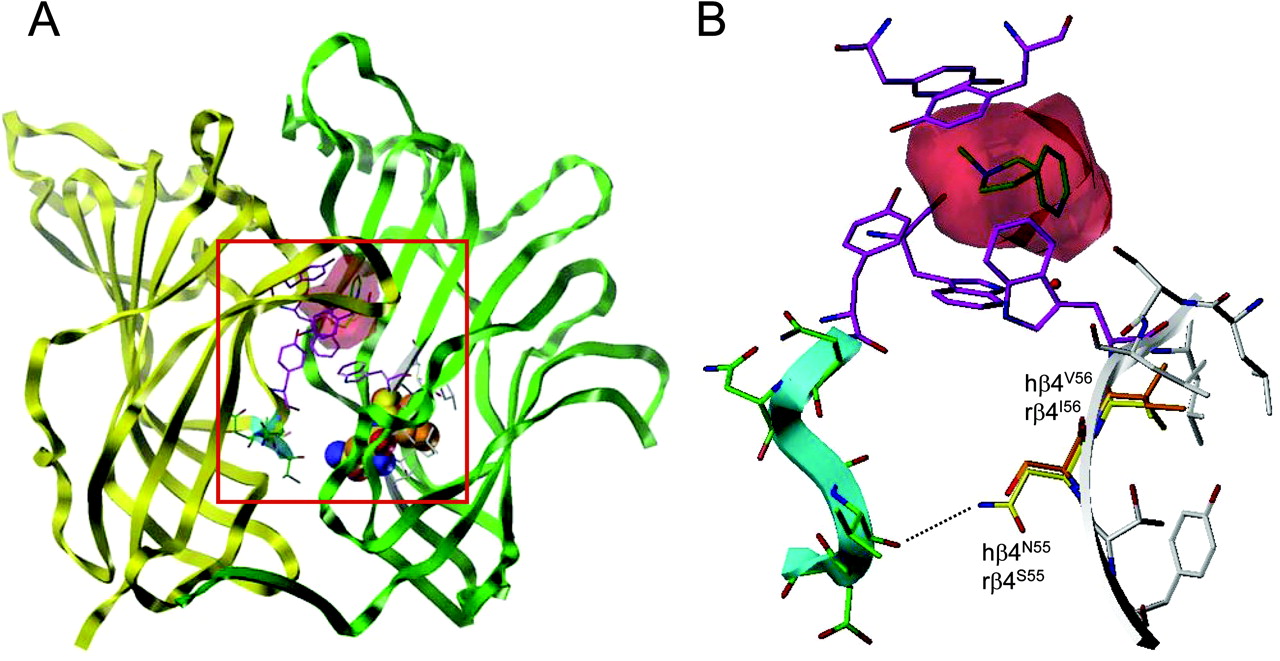
Homology model of the human α3β4 nAChR. A, model of the extracellular domain of the human α3β4 nAChR based on the atomic resolution structure of the L. stagnalis AChBP cocrystallized with nicotine (Protein Data Base code 1UW6). The location of nicotine, superimposed at the agonist binding site, is shown (red surface), and aromatic residues lining the binding site are shown in magenta. The α3 subunit is shown as a yellow ribbon, whereas the β4 subunit is shown as a green ribbon. An enlarged view of the region bordered be the red box is shown in B. B, enlarged view of the α3β4 homology model showing the positions of amino acids 55 and 56 (in loop D of the β4 subunit). Rat amino acids (rβ4S55 and rβ4I56) are shown in orange and human amino acids (hβ4N55 and hβ4V56) in yellow. The β4to β5 loop of the human α3 subunit is colored in cyan, and the postulated hydrogen bond is represented as a broken line.
The results presented in the present study provide further evidence that residues within loop D have a profound effect upon agonist sensitivity. In particular, we have identified two adjacent amino acids (at positions 55 and 56) as being major determinants of nAChR sensitivity to the agonist TMAQ. Note that we have numbered amino acid residues within the β4 subunit with reference to the predicted signal sequence cleavage site reported previously (Couturier et al., 1990; Gerzanich et al., 1997). Evidence for the involvement of amino acids 55 and 56 in conferring species-selective agonist activity of TMAQ has been obtained both by agonist-induced changes in intracellular calcium and by two-electrode voltage-clamp techniques. The differences in these two adjacent amino acids, located within loop D of the β4 subunit, seem to be sufficient to explain the marked difference in sensitivity of human and rat nAChRs to this agonist.
In nAChRs containing the rat β4 subunit, TMAQ retains the ability to bind (as illustrated by competition binding experiments with [3H]epibatidine; see Fig. 7B) but shows little or no agonist activity. Indeed, when TMAQ is coapplied with ACh, TMAQ displays antagonist activity on rβ4-containing nAChRs (Fig. 8). In this respect, TMAQ seems to be behaving similarly to the way nicotine does on chick and rat α3β2 nAChRs (Hussy et al., 1994). Nicotine has been shown to act as an agonist of rat α3β2 nAChRs but as a potent competitive antagonist of chick α3β2 nAChRs (Hussy et al., 1994). As we observed for TMAQ acting on human and rat α3β4 nAChRs, it seems that the species-selective behavior of nicotine on chick and rat α3β2 nAChRs can be attributed in large part to a single amino acid difference. In contrast, however, the amino acids critical for conferring species-selectivity and agonist/antagonist activity of nicotine are located within the α3 subunit and are located close to the first transmembrane domain (Hussy et al., 1994). Other studies have identified single amino acid mutations within the nAChR M2 transmembrane domain (e.g., L247T in the α7 subunit), for which antagonists of the wild-type receptor act as agonist (Bertrand et al., 1992).
The atomic-resolution structure of the AChBP from the snail L. stagnalis (Brejc et al., 2001; Celie et al., 2004) has provided a powerful tool with which to predict the structure of nAChRs. The close sequence similarity between the extracellular domain of nAChR subunits and the AChBP has enabled homology modeling and has been useful in interpreting site-directed mutagenesis studies performed with nAChRs subunits. Comparison of the AChBP and nAChR subunits indicates that the β4 amino acids 55 and 56 are located at positions analogous to Val51 and Phe52 in the AChBP (Brejc et al., 2001). Although residues within loop D of nAChRs are predicted to contribute to the agonist binding site (Corringer et al., 2000), Val51 and Phe52 do not lie in immediate proximity to the AChBP ligand binding site (Celie et al., 2004). Analysis of the homology models of the rat and human α3β4 nAChRs (the present study, and Costa et al., 2003), suggest that the β4 amino acids 55 and 56 are not located within the interaction shell of the agonist binding site and, therefore, may not be involved directly in agonist binding. Our data indicate that mutation of amino acids 55 and 56 has a much more profound effect upon TMAQ agonist efficacy (e.g., Figs. 6 and 8) than upon TMAQ binding (Fig. 7B). It is plausible that mutation of these amino acids may induce a conformational change that influences the affinity of TMAQ binding but a more profound and direct influence upon the coupling of agonist binding to channel opening. It is interesting that amino acids 55 and 56 lie within one of the β-strand regions (β2) of the AChBP (Brejc et al., 2001) adjacent to the β1-β2 loop domain, which has been implicated previously in the coupling of binding and gating (Bouzat et al., 2004). Our results, together with previous studies of mutated nAChRs (Campos-Caro et al., 1996; Bouzat et al., 2004; Criado et al., 2005; Lee and Sine, 2005; Mukhtasimova et al., 2005; Sala et al., 2005; Castillo et al., 2006), indicate that amino acids located at diverse locations within the extracellular domain are able to influence the coupling of agonist binding and channel gating.
Acknowledgments
We thank Sam Ranasinghe for assistance with FLIPR experiments and Anesh Chavda for assistance with site-directed mutagenesis.
Footnotes
-
Funding was provided by the Biotechnology and Biological Sciences Research Council Industrial Partnership CASE studentship (to G.T.Y. and N.S.M.).
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; AChBP, acetylcholine binding protein; FLIPR, fluorometric imaging plate reader; TMAQ, 5-(trifluoromethyl)-6-(1-methyl-azepan-4-yl)methyl-1H-quinolin-2-one; HEK, human embryonic kidney; ACh, acetylcholine.
- Received September 12, 2006.
- Accepted October 25, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}