


Abstract
We demonstrated recently that opioid-induced activation of phospholipase D2 (PLD2) enhances μ- (MOPr) and δ-opioid receptor endocytosis/recycling and thus reduces the development of opioid receptor desensitization and tolerance. However, the mechanistic basis for the PLD2-mediated induction of opioid receptor endocytosis is currently unknown. Here we show that PLD2-generated phosphatidic acid (PA) might play a key role in facilitating the endocytosis of opioid receptors. However, PLD2-derived PA is known to be further converted to diacylglycerol (DAG) by PA phosphohydrolase (PPAP2). In fact, blocking of PA phosphohydrolase activity by propranolol or PPAP2-short interfering RNA (siRNA) transfection significantly attenuated agonist-induced opioid receptor endocytosis. The primary importance of PA-derived DAG in the induction of opioid receptor endocytosis was further supported by the finding that increasing the DAG level by inhibiting the reconversion of DAG into PA with the DAG kinase inhibitor 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2,3-dihydro-2-thioxo-4(1H)quinazolinone (R59949) or the addition of the synthetic cell-permeable DAG analog 1,2-dioctanoyl-sn-glycerol (DOG), further increased the agonist-induced opioid receptor endocytosis. Moreover, the addition of DOG bypasses the PLD2-siRNA- or PPAP2-siRNA-mediated impairment of DAG synthesis and resulted in a restoration of agonist-induced opioid receptor internalization. Further studies established a functional link between PA-derived DAG and the activation of p38 mitogen-activated protein kinase (MAPK) and the subsequent phosphorylation of the Rab5 effector early endosome antigen 1, which has been demonstrated recently to be required for the induction of MOPr endocytosis. Taken together, our results revealed that the regulation of opioid receptor endocytosis by PLD2 involves the conversion of its product PA to DAG resulting in an activation of the p38 MAPK pathway.
Opioids are characteristically associated with phenomena such as analgesia, respiratory depression, and addiction. To date, three opioid receptor subtypes, μ, δ, and κ, have been identified, which belong to the G protein-coupled receptor (GPCR) family. Agonist-induced internalization of opioid receptors via clathrin-dependent pathway is one important regulatory event of opioid receptor signal transduction. Our group has found that phospholipase D2 (PLD2) modulates agonist-induced MOPr endocytosis (Koch et al., 2003). Phospholipase D (PLD) is a ubiquitous enzyme present in organisms ranging from bacteria, plants, and animals to humans. It hydrolyzes phosphatidylcholine to release choline and phosphatidic acid (PA), which is an important bioactive lipid. During recent years, two mammalian PLD subtypes, PLD1 and PLD2, have been cloned (Frohman et al., 1999). Subcellular fractionation studies showed that PLD1 is present predominantly in intracellular membranes (e.g., endoplasmic reticulum, Golgi, and vesicular compartments), whereas PLD2 is mainly distributed within the plasma membrane (Du et al., 2004). PLD plays an important role in basic cell functions, such as signal transduction, cytoskeletal reorganization, and cellular proliferation (Ghosh et al., 2003; Jenkins and Frohman, 2005; Zouwail et al., 2005). In addition, PLD and its product PA have been shown also to be involved in endocytosis and vesicular trafficking of other receptors, such as angiotensin II receptor endocytosis (Du et al., 2004) and epidermal growth factor endocytosis (Shen et al., 2001). It is also known that PA is converted into diacylglycerol (DAG) by PA phosphohydrolase; however, little is known about the role of PA-derived DAG in the regulation of GPCR endocytosis.
Therefore, in the present study, we investigated the role of PLD2-derived PA and DAG in the regulation of agonist-induced opioid receptor endocytosis. We first provide evidence that PLD2 activity is required for agonist-induced MOPr and DOPr internalization and that its product PA plays a crucial role in this process. Then we found that the conversion of PA into DAG is a prerequisite for the PLD2-mediated enhancement of the agonist-induced MOPr and DOPr endocytosis and that this DAG effect is protein kinase C (PKC)-independent. Further evidence demonstrates that the PLD2-PA-DAG pathway is involved in the opioid receptor-mediated activation of p38 mitogen-activated protein kinase (MAPK). Thus, our results are in line with previous findings demonstrating an important role of p38 MAPK activation in the induction of endocytotic receptor trafficking (Cavalli et al., 2001; Macé et al., 2005; McLaughlin et al., 2006; Vergarajauregui et al., 2006).
Materials and Methods
Reagents.
[d-Ala2,N-Me-Phe4,Gly5-ol]-Enkephalin (DAMGO) and [d-Pen2,d-Pen5]-enkephalin (DPDPE) were purchased from Bachem (Heidelberg, Germany). [3H]Naltrindol was obtained from Perkin-Elmer Life and Analytical Sciences (Rodgau, Germany). 1-Butanol was from Merck (Darmstadt, Germany), and chelerythrine chloride was from Sigma-Aldrich (St. Louis, MO). Other substances were purchased from Calbiochem (San Diego, CA) unless otherwise indicated.
Epitope Tagging and Cloning of cDNA.
An HA epitope tag sequence MYPYDVPDYA was added to the N terminus of the μ- and δ-opioid receptor cDNA using polymerase chain reaction (PCR) and then subcloned into pEAK10 expression vector containing the puromycin resistance gene (Edge Biosystems, Gaithersburg, MD), which generated the plasmids pEAK10-HAMOPr and pEAK10-HADOPr.
Generation of Stable Cell Lines Expressing μ- or δ-Opioid Receptor.
To generate MOPr- or DOPr-receptor expressing cell line, HEK293 cells were transfected with the pEAK10-HAMOPr or pEAK10-HADOPr plasmid by the calcium phosphate precipitation method and selected with 1.25 μg/ml puromycin (Sigma-Aldrich, Deisenhofen, Germany) as described previously (Koch et al., 2003, 2004, 2006). Stable transfectants were selected with 1.25 μg/ml puromycin and 500 μg/ml G418 (Invitrogen, Darmstadt, Germany) in Dulbecco's modified Eagle's medium (DMEM; Lonza Verviers SPRL, Verviers, Belgium) containing 10% fetal calf serum. Multiclones were used for the further studies. Receptor expression of the stable transfectants were monitored and selected by radioligand binding assay and confocal microscopy as described previously (Koch et al., 2003, 2004, 2006).
Transient siRNA Transfection Method.
HEK293 or SHSY-5Y cells (5 × 104 cells) were seeded in six-well dishes and incubated overnight. Lipofectamine 2000 was used for siRNA transfection according to the manufacturer's instructions. For down-regulation of PLD2-expression the cells were transfected with 150 ng of PLD2-siRNA (5′-GAUGCAGGCAACAGAGAGATT-3′) and 1.5 μg of PLD2-siRNA containing pGFR-PLD2 vector (Kim et al., 2005). For down-regulation of PPAP2A and PPAP2B expression the cells were transfected with 150 ng of PPAP2A-siRNA (5′-AGGAAUAACUACAUAGCCATT-3′) and 150 ng of PPAP2B-siRNA (5′-ACUGAAAACUGGUGAGACATT-3′). Six hours after transfection medium was changed, and cells were incubated for an additional 36 h. Then cells were treated as indicated and prepared for Western blot analyses or analyses of receptor internalization.
RNA Preparation and Real-Time RT-PCR.
Isolation of total cellular RNA, synthesis of cDNA, and real-time PCR analysis was performed as described previously (Seifert et al., 2008). For RT-PCR reactions, 2 μl of cDNA was used, and quantitative RT-PCR was performed in a total volume of 20 μl on a LightCycler instrument using the LightCycler Fast Start DNA Master SYBR Green I kit (both from Roche Diagnostics, Indianapolis, IN). Conditions were as follows: PPAP2A-primers: forward, 5′-GGGAATCACTATCCGAGCAA-3′, and reverse, 5′-TGTGCATCCAAGAGGCATAG-3′ (preincubation, 8 min at 95°C, 50 cycles, 5 s at 95°C, 10 s at 60°C, and 6 s at 72°C); and PPAP2B-primers: forward, 5′-CGACTTCGGTTACTGCCTTC-3′, and reverse, 5′-GCTTCTCTGGCTCCTTCTGA-3′ (preincubation, 8 min at 95°C, 50 cycles, 5 s at 95°C, 10 s at 60°C, and 6 s at 72°C); ribosomal 18S (Seifert et al., 2008) (preincubation, 8 min at 95°C, 50 cycles, 5 s at 95°C, 6 s at 60°C, and 6 s at 72°C). Melting curves were generated for verification of the PCR product. In addition, gel electrophoresis was performed to proof the quality of the PCR product. Ct values for all genes analyzed were determined three times, averaged, and normalized to values for the endogenous control 18S. The relative mRNA amounts were calculated by using the ΔΔCt method (Seifert et al., 2008).
Radioligand Binding Assay.
The binding characteristics of the MOPr and DOPr were determined by saturation binding assays on membranes prepared from stably transfected cells. Dissociation constant (Kd) and number of [3H]DAMGO or [3H]naltrindol binding sites (Bmax) were calculated by Scatchard analysis using at least seven concentrations of radioligand in a range from 0.025 to 25 nM. Nonspecific binding was determined as radioactivity bound in the presence of 1 μM unlabeled DAMGO or naltrindol. By the ratio of specific bound radioligand per milligram of protein, the number of [3H]DAMGO or [3H]naltrindol binding sites in membrane binding assay reflects the amount of total MOPr or DOPr, respectively.
Quantitative Analysis of Receptor Internalization.
Cells were seeded at a density of 2 × 105/well and grown overnight in poly(l-lysine)-coated 24-well plates. They were treated as indicated with or without agonist in UltraMEM (Lonza Verviers SPRL) at 37°C for 30 min. Then, cells were incubated with rabbit anti-HA antibody (1 μg/ml) in UltraMEM at 4°C for 1.5 h to label HA-tagged surface receptors. Subsequently, cells were fixed with 4% paraformaldehyde for 40 min and incubated with peroxidase-conjugated anti-rabbit antibody (1:1000; GE Healthcare, Munich, Germany) for 2 h at room temperature. Color was developed with 250 μl of 2,2′-azino-di-[3-ethylbenzthiazoline sulfonate (6)] solution (Roche Molecular Biochemicals, Indianapolis, IN). After 10 to 20 min of rotation, 200 μl of the substrate solution from each well was transferred to a 96-well plate and measured at 405 nm with a microtiter plate reader. Agonist-induced receptor internalization was calculated as the percentage loss of surface opioid receptors compared with control cells without agonist treatment.
Preparation of Cell Lysates and Western Blotting Analysis.
Cells were seeded in 12-well plate at a density of 1.8 × 105/well. After 24 h, cells were further serum-starved for 16 h in Dulbecco's modified Eagle's medium containing 1% fetal calf serum. After treatment as indicated, cells were washed with ice-cold phosphate-buffered saline and lysed on ice with radioimmunoprecipitation assay buffer containing fresh-prepared proteinase inhibitors and phosphatase inhibitors for 1 h. Cell lysate was centrifuged at 14,000 rpm for 20 min, and the total protein in supernatant was quantified. Supernatants containing the same amount of total proteins were subjected to 10% SDS-polyacrylamide gel electrophoresis. After electroblotting, the nitrocellulose membrane was blocked with 3% nonfat milk for 30 min and incubated with 1 μg/ml polyclonal rabbit anti-human PLD2 antibody (Sigma-Aldrich), 1 μg/ml phospho-p38 MAPK (Thr180/Tyr182) rabbit antibody (Cell Signaling Technology, Inc., Danvers, MA), 5 μg/ml phospho-threonine mouse antibody (Sigma-Aldrich) or 1 μg/ml EEA1 goat antibody (Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. Binding of primary antibody was detected with secondary anti-rabbit antibody or anti-mouse antibody for 1 h at room temperature, followed by detection using enhanced chemiluminescence detection system. To detect other protein or β-actin, the same membrane was treated with 1% sodium azide in Tris-buffered saline for 45 min and then subjected to standard immunoblotting procedure as described previously (Koch et al., 2003, 2004).
Results
Agonist-Induced Opioid Receptor Endocytosis Requires PLD2-Mediated PA Synthesis.
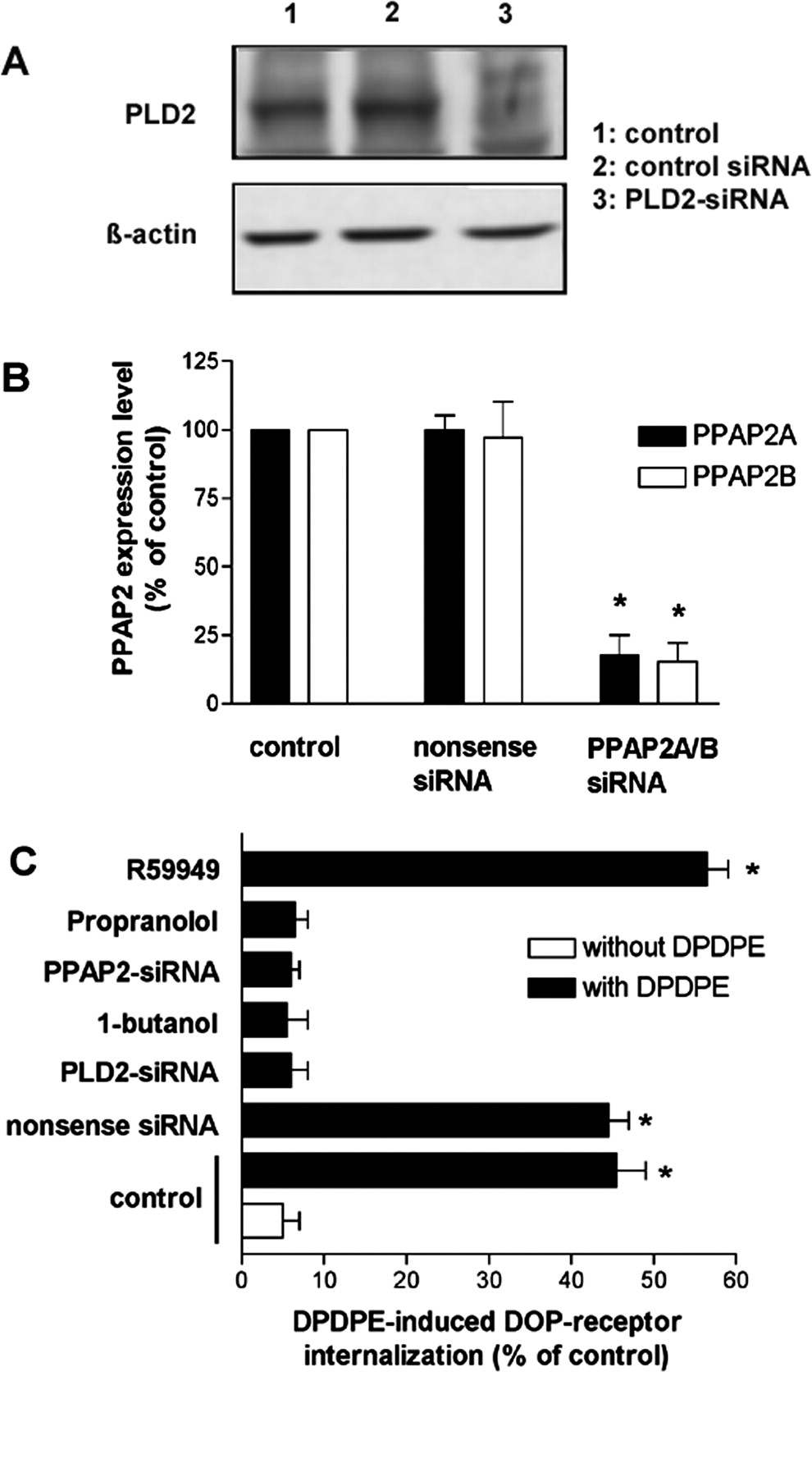
By using a dominant-negative PLD2 mutant, we have shown previously that PLD2 activity is required for agonist-induced MOPr and DOPr endocytosis (Koch et al., 2003, 2006). To confirm these findings, in the present study, we reduced the endogenous PLD2 expression level by transfecting MOPr- and DOPr-expressing HEK293 cells with PLD2-siRNA and determined the effect on the agonist-induced opioid receptor endocytosis (Figs. 1, A and C, and 2, A and C). The stable expression of MOPr or DOPr in HEK293 cells was monitored by ligand binding experiments. These saturation binding experiments revealed no substantial differences between MOPr and DOPr expressing cells with respect to their numbers of binding sites (Bmax; 1329 ± 399 and 1247 ± 352 fmol/mg protein for MOPr and DOPr, respectively). The affinities (Kd) of the MOPr for [3H]DAMGO and DOPr for [3H]naltrindol were 1.4 ± 0.3 and 0.142 ± 0.048 nM, respectively. In Figs. 1C and 2C, it is demonstrated that blocking PLD2 activity by cotransfection of PLD2-siRNA completely blocked the agonist-induced MOPr and DOPr endocytosis, which is in line with our previous findings (Koch et al., 2003, 2006). It is further known that PLD2 catalytic activity results in the production of PA by hydrolyzing phosphatidylcholine. Therefore, we next investigated the effect of blocking PLD2-mediated PA production for the induction of opioid receptor endocytosis. For inhibition of PA production, the primary alcohol 1-butanol was used, which is known to be preferentially used over water by PLD2 in the transphosphatidylation reaction to generate phosphatidylalcohol instead of phosphatidic acid. Quantitative internalization analysis shows that in the presence of 1-butanol, the agonist-induced MOPr and DOPr endocytosis was strongly reduced (Figs. 1C and 2C). To ensure that the 1-butanol effect was not simply an alcohol artifact, the secondary alcohol isobutanol was used in control experiments and showed no effect on the agonist-induced opioid receptor internalization (data not shown). These results strongly suggest that PLD2-mediated PA production is required for agonist-induced opioid receptor endocytosis.

Role of PLD2-derived PA and DAG in the μ-opioid receptor endocytosis. A, effect of PLD2-siRNA transfection on PLD2 expression in HEK293 cells. Cells stably expressing MOPr were transfected or not with PLD2-siRNA as described under Materials and Methods. After 48 h, membrane proteins from MOPr-expressing cells transfected or not with PLD2-siRNA were extracted and immunoblotted. PLD2 expression was monitored by Western blot analysis using rabbit anti-human PLD2 antibody as described previously (Koch et al., 2001, 2003). Densitometric measurement revealed that 48 h after PLD2-siRNA transfection, the PLD2 level was reduced to approximately 44% compared with untransfected control cells. Two additional experiments gave similar results. B, effect of PPAP2A/B-siRNA transfection on PPAP2A and PPAP2B expression in HEK293 cells. Cells stably expressing MOPr were transfected or not with PPAP2A/B-siRNA as described under Materials and Methods. After 48 h, RNA from MOPr-expressing cells transfected or not with PPAP2A/B-siRNA was extracted and PPAP2A and PPAP2B expression level was quantified via real-time RT-PCR as described under Materials and Methods. C, HEK293 cells stably expressing MOPr were transfected with PLD2-siRNA or treated with 0.5% 1-butanol for 30 min to block PLD2-expression or PLD2-mediated PA synthesis, respectively. Furthermore, PPAP2-mediated conversion of PA into DAG was blocked by transfection with PPAP2-siRNA or treatment with 250 μM propranolol. The reconversion of DAG to PA was inhibited by application of the DGK-inhibitor R59949 (40 μM). Cell surface receptors were determined by a quantitative enzyme immunoassay as described under Materials and Methods. Agonist-induced MOPr internalization was quantified as the percentage loss of surface receptors compared with the cells that were treated without agonist. Data presented are means ± S.E. from at least three independent experiments performed in triplicate. *, significant difference (p < 0.05) between DAMGO-treated and untreated control cells (ANOVA followed by Bonferroni test).
The Conversion of PLD2-Derived PA into DAG Is Involved in Agonist-Induced MOPr and DOPr Endocytosis in a PKC-Independent Way.
PLD2 hydrolyzes phosphatidylcholine to form PA, which can be further dephosphorylated to DAG by PA phosphohydrolase (PAP) (Sciorra and Morris, 2002). DAG in turn can be regenerated to PA by DAG kinase (DGK) (Jenkins and Frohman, 2005). Therefore, here we investigated the role of PA-derived DAG for opioid receptor endocytosis.
First, the conversion from PA to DAG was inhibited by using propranolol, an effective PAP blocker that has been used in numerous studies (Albert et al., 2005; Grkovich et al., 2006). As seen in Figs. 1C and 2C, when the conversion from PA to DAG was inhibited by propranolol, agonist-induced MOPr and DOPr endocytosis was drastically decreased.

Role of PLD2-derived PA and DAG in the δ-opioid receptor endocytosis. A, effect of PLD2-siRNA transfection on PLD2 expression in HEK293 cells. Cells stably expressing DOPr were transfected or not with PLD2-siRNA as described under Materials and Methods. After 48 h, membrane proteins from DOPr-expressing cells transfected or not with PLD2-siRNA were extracted and immunoblotted. PLD2 expression was monitored by Western blot analysis using rabbit anti-human PLD antibody as described previously (Koch et al., 2001, 2003). Densitometric measurement revealed that 48 h after PLD2-siRNA transfection, the PLD2 level was reduced to approximately 32% compared with untransfected control cells. Two additional experiments gave similar results. B, effect of PPAP2A/B-siRNA transfection on PPAP2A and PPAP2B expression in HEK293 cells. Cells stably expressing DOPr were transfected or not with PPAP2A/B-siRNA as described under Materials and Methods. After 48 h, RNA from DOPr-expressing cells transfected or not with PPAP2A/B-siRNA was extracted, and PPAP2A and PPAP2B expression level was quantified via real-time RT-PCR as described under Materials and Methods. C, HEK293 cells stably expressing DOPr were transfected with PLD2-siRNA or treated with 0.5% 1-butanol for 30 min to block PLD2-expression or PLD2-mediated PA synthesis, respectively. Furthermore, PPAP2-mediated conversion of PA into DAG was blocked by transfection with PPAP2-siRNA or treatment with 250 μM propranolol. The reconversion of DAG to PA was inhibited by application of the DGK inhibitor R59949 (40 μM). Cell surface receptors were determined by a quantitative enzyme immunoassay as described under Materials and Methods. Agonist-induced DOPr internalization was quantified as the percentage loss of surface receptors compared with the cells that were treated without agonist. Data presented are means ± S.E. from at least three independent experiments performed in triplicate. *, significant difference (p < 0.05) between DPDPE-treated and untreated control cells (ANOVA followed by Bonferroni test).
However, to exclude potential unspecific effects of the PAP blocker propranolol, we next used siRNA technology to confirm the essential role of PA-derived DAG in the induction of MOPr and DOPr internalization. Thus, in MOPr- and DOPr-expressing cells, we cotransfected PPAP2A/B-siRNA to down-regulate the expression of phosphatidic acid phosphatase 2, which mediates the conversion of PA into DAG (Figs. 1B and 2B). As shown in Figs. 1C and 2C, transfection of PPAP2A/B-siRNA led to a complete inhibition of the agonist-induced MOPr and DOPr internalization. Together, these data revealed that the conversion from PLD2-derived PA to DAG is important for the PLD2-mediated induction of opioid receptor endocytosis.
To further verify the stimulatory effect of DAG on opioid receptor endocytosis, we increased the PA-derived DAG accumulation by inhibiting the reconversion of DAG to PA with the most commonly used DGK inhibitor 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2,3-dihydro-2-thioxo-4(1H)quinazolinone (R59949). R59949 specifically acts on the catalytic domain of DGK and inhibits its catalytic activity (Jiang et al., 2000). As shown in Figs. 1C and 2C, the inhibition of the conversion from DAG to PA augmented the DAMGO-induced MOPr endocytosis and DPDPE-induced DOPr endocytosis significantly.
To obtain direct evidence that agonist-induced opioid receptor endocytosis is facilitated by DAG, we incubated the MOPr- or DOPr-expressing cells with the cell-permeable DAG analog 1,2-dioctanoyl-sn-glycerol (DOG) (Lucas et al., 2003). In fact, compared with the treatment with DAMGO or DPDPE alone, the presence of DOG increased the agonist-induced MOPr and DOPr endocytosis (Fig. 3, A and B). Moreover, DOG application overcomes the inhibition of agonist-induced opioid receptor endocytosis by PLD2-siRNA or PPAP2-siRNA transfection (Fig. 3, A and B), indicating the key role of DAG in the induction of opioid receptor endocytosis.

The DAG analog DOG enhances agonist-induced opioid receptor endocytosis in a PKC-independent manner and bypasses the PLD2-siRNA- or PPAP2-siRNA-mediated impairment of opioid-receptor endocytosis. A, quantitative analysis of MOPr internalization in MOPr-expressing HEK293 cells, which were stimulated with or without 1 μM DAMGO for 30 min in the presence or absence of 150 μM DOG. Note that the stimulatory effect of DOG on the receptor endocytosis was influenced neither by treatment with PKC inhibitors [5 μM chelerythrine chloride (Che) or 1 μM calphostin C (Calph)] nor by transfection of MOPr-expressing cells with PLD2-siRNA or PPAP2-siRNA. B, quantitative analysis of DOPr internalization in DOPr-expressing HEK293 cells, which were stimulated with or without 0.1 μM DPDPE for 30 min in the presence or absence of 150 μM DOG. Note that the stimulatory effect of DOG on the receptor endocytosis was influenced neither by treatment with PKC inhibitors (5 μM Che or 1 μM Calph) nor by transfection of DOPr-expressing cells with PLD2-siRNA or PPAP2-siRNA. For quantitative analysis of receptor internalization in A and B, agonist-induced opioid receptor internalization was measured and calculated as described under Materials and Methods. Data are presented as means ± S.E. from three independent experiments performed in triplicate. *, significant difference (p < 0.05) compared with the treatment with agonist alone (ANOVA followed by Bonferroni test).
Because DAG is known to activate PKC, we next investigated whether the activation of PKC is involved in DAG-mediated opioid receptor endocytosis. Therefore, we used two specific PKC inhibitors, chelerythrine chloride and calphostin C to block DAG-mediated PKC activation. The inhibition of PKC by either chelerythrine chloride or calphostin C did not impair DOG-induced MOPr and DOPr endocytosis (Fig. 3, A and B), ruling out that PKC is involved in the DAG-effect on MOPr and DOPr endocytosis.
The Function of PLD2 in Opioid Receptor Endocytosis Involves Activation of p38 MAP Kinase.
p38 MAPK has been identified to regulate the endocytic trafficking of various receptors, such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, MOPr, platelet-activating factor receptor, and epidermal growth factor receptor (Cavalli et al., 2001; Macé et al., 2005; McLaughlin et al., 2006; Vergarajauregui et al., 2006). For the MOPr, it has been shown that the receptor-internalizing μ-agonist DAMGO can rapidly stimulate p38 MAPK activation and that this activation is a prerequisite for the subsequent MOPr endocytosis (Macé et al., 2005). In contrast, the noninternalizing μ-agonist morphine was shown not to activate p38 MAPK (Macé et al., 2005). However, the mechanisms through which opioid receptors mediate the activation of p38 MAPK are still unknown. We have shown previously that receptor-internalizing μ-agonists, such as DAMGO, activate PLD2, whereas noninternalizing agonists, such as morphine, cannot, and that this agonist-selective PLD2 activation is essential for opioid receptor endocytosis. To investigate whether the opioid-mediated PLD2 activation is involved in the activation of p38 MAPK, we first tested the effect of blocking PLD2-derived PA synthesis on the opioid-induced p38 MAPK activation.
Using a specific antibody against phosphorylated p38 MAPK (Thr180/Tyr182), we first tested the activation of p38 MAPK by the agonist DAMGO in MOPr-expressing cells and by DPDPE in DOPr-expressing cells (Fig. 4A). The increase of p38 MAPK phosphorylation was readily detected after 5 min of stimulation with DPDPE in DOPr-expressing cells or with DAMGO in MOPr-expressing cells. For inhibition of PLD-derived PA synthesis, we used the primary alcohol 1-butanol, which is known to be preferentially used over water by PLD in the transphosphatidylation reaction forming phosphatidylalcohols at the expense of PA. This reaction is highly specific for primary alcohols, whereas secondary alcohols (such as isobutanol) are not used by PLD. After blocking PLD-derived PA synthesis by 1-butanol, the opioid-induced p38 MAPK phosphorylation was blocked, and this effect was not due simply to an unspecific alcohol effect, because the secondary alcohol isobutanol did not block the opioid-induced p38 MAPK activation. These results indicate that PLD activity and PA production are essential for p38 MAPK activation by both μ- and δ-opioid receptors (Fig. 4A).

PLD2 is involved in the activation of p38 MAPK, which is required for agonist-induced opioid receptor endocytosis. A, blocking of opioid receptor-mediated p38 MAPK activation by the PLD inhibitor 1-butanol. As indicated, MOPr- or DOPr-expressing HEK293 cells were stimulated with agonist in the presence or absence of 0.5% 1-butanol or isobutanol for 5 min. Subsequently, cells were lysed and subjected to Western blot to analyze phosphorylated p38 MAPK kinase as described under Materials and Methods. B, agonist-induced opioid receptor endocytosis was attenuated by inhibition of p38 MAPK. HEK293 cells expressing MOPr or DOPr were treated with agonist for 30 min in the presence or absence of 10 μM p38 MAPK inhibitor SB203580. Cell surface opioid receptors were determined by quantitative enzyme immunoassay as described under Materials and Methods. Data are presented as means ± S.E. from three independent experiments performed in triplicate. *, significant difference (p < 0.05) between SB203580-treated cells and untreated controls (ANOVA followed by Bonferroni test).
But is the opioid-mediated activation of p38 MAPK via PLD2 really involved in the induction of opioid receptor endocytosis? To test this, we blocked p38 MAPK activation with the specific p38 MAPK inhibitor 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole (SB203580). We found that the agonist-induced endocytosis was attenuated by the pretreatment with the p38 MAPK inhibitor SB203580 for both MOPr and DOPr (Fig. 4B), which is consistent with the observations from Macé et al. (2005).
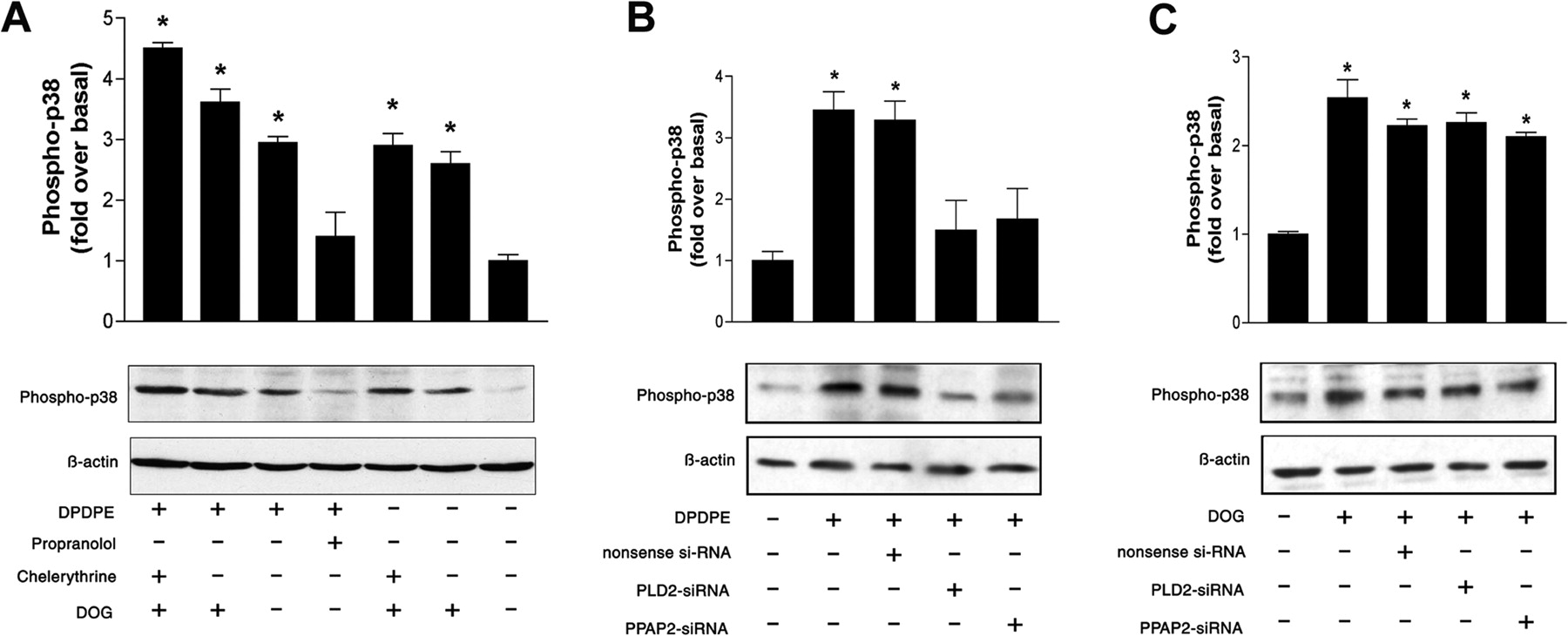
Our data revealed that inhibition of PLD2-activity by primary alcohol or PLD2-siRNA transfection blocks opioid-induced p38 MAPK activation for both MOPr and DOPr (Figs. 4A , 5B, and 6B). Because PLD2 activation results in an increase of PA and subsequent in PA-derived DAG, it is possible that the conversion of PA into DAG is involved in opioid receptor-mediated p38 MAPK activation. To address this issue, we inhibited the dephosphorylation of PA to DAG with the PAP inhibitor propranolol or PPAP2-siRNA transfection. The opioid-induced p38 MAPK phosphorylation in MOPr- or DOPr-expressing cells was remarkably impaired by propranolol (Figs. 5A and 6A) or PPAP2-siRNA (Figs. 5B and 6B), indicating an important role of the PA-derived DAG in the opioid receptor-mediated p38 MAPK activation.

MOPr-mediated activation of p38 MAPK can be blocked by inhibition of PLD2 and is PKC-independent. A, effect of agonist-induced MOPr activation on the p38 MAPK activity in the presence or absence of DOG, propranolol, or chelerythrine. Cells were treated with or without 150 μMDOG, 5 μM chelerythrine, or 250 μM propranolol as indicated. As a marker for p38 MAPK activation, we determined the level of phosphorylated p38 MAPK by Western blot analysis as described under Materials and Methods. B, effect of transfection of MOPr-expressing cells with PLD2-siRNA or PPAP2-siRNA on the DAMGO-induced p38 MAPK activation. C, the stimulatory effect of DOG on the p38 MAPK activation in MOPr-expressing cells was not influenced by the transfection with PLD2-siRNA or PPAP2-siRNA. Quantitative data from Fig. 5 are presented as means ± S.E. from three independent experiments performed in triplicate. *, significant difference (p < 0.05) compared with untreated control cells (ANOVA followed by Bonferroni test).
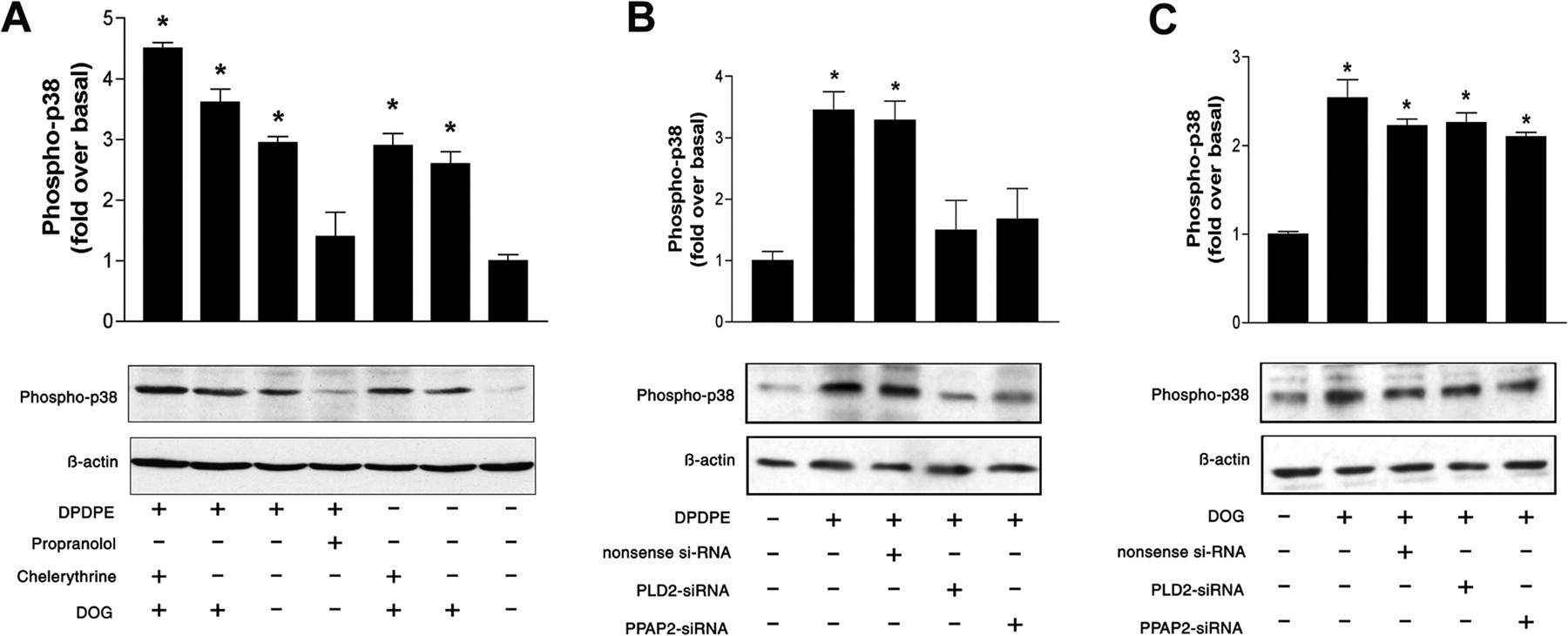
DOPr-mediated activation of p38 MAPK can be blocked by inhibition of PLD2 and is PKC-independent. A, effect of agonist-induced DOPr activation on the p38 MAPK activity in the presence or absence of DOG, propranolol, or chelerythrine. Cells were treated with or without 150 μMDOG, 5 μM chelerythrine, or 250 μM propranolol as indicated. As a marker for p38 MAPK activation, we determined the level of phosphorylated p38 MAPK by Western blot analysis as described under Materials and Methods. B, effect of transfection of DOPr-expressing cells with PLD2-siRNA or PPAP2-siRNA on the DPDPE-induced p38 MAPK activation. C, the stimulatory effect of DOG on the p38 MAPK activation in DOPr-expressing cells was not influenced by the transfection with PLD2-siRNA or PPAP2-siRNA. Quantitative data from Fig. 6 are presented as means ± S.E. from three independent experiments performed in triplicate. *, significant difference (p < 0.05) compared with untreated control cells (ANOVA followed by Bonferroni test).
In addition, we analyzed the effect of the DAG analog DOG on p38 MAPK activation. As shown in Figs. 5A and 6A, in MOPr- or DOPr-expressing cells, treatment with DOG alone was sufficient to induce p38 MAPK phosphorylation, which could not be blocked by the inhibition of PKC with chelerythrine chloride. When the cells were stimulated with the DOPr agonist DPDPE or the MOPr agonist DAMGO, respectively, the presence of DOG augmented the agonist-induced phosphorylation of p38 MAPK. Inhibition of PKC with chelerythrine chloride could not impair this augmented phosphorylation of p38 MAPK as well. Taken together, these data indicate that PA-derived DAG can activate p38 MAPK in a PKC-independent manner. Furthermore, the inhibition of p38 MAPK activation by PLD2-siRNA or PPAP2-siRNA could be bypassed by treatment with DOG, indicating the important role of DAG in the opioid-mediated activation of p38 MAPK (Figs. 5C and 6C).
Opioid-Induced Activation of p38 MAPK in Neuronal Cells Is PLD2-Mediated.
To ensure that the requirement of PLD2 activity for the opioid-induced p38 MAPK activation was not an artifact of the HEK293 cell model, we examined whether this regulation also occurred in neuronal SHSY-5Y cells endogenously expressing MOPr and DOPr. Therefore, the SHSY-5Y cells were transfected or not with nonsense-siRNA, PLD2-siRNA, or PPAP2-siRNA, and the effect of the μ-agonist DAMGO and δ-agonist DPDPE on the p38 MAPK activation was determined by Western blot analyses. Our data revealed that inhibition of endogenous PLD2 activity by PLD2-siRNA transfection or inhibition of the PPAP2-mediated conversion of PLD2-derived PA into DAG by PPAP2-siRNA resulted in an inhibition of the DAMGO- and DPDPE-induced p38 MAPK activation (Fig. 7). These results emphasize the important physiological role of PLD2 activity and signal transduction in the opioid-mediated activation of p38 MAPK.

Effect of PLD2-siRNA or PPAP2-siRNA transfection on the DAMGO- or DPDPE-induced p38 MAPK activation in neuronal SHSY-5Y cells. SHSY-5Y cells endogenously expressing MOPr and DOPr were transfected or not with PLD2-siRNA or PPAP2-siRNA as described under Materials and Methods. Forty-eight hours after transfection, membrane proteins from SHSY-5Y cells were extracted and immunoblotted. As a marker for p38 MAPK activation, we determined the level of phosphorylated p38 MAPK by Western blot analysis as described under Materials and Methods. Representative immunoblots from one of three independent experiments are shown. Quantitative data from Fig. 7 are expressed as the fold p38 MAPK phosphorylation over basal in untreated cells and are presented as means ± S.E. from three independent experiments. *, significant difference (p < 0.05), and **, significant difference (p < 0.01) compared with untreated control cells (ANOVA followed by Bonferroni test).
Opioid-Induced EEA1 Activation in Neuronal Cells Requires PLD2 Activity.
Previous studies demonstrated that p38 MAPK regulates MOPr endocytosis by phosphorylating the Rab5 effector EEA1 to regulate its recruitment to the plasma membrane (Macé et al., 2005). To test whether this opioid-induced and p38 MAPK-mediated phosphorylation of EEA1 requires PLD2 activity, we examined the effect of PLD2 and PPAP2 inhibition on the opioid-induced phosphorylation of endogenous EEA1 in neuronal SHSY-5Y cells. Therefore, the SHSY-5Y cells were transfected or not with nonsense-siRNA, PLD2-siRNA or PPAP2-siRNA, and the effect of the μ-agonist DAMGO and δ-agonist DPDPE on the EEA1 phosphorylation was determined by Western blot analyses. Our data revealed that inhibition of endogenous PLD2 activity by PLD2-siRNA transfection or inhibition of the PPAP2-mediated conversion of PLD2-derived PA into DAG by PPAP2-siRNA resulted in an inhibition of the DAMGO- and DPDPE-induced EEA1 phosphorylation (Fig. 8). These results emphasize the important physiological role of PLD2 activity and signal transduction in the opioid-mediated activation of the Rab5 effector EEA1. Together with the previous findings, these results also clearly demonstrate that the conversion of PLD2-derived PA into DAG is directly linked with EEA1/Rab5 function in the regulation of MOPr endocytosis.

Effect of PLD2-siRNA or PPAP2-siRNA transfection on the DAMGO- or DPDPE-induced EEA1 phosphorylation in neuronal SHSY-5Y cells. SHSY-5Y cells endogenously expressing MOPr and DOPr were transfected or not with PLD2-siRNA or PPAP2-siRNA as described under Materials and Methods. Forty-eight hours after transfection, proteins from SHSY-5Y cells were extracted, and endogenous EEA1 protein was immunoprecipitated and blotted with antibodies against EEA1 or phospho-threonine. As a marker for EEA1 activation, we determined the level of phosphorylated EEA1 by Western blot analysis as described under Materials and Methods. Representative immunoblots from one of three independent experiments are shown. Quantitative data from Fig. 8 are expressed as the fold EEA1 phosphorylation over basal in untreated cells and are presented as means ± S.E. from three independent experiments. *, significant difference (p < 0.05) compared with untreated control cells (ANOVA followed by Bonferroni test).
Discussion
PLD hydrolyzes phosphatidylcholine to generate choline and PA, a bioactive lipid that has various functions in signal transduction, membrane trafficking, transformation, and cytoskeletal dynamics. PLD activity has been shown to be regulated by a number of GPCRs, including VPAC 1 and 2 and PAC1 receptor (McCulloch et al., 2001), metabotropic glutamate receptors (Shinomura et al., 2000; Kanumilli et al., 2002; Bhattacharya et al., 2004), m1 to m4 muscarinic receptors (Sandmann et al., 1991; Mitchell et al., 2003), the endothelin receptor (Ambar and Sokolovsky, 1993), the α2-adrenergic receptor (MacNulty et al., 1992), the D2 dopamine receptor (Senogles, 2000), the somatostatin sstr2 receptor (Grodnitzky et al., 2007), the 5-hydroxytryptamine-2A receptor (Robertson et al., 2003), and the cannabinoid receptor isoform 1(Koch et al., 2006). We have demonstrated recently that PLD2 is also activated by the μ- and δ-opioid receptors and that this PLD2 activation is ARF6-dependent and essential for the agonist-induced opioid receptor endocytosis (Koch et al., 2003, 2006; Rankovic et al., 2009). Our results also revealed that a direct interaction between the PLD2 and opioid receptor is required for a sufficient opioid-induced PLD2 activation (Koch et al., 2003, 2006). Our results are consistent with other findings demonstrating that PLD activity is involved in the endocytosis of various receptors such as the angiotensin II receptor (Du et al., 2004), B-cell antigen receptor (Snyder and Pierce, 2006), class 1 metabotropic glutamate receptors (Bhattacharya et al., 2004), and epidermal growth factor receptor (Lee et al., 2006). However, the mechanistic basis for the PLD2-mediated regulation of receptor endocytosis is only barely understood. It is suggested that the PLD2-derived PA plays a key role in the induction of receptor endocytosis via activation of type I phosphoinositide-4-phosphate 5-kinases, which results in an increased phosphatidylinositol-4,5-bisphosphate level of the membrane (Jones et al., 2000). Clathrin, dynamin, and proteins of the AP-2 adapter complex contain domains that mediate their binding to phosphatidylinositol-4,5-bisphosphate-harboring membranes (Mousavi et al., 2004). In addition, an increase in the level of PA after PLD2 activation results in a change of the physical properties (e.g., charge and pH) of cellular membranes, thereby facilitating vesicle formation.
However, an alternative explanation arises from the data presented here. We found that agonist-induced opioid receptor endocytosis involves PLD2-mediated p38 MAPK activation. This is in line with previous publications demonstrating that p38 MAPK is a regulator of receptor endocytic trafficking (Cavalli et al., 2001; Macé et al., 2005; McLaughlin et al., 2006; Vergarajauregui et al., 2006). The function of p38 MAPK in receptor endocytosis seems to be tightly related to the small GTPase Rab5, one of the key regulators of clathrin-dependent endocytosis. Rab5 coordinates multiple processes, such as the formation of clathrin-coated vesicles, their fusion with early endosomes and homotypic early endosome fusion, and motility of endosomes (Seachrist and Ferguson, 2003). It has been found that p38 MAPK regulates MOPr endocytosis by phosphorylating the Rab5 effectors EEA1 and Rabenosyn-5 to regulate their recruitment to the plasma membrane (Macé et al., 2005). This is in line with the findings from the present study showing that blocking p38 MAPK activity results in a complete block of agonist-induced opioid receptor endocytosis.
Therefore, opioid-induced activation of p38 MAPK seems to be required for the opioid receptor endocytosis. Furthermore, several lines of evidence suggest that opioid-induced p38 MAPK activation requires PLD2 activity. First, inhibition of PLD2 expression by PLD2-siRNA completely blocks not only opioid receptor endocytosis but also opioid-induced p38 MAPK activation. Second, opioids that cannot induce receptor endocytosis, such as morphine, failed to activate both PLD2 and p38 MAPK (Koch et al., 2003; Macé et al., 2005). Third, inhibition of the PLD2-mediated PA synthesis by primary alcohols completely blocks not only opioid receptor endocytosis but also p38 MAPK activation. These observations are supported by other publications demonstrating a crucial role of PLD in the p38 MAPK activation pathways (Grab et al., 2004; Azuma et al., 2007). However, the mechanism of how PLD activates p38 MAPK still needs to be elucidated.
Our findings indicate that PLD2-generated PA might play a key role in the activation of p38 MAPK and subsequent opioid receptor endocytosis. As mentioned above, PLD2-generated PA can be further converted into DAG by the family of enzymes known as PAPs, which are highly active under physiological conditions (Sciorra and Morris, 2002). Furthermore, the PA-derived DAG can be reconverted to PA via phosphorylation by DGKs. Thus, the regulation of PA and DAG levels seems to be tightly controlled via the activities of PAP and DGK. In fact, we found that agonist-induced MOPr and DOPr endocytosis and the p38 MAPK activation were remarkably reduced when the PAP activity was inhibited. Conversely, DGK inhibition significantly augmented both agonist-induced MOPr and DOPr endocytosis. These experiments indicate that the PA-derived DAG is involved in the endocytosis of opioid receptors and in the opioid-mediated p38 MAPK activation. This finding was further supported by the addition of the cell-permeable DAG analog DOG. The presence of DOG strongly enhanced both agonist-induced MOPr and DOPr endocytosis and the p38 MAPK activation. Our finding that the DOG treatment overcomes the inhibitory effect of PLD2-siRNA or PPAP2-siRNA transfection on the agonist-induced opioid receptor endocytosis and p38 MAPK activation indicated that DAG might have a key function upstream of p38 MAPK activation. Because DAG is a lipid second messenger that is known to activate lipid-dependent kinases such as classic and novel PKC families, it was reasonable to suggest that this DAG effect on the opioid receptor endocytosis and p38 MAPK activation is PKC-mediated. However, our data revealed that this is not the case. In fact, the observed effects of PA-derived DAG on receptor endocytosis and p38 MAPK activation seemed to be PKC-independent because they could not be blocked by PKC inhibitors such as calphostin C or chelerythrine.
In addition to the findings with the HEK293 cell model, we and others clearly demonstrate the physiological importance of PLD2 and p38 MAPK activation for the opioid receptor endocytosis in neuronal cells under more physiological conditions (Macé et al., 2005; Rankovic et al., 2009). This is further supported by our present results indicating that the opioid-induced p38 MAPK activation is also PLD2-mediated in neuronal SHSY-5Y cells, which endogenously express the MOPr and DOPr. We also provide evidence, that PLD2-derived PA and DAG synthesis is required for the opioid-induced p38 MAPK-mediated activation of the Rab5 effector EEA1 in neuronal SHSY-5Y cells, emphasizing the important role of EEA1/Rab5 activation in the opioid-mediated endocytosis. In summary, our results reveal that the metabolism from PLD2-generated PA to DAG influences agonist-induced opioid receptor endocytosis via activation of p38 MAPK.
Acknowledgments
We thank Evelyn Kahl, Dana Meyer, and Karina Schäfer for excellent technical assistance.
Footnotes
-
This work was supported by the Deutsche Forschungsgemeinschaft [Grant KR1740/10-1]; and the Forschungszentrum “Center for Behavioral Brain Sciences” Land Sachsen-Anhalt.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.109.063107.
-
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- DOPr
- δ-opioid receptor
- MOPr
- μ-opioid receptor
- DPDPE
- [d-Pen2,d-Pen5]-enkephalin
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin
- PLD
- phospholipase D
- PA
- phosphatidic acid
- DAG
- diacylglycerol
- PAP
- phosphatidic acid phosphohydrolase
- DAG
- diacylglycerol
- DGK
- diacylglycerol kinase
- DOG
- 1, 2-dioctanoyl-sn-glycerol
- PKC
- protein kinase C
- MAPK
- mitogen-activated protein kinase
- EEA1
- early endosome antigen 1
- PCR
- polymerase chain reaction
- RT-PCR
- reverse transcription polymerase chain reaction
- HA
- influenza virus hemagglutinin epitope
- HEK
- human embryonic kidney
- siRNA
- short interfering RNA
- PPAP2
- phosphatidic acid phosphohydrolase type 2
- ANOVA
- analysis of variance
- R59949
- 3-[2-(4-[bis-(4-fluorophenyl)methylene]-1-piperidinyl)ethyl]-2,3-dihydro-2-thioxo-4(1H)quinazolinone
- SB203580
- 4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)1H-imidazole.
- Received December 15, 2009.
- Accepted March 30, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}