


Abstract
The liver X receptor (LXR) and constitutive androstane receptor (CAR) are two nuclear receptors postulated to have distinct functions. LXR is a sterol sensor that promotes lipogenesis, whereas CAR is a xenosensor that controls xenobiotic responses. Here, we show that LXRα and CAR are functionally related in vivo. Loss of CAR increased the expression of lipogenic LXR target genes, leading to increased hepatic triglyceride accumulation, whereas activation of CAR inhibited the expression of LXR target genes and LXR ligand-induced lipogenesis. On the other hand, a combined loss of LXR α and β increased the basal expression of xenobiotic CAR target genes, whereas activation of LXR inhibited the expression of CAR target genes and sensitized mice to xenobiotic toxicants. The mutual suppression between LXRα and CAR was also observed in cell culture and reporter gene assays. LXRα, like CAR, exhibited constitutive activity in the absence of an exogenously added ligand by recruiting nuclear receptor coactivators. Interestingly, although CAR competed with LXRα for coactivators, the constitutive activity and recruitment of coactivators was not required for CAR to suppress the activity of LXRα. In vivo chromatin immunoprecipitation assay showed that cotreatment of a CAR agonist compromised the LXR agonist responsive recruitment of LXRα to Srebp-1c, whereas an LXR agonist inhibited the CAR agonist-responsive recruitment of CAR to Cyp2b10. In conclusion, our results have revealed dual functions of LXRα and CAR in lipogenesis and xenobiotic responses, establishing a unique role of these two receptors in integrating xenobiotic and endobiotic homeostasis.
Introduction
Metabolic homeostasis, including those of the endogenous chemicals (endobiotics) and foreign substances (xenobiotics), are essential for the survival of mammals. Nuclear hormone receptors play an important role in metabolic homeostasis. These include the sterol sensors liver X receptors (LXRs) that control lipid homeostasis (Tontonoz and Mangelsdorf, 2003) and the xenosensor constitutive androstane receptor (CAR) (Honkakoski et al., 1998; Wei et al., 2000) that regulates the expression of drug-metabolizing enzymes and transporters.
LXRs, both the α and β isoforms, are defined as sterol sensors. LXRα is highly expressed in the liver, whereas LXRβ is ubiquitously expressed. In addition to being activated by endogenous oxysterols, LXRs are also activated by synthetic agonists, such as N-methyl-n-[4-(2,2,2-trifluoro-1-hydroxy-1-trifluoromethylethyl)-phenyl]-benzenesulfonamide (T0901317, TO1317) (Schultz et al., 2000) and 3-[3-[[[2-chloro-3-(trifluoromethyl)phenyl]methyl](2,2-diphenylethyl)amino]propoxy]benzeneacetic acid hydrochloride (GW3965) (Collins et al., 2002). In rodents, LXRs increase hepatic cholesterol catabolism and formation of bile acids by inducing cholesterol 7α-hydroxylase (Peet et al., 1998). LXRs were later found to promote hepatic lipogenesis by activating SREBP-1c (Repa et al., 2000), a transcriptional factor that regulates the expression of lipogenic enzymes ACC-1, FAS, and SCD-1. ACC-1, FAS, and SCD-1 can also be directly regulated by LXR (Joseph et al., 2002; Chu et al., 2006; Talukdar and Hillgartner, 2006). Loss of both LXR isoforms in mice resulted in an increased expression of Cyp3a11 and 2b10 (Gnerre et al., 2005), two drug-metabolizing enzymes and primary target genes of CAR and pregnane X receptor (PXR) (Blumberg et al., 1998; Kliewer and Willson, 2002). However, the mechanism by which LXRs affect the expression of drug-metabolizing enzymes remains unknown.
CAR, along with its sister receptor PXR, has been shown to function as a master xenosensor by its coordinated transcriptional regulation of drug-metabolizing enzymes and transporters (Swales and Negishi, 2004). CAR(−/−) mice showed defective basal and inducible expression of xenobiotic enzymes and altered responses to drugs (Wei et al., 2000). CAR has been suggested recently to play a role in energy metabolism, ranging from thyroid hormone metabolism (Maglich et al., 2004; Qatanani et al., 2005) to lipogenesis (Roth et al., 2008a,b; Gao et al., 2009; Maglich et al., 2009), gluconeogenesis (Ueda et al., 2002; Kodama et al., 2004; Miao et al., 2006; Gao et al., 2009; Wada et al., 2009), and obesity and diabetes (Dong et al., 2009; Gao et al., 2009). It is unclear whether the effect of CAR on lipogenesis involves the cross-talk with LXRs.
In this report, we found that LXRα and CAR are mutually suppressive in their target gene regulation, which could be translated into their effects on lipogenesis and xenobiotic responses. Our results suggest dual and unique roles of LXRα and CAR in integrating xenobiotic and endobiotic homeostasis.
Materials and Methods
Animals and Drug Treatment.
The creation of PXR(−/−) (Xie et al., 2000a), CAR(−/−) (Wei et al., 20000), PXR/CAR double knockout (PC DKO) (Saini et al., 2004), LXR DKO (Peet et al., 1998), VP-CAR transgenic (Saini et al., 2004), and fatty acid binding protein-VP-LXRα transgenic (Uppal et al., 2007) mice has been described previously. All transgenic and their wild-type control mice were maintained in a mixed background of C57BL/6J and 129/SvJ, except for the wild-type mice used in Fig. 2, which were C57BL/6J mice purchased from The Jackson Laboratory (Bar Harbor, ME). TO1317 (50 mg/kg) and GW3965 (20 mg/kg) were given by gavage (Joseph et al., 2002; Laffitte et al., 2003). TCPOBOP (3 mg/kg) was given by intraperitoneal injection (Wei et al., 2000). The drug treatment lasted for 3 days. Tribromoethanol tolerance experiment was performed as we have described previously (Xie et al., 2000a). The use of mice in this study complied with all relevant federal guidelines and institutional policies.
Measurement of Liver and Circulating Lipids.
To measure circulating lipid levels, mice were fasted for 16 h before sacrificing and blood collection. Lipid tissue lipids were extracted as described previously (Zhou et al., 2006). Triglyceride and cholesterol levels were measured by assay kits from Stanbio (Boerne, TX).
Real-Time Reverse Transcriptase-PCR.
Total RNA was isolated using TRIzol reagent from Invitrogen (Carlsbad, CA). SYBR Green-based real-time reverse transcriptase-PCR was performed with the ABI 7300 real-time PCR System (Applied Biosystems, Foster City, CA). The gene expression was normalized against the expression of cyclophilin. PCR primer sequences are listed in Supplementary Table 1.
DNA Constructs, Transient Transfection, and GST Pull-Down Assay.
tk-SCD1/LXRE (Chu et al., 2006), tk-MRP2 (Mu et al., 2005), pGL-SCD1 (Chu et al., 2006), pGL-Cyp2b10 (Xie et al., 2000b), and Gal-SRC1 (Saini et al., 2005) constructs were described previously. pCMX-Flag-hLXRα and pCMX-HA-mCAR were cloned by PCR. Transfection of HepG2 or CV-1 cells on 48-well plates was performed as described previously (Uppal et al., 2007). When necessary, cells were treated with drugs for 24 h before luciferase assay. Transfection efficiency was normalized against the β-gal activities from a cotransfected CMX-β gal vector. GST pull-down using bacteria-expressed GST-SRC1, and in vitro-translated and 35S-labeled receptor proteins were performed as described previously (Saini et al., 2005).
Chromatin Immunoprecipitation Assay.
Eight-week-old wild-type female mice were pretreated with an intraperitoneal injection of DMSO or TCPOBOP (3 mg/kg) and/or a gavage of vehicle or GW3965 (20 mg/kg) 1 h before being liver-transfected with pCMX-Flag-hLXRα and pCMX-HA-mCAR plasmids by a hydrodynamic gene delivery method (Zhou et al., 2006). Mice were sacrificed 24 h after transfection, and the liver tissues were harvested for chromatin immunoprecipitation (ChIP) assay and Western blot analysis. The ChIP procedures followed the Millipore protocol (Billerica, MA) and were essentially as described previously (Zhou et al., 2006). Antibodies used for immunoprecipitation include an anti-HA antibody from Cell Signaling Technology (Danvers, MA), an anti-Flag antibody from Sigma-Aldrich (St. Louis, MO), and a normal mouse IgG antibody from Millipore. PCR was carried out with Cyp2b10-specific primers encompassing the phenobarbital response element (PBRE) (5′-CTCCAGTGACTTAGGAGGAAG-3′; 5′-AAGTATTGTGCCAGTTGCTG-3′), and Srebp-1c-specific primers encompassing the direct repeat spaced by four nucleotides (DR-4) site (5′-TCCAGGCAAGTTCTGGGTGTGTGCG-3′; 5-CGGGTTTCTCCCGGTGCTCTGAATG-3′). The sequences for Cyp2b10/PBRE and Srebp-1c/LXRE are 5′-TCTGTACTTTCCTGACCTTGGCACAGTGCCACCATCAACTTGCCTGACACC-3′ (Sueyoshi et al., 1999) and 5′-ACAGTGACCGCCAGTAACCCCAGC-3′ (Yoshikawa et al., 2001), respectively.
Results
Reciprocal Activation of Target Gene Expression in Mice Deficient of CAR and LXR.
We have reported recently that PXR(−/−) mice had increased basal expression of the LXR target genes Scd-1 (Zhou et al., 2006). This observation prompted us to examine the effect of loss of CAR on LXR target gene expression compared with PXR(−/−) mice and PC DKO mice. Loss of PXR induced the expression of Scd-1 but had little effect on the expression of Srebp-1c, Acc-1, Fas, Abcg5, and Abcg8 (Fig. 1A), consistent with our previous finding (Zhou et al., 2006). In contrast, CAR(−/−) mice showed significantly increased expression of all of these LXR target genes (Fig. 1A). Combined loss of CAR and PXR (PC DKO) had a synergistic effect in inducing Scd-1, Srebp-1c, and Abcg8. Interestingly, the synergistic effect of PC DKO seemed to be gene-specific. Compared with CAR(−/−) mice, the induction of Abcg5 remained unchanged, and the induction of Acc-1 and Fas was actually decreased in PC DKO mice. The mechanism for this gene-specific effect remains to be determined. Loss of PXR and/or CAR had little effect on the expression of LXRα or LXRβ (Fig. 1A). Despite their higher basal expression, LXR target genes remained inducible by TO1317 in CAR(−/−) mice (Fig. 1B, left). Compared with their wild-type counterparts (Fig. 1B, right), the TO1317-responsive induction of Fas, Scd-1, and Abcg5 was more dramatic in CAR(−/−) mice. TO1317 at 50 mg/kg has been reported to activate PXR in vivo (Mitro et al., 2007). We showed that PC DKO mice responded to TO1317 in a similar fashion as the CAR(−/−) mice (Supplementary Fig. 1), suggesting that the TO1317 effect on the expression of LXR target genes in CAR(−/−) mice can be PXR-independent. Consistent with the patterns of gene expression, we found the triglyceride content in the liver of CAR(−/−) and PC DKO mice was nearly three times that of the wild-type and PXR(−/−) mice (Fig. 1C). No significant changes in the hepatic cholesterol levels were observed (Fig. 1C). The circulating levels of triglycerides increased in CAR(−/−) but not PC DKO mice (Fig. 1D).

Reciprocal activation of target gene expression in mice deficient of CAR and LXR. A, real-time PCR analysis on the hepatic expression of LXR target genes in wild-type, PXR(−/−), CAR(−/−), and double knockout (PC DKO) mice. B, real-time PCR analysis on the hepatic expression of LXR target genes in CAR(−/−) mice (left) or WT mice (right) in the presence or absence of TO1317. C and D, measurements of triglycerides and cholesterol in the liver (C) and plasma (D). E, real-time PCR analysis on the hepatic expression of CAR target genes in wild-type and LXR DKO mice. F, real-time PCR analysis on the hepatic expression of CAR target genes in LXR DKO mice in the presence or absence of TCPOBOP. The fold inductions in F are labeled. All mice shown are males. Results represent the averages and standard deviation from four to six mice per group. *, P < 0.05; **, P < 0.01, compared with the WT (A, C–E) or DMSO control (B).
The reciprocal effect of loss of LXR on CAR target gene expression was evaluated in LXR α and β double-knockout mice (LXR DKO). The expression of Cyp2b10 and Cyp3a11, two CAR target genes, was induced in LXR DKO mice (Fig. 1E) as expected (Gnerre et al., 2005). Loss of LXRs had little effect on the expression of PXR or CAR (Fig. 1E). Despite their high basal expression, Cyp2b10 and Cyp3a11 remained inducible by TCPOBOP in LXR DKO mice (Fig. 1F).
Mutual Repression of Ligand-Dependent Target Gene Expression by Pharmacological Activation of LXR and CAR.
This was evaluated in wild-type C57BL/6J mice treated with the LXR agonist GW3965 and CAR agonist TCPOBOP individually or in combination. GW3965 alone induced the hepatic expression of Srebp-1c, Acc-1, Fas, Scd-1, Abcg5, and Abcg8 as expected (Fig. 2A). TCPOBOP alone, on the other hand, suppressed the basal expression of Srebp-1c, Acc-1, Fas, Scd-1, and Abcg5 (Fig. 2A). The most notable phenotype, however, is that the GW3965-induced LXR target gene activation was largely abolished in mice treated with both drugs (Fig. 2A). Consistent with the pattern of gene expression, the hepatic content of triglycerides in dual-treated mice was lower than in mice treated with GW3965 alone (Fig. 2B). Interestingly, TCPOBOP alone caused a modest but significantly increased triglyceride level (Fig. 2B), despite the suppression of lipogenic enzymes in this group (Fig. 2A). This mild steatosis might be secondary to TCPOBOP-induced hepatomegaly (Wei et al., 2000). Treatment with GW3965 increased serum concentration of triglycerides, but this effect was abolished in dual-treated mice (Fig. 2C). This regimen of drug treatment had little effect on cholesterol levels (Fig. 2, B and C). When the expression of CAR target genes was analyzed, we found that GW3965 suppressed the basal expression of both Cyp2b10 and Cyp3a11 (Fig. 2D). The expression of Cyp2b10 and Cyp3a11 was induced by TCPOBOP as expected, and the TCPOBOP effect was largely intact in dual-treated mice (Fig. 2D), suggesting that the CAR agonist plays a dominant role in regulating xenobiotic enzymes when ligands for both CAR and LXR are present.

Mutual repression of target gene expression by pharmacological activation of LXR and CAR. A, real-time PCR analysis on the hepatic expression of LXR target genes in wild-type C57BL/6J male mice treated with GW3965 and TCPOBOP individually or in combination. The expression of individual genes in vehicle (DMSO) treated mice is arbitrarily set as 1. B and C, measurements of triglycerides and cholesterol in the liver (B) and plasma (C). D, real-time PCR analysis on the hepatic expression of CAR target genes. All mice shown are males. Results represent the averages and standard deviation from four to six mice per group. *, P < 0.05; **, P < 0.01, compared with the DMSO control.
Reciprocal Repression of Target Gene Expression by Genetic Activation of LXR and CAR in Transgenic Mice.
We have created recently transgenic mice expressing the activated LXRα (VP-LXRα) in the liver (Uppal et al., 2007; Gong et al., 2008; Lee et al., 2008). VP-LXRα was created by fusing the VP16 activation domain of the herpes simplex virus to the amino-terminal of mouse LXRα. In VP-LXRα transgenic mice, in addition to the expected activation of LXR target genes, we observed the suppression of Cyp2b10 and Cyp3a11 (Fig. 3A). The expression of Cyp2b10 and Cyp3a11 remained inducible by TCPOBOP in VP-LXRα transgenic mice (Fig. 3B), but the magnitude of Cyp2b10 induction was markedly lower than that observed in TCPOBOP-treated LXR DKO (Fig. 1F) or wild-type (Fig. 2D) mice. VP-LXRα transgenic mice were more sensitive to the anesthetic effect of tribromoethanol, consistent with the notion that Cyp2b10 and Cyp3a11 play a role in the detoxification of this drug (Xie et al., 2000a, 2001). Loss of PXR or CAR sensitized mice to tribromoethanol-induced sleep (Fig. 3C). When the LXR effect was evaluated, we found that wild-type mice slept for an average of 20 min, whereas the VP-LXRα transgenic mice slept for nearly 50 min (Fig. 3D). The tribromoethanol-sensitizing effect was also observed in wild-type mice pretreated with GW3965 (Fig. 3D).
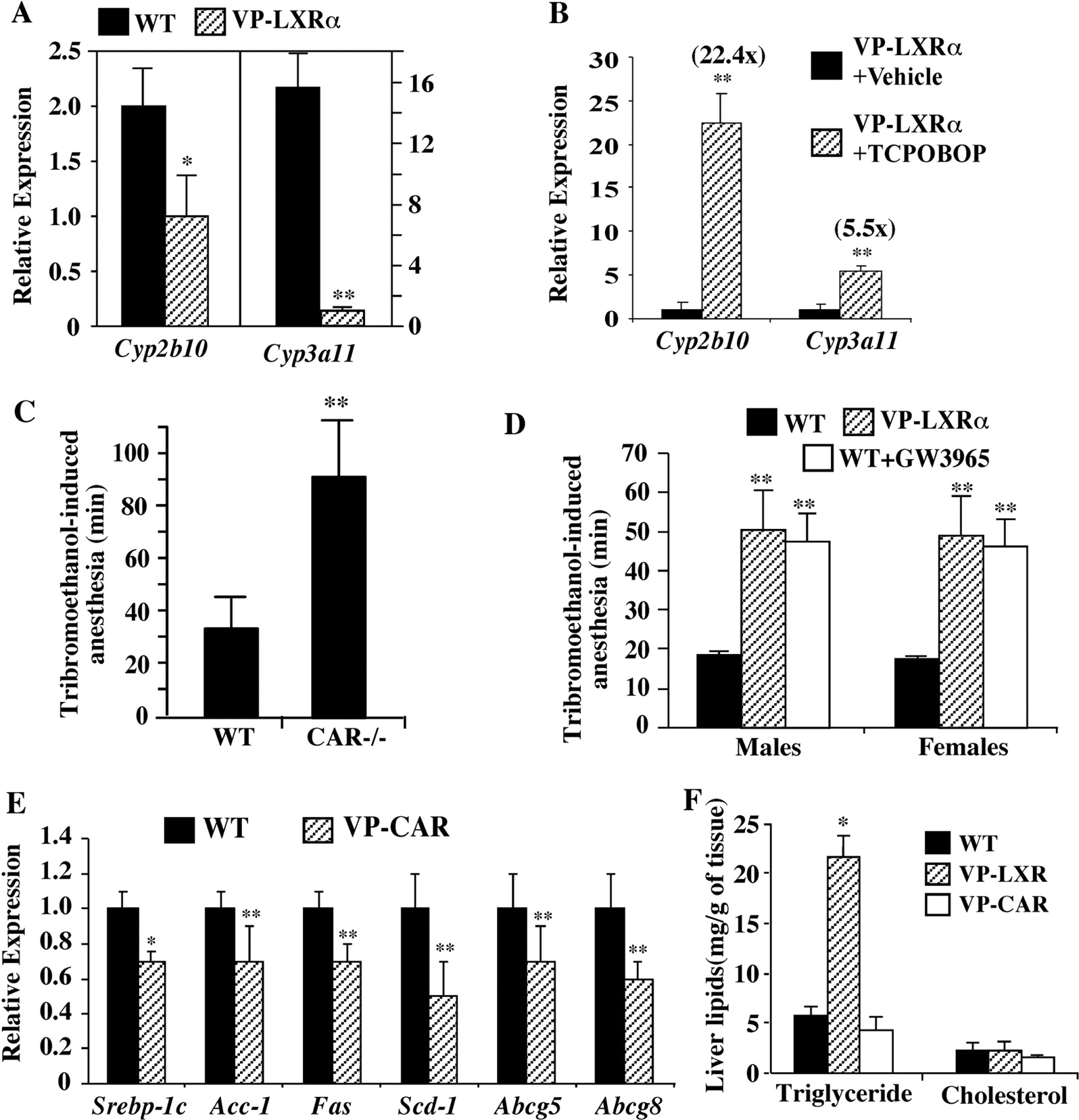
Mutual repression of target gene expression by genetic activation of LXRα and CAR in transgenic mice. A, real-time PCR analysis on the hepatic expression of CAR target genes in the wild-type (WT) and VP-LXRα transgenic mice. B, real-time PCR analysis on the hepatic expression of CAR target genes in VP-LXRα transgenic mice in the presence or absence of TCPOBOP. The fold inductions are labeled. C, wild-type and CAR(−/−) mice were subjected to the tribromoethanol anesthesia test. D, the VP-LXRα transgene or treatment with GW3965 sensitized mice to the tribromoethanol anesthesia test. E, real-time PCR analysis on the hepatic expression of LXR target genes in WT and VP-CAR transgenic mice. F, measurements of triglycerides and cholesterol in the liver of WT and VP-CAR transgenic mice. Mice are males if not specified. Results represent the averages and standard deviation from four to six mice per group. *, P < 0.05; **, P < 0.01, all compared with the WT.
We have also reported the creation and characterization of transgenic mice that bear the expression of activated CAR (VP-CAR) in the liver (Saini et al., 2004, 2005). VP-CAR transgenic mice showed decreased expression of LXR target genes Srebp-1c, Acc-1, Fas, Scd-1, Abcg5, and Abcg8 (Fig. 3E). VP-LXRα transgenic mice had a spontaneous hepatic accumulation of triglycerides, whereas the VP-CAR transgene had little effect (Fig. 3F).
Mutual Suppression between LXRα and CAR in Promoter Reporter Gene Assays.
tk-Scd1/LXRE-Luc and tk-PBRE-Luc reporter genes contain a DR-4-type LXRE from the Scd-1 gene promoter and PBRE from the Cyp2b10 gene promoter, respectively. The tk-Scd1 report had a 4-fold activation by LXRα in the absence of an exogenously added ligand (Fig. 4A). The activity of LXRα increased in the presence of TO1317 or GW3965 but not TCPOBOP. Cotransfection of CAR inhibited both the constitutive and TO1317/GW3965-inducible activities of LXRα, and this inhibition was enhanced by TCPOBOP (Fig. 4A). CAR itself had little effect on the activity of tk-Scd1 (Fig. 4A). On the other hand, when tk-PBRE reporter was used, the mouse CAR exhibited constitutive activity, which can be further activated by TCPOBOP (Fig. 4B). Both the constitutive and TCPOBOP-dependent CAR activities were inhibited by the cotransfection of LXRα, and this inhibition was enhanced by TO1317 or GW3965 (Fig. 4B). LXRα itself had little effect on the activity of tk-PBRE (Fig. 4B). When the Scd-1 natural gene promoter reporter pGL-Scd1 was transfected, both the constitutive and GW3965-dependent activities of LXRα were inhibited by the cotransfected CAR in a dose-dependent manner (Fig. 4C). On the other hand, the constitutive and TCPOBOP-dependent activities of CAR on the Cyp2b10 natural promoter reporter pGL-Cyp2b10 were inhibited by the cotransfected LXRα (Fig. 4D). The LXRα-CAR mutual suppression was also observed when the Gal-LXRα and Gal-CAR chimeric receptors and the Gal4-responsive tk-UAS report were used. The constitutive and GW3965-dependent activities of Gal-LXRα were inhibited by the cotransfection of wild-type CAR (Fig. 4E), whereas the constitutive and TCPOBOP-dependent activities of Gal-CAR were inhibited by the cotransfection of wild-type LXRα (Fig. 4F).

Mutual suppression between LXRα and CAR in reporter gene assays. The tk-LXRE (A), tk-PBRE (B), pGL-Scd1 (C), pGL-Cyp2b10 (D), and tk-UAS (E and F) luciferase reporter genes were transiently transfected into cells in the presence of expression vectors for indicated receptors or their combinations. Where applicable, transfected cells were treated with indicated drugs for 24 h before luciferase assay. The transfection efficiency was normalized against the β-gal activity from the cotransfected CMX-β-gal vector. Results shown are fold induction over vector control and represent the averages and standard deviation from triplicate assays. Drug concentrations are the following: androstenol, 5 μM; TCPOBOP, 250 nM; TO1317, 10 μM; and GW3965, 10 μM.
Mechanistic Studies for the Mutual Suppression between LXRα and CAR in Cell Cultures.
CAR exhibits constitutive activity because of its ligand-independent recruitment of nuclear receptor coactivators (Forman et al., 1998). The high basal activity of LXRα prompted us to examine whether this receptor can also recruit coactivators in the absence of a ligand. We first used a mammalian two-hybrid assay to examine the recruitment of steroid receptor coactivator 1 (SRC1) by LXRα. Cells were transfected with tk-UAS reporter together with the expression vectors for Gal-hSRC1 and VP-LXRα. The activation of reporter in the absence of an exogenously added ligand suggested the constitutive recruitment of SRC1 by LXRα (Fig. 5A). The LXRα-SRC1 interaction was enhanced by TO1317 but not by TCPOBOP (Fig. 5A). Cotransfection of CAR inhibited the constitutive recruitment of SRC1 by LXRα. The inhibitory effect of CAR was relieved by the treatment of TO1317 but was exacerbated by TCPOBOP (Fig. 5A). The inhibition of the LXRα-SRC1 interaction seemed to be CAR-specific, because cotransfection of PPARγ had little effect in the absence of a PPARγ agonist (Fig. 5A). The addition of the PPARγ agonist rosiglitazone (BRL49653) decreased the LXRα-SRC1 interaction (Fig. 5A). The ligand-independent recruitment of SRC1 by LXRα was confirmed by GST pull-down assays. As shown in Fig. 5B, GST-SRC1 interacted with both [35S]LXRα and [35S]CAR but not [35S]PPARγ in the absence of an exogenously added ligand. The PPARγ-SRC1 interaction was induced by BRL49653. Consistent with their patterns of coactivator recruitment in the absence of exogenously added ligands, CAR was more efficient than PPARγ in suppressing the constitutive and GW3965-dependent activities of LXRα in reporter gene assays (data not shown). These results suggest that competition for coactivators is a plausible mechanism for the mutual suppression between LXRα and CAR. Indeed, the increasing concentration of Gal-SRC1 was able to titrate the inhibitory effect of CAR in the mammalian two-hybrid assay (Fig. 5C). Reciprocally, the constitutive and TCPOBOP-dependent SRC1-CAR interaction was inhibited by the cotransfection of LXRα (Fig. 5D).

Mechanistic studies for the mutual suppression between LXRα and CAR in cell cultures. A, mammalian two-hybrid assay to demonstrate the SRC1-LXRα interaction and the effect of CAR cotransfection. HepG2 cells were transfected with Gal-SRC1 and the tk-UAS-Luc reporter gene in the presence of indicated receptors. Transfected cells were treated with indicated drugs for 24 h before luciferase assay. Results shown are fold induction over vector control and represent the averages and standard deviation from triplicate assays. B, GST pull-down assay to demonstrate the SRC1-LXRα, SRC1-CAR, and SRC1-PPARγ interactions. Equal volumes of 35S-labeled proteins were loaded to demonstrate the efficiency of protein translation. C, the inhibition of SRC1-LXRα interaction by CAR was relieved by increased concentration of SRC1 in a mammalian two-hybrid assay. D, the inhibition of SRC1-CAR interaction by LXRα. E, CARΔ8 and CARΔ37 lacked transcriptional activity on the tk-PBRE report gene. F, CARΔ37 failed to interact with GST-SRC1 in the GST pull-down assay. G, CARΔ37, but not CARΔ8, failed to suppress the constitutive and GW3965-inducible activities of LXRα on the tk-LXRE reporter gene. H, a forced expression of RXR did not abolish the inhibitory effect of CAR on LXRα. Drug concentrations are the following: TO1317, 10 μM; TCPOBOP, 250 nM; BRL49653, 5 μM; and GW3965, 10 μM.
We then used two CAR mutants CARΔ8 and CARΔ37 to determine whether the recruitment of coactivators was necessary for the inhibitory effect of CAR on LXRα. CARΔ8 and CARΔ37 lack the C-terminal 8 and 37 amino acids, respectively (Choi et al., 1997). CARΔ8 has the disruption of the activator function (AF)-2 region and thus fails to bind to coactivators (Choi et al., 1997; Min et al., 2002). CARΔ37 also lacks the C terminus of the helix 10 that is important for heterodimerization with RXR and consequently fails to bind to DNA (Choi et al., 1997). As expected, neither CARΔ8 nor CARΔ37 can activate tk-PBRE reporter (Fig. 5E). Both CARΔ8 and CARΔ37 failed to interact with GST-SRC1 in a GST pull-down assay (Fig. 5F). Interestingly and surprisingly, CARΔ8 was effective in suppressing the constitutive and GW3965-dependent LXRα activities on the tk-Scd1 reporter (Fig. 5G), whereas CARΔ37 completely lost its inhibitory effect. The intact suppression by CARΔ8 suggests that the recruitment of coactivator is not required for the inhibitory effect of CAR. Because RXR is a shared heterodimerization partner for CAR and LXR, we also evaluated whether the inhibition of LXRα activity by CAR can be relieved by the overexpression of RXR. As shown in Fig. 5H, cotransfection of RXR increased the basal activity of LXRα but did not abolish the suppressive effect of CAR.
Effects of CAR and LXR Agonists on Receptor Recruitment to Target Gene Promoters In Vivo.
To understand the in vivo mechanism for the mutual suppression between LXRα and CAR, we performed ChIP assay to determine the effects of individual and combined treatment of GW3965 and TCPOBOP on the respective recruitment of LXRα to Srebp-1c gene promoter and CAR to Cyp2b10 gene promoter. In this experiment, mouse livers were transfected with both Flag-tagged LXRα (Flag-LXRα) and HA-tagged CAR (HA-CAR). Anti-Flag and anti-HA antibodies were used for chromatin immunoprecipitation. As shown in Fig. 6A, Flag-LXRα was specifically recruited onto the Srebp-1c gene promoter in response to GW3965, but the recruitment was largely abolished in mice coadministered with TCPOBOP. When the recruitment of HA-CAR onto the Cyp2b10 gene promoter was evaluated, we found that TCPOBOP enhanced the recruitment of HA-CAR, which was modestly inhibited by the cotreatment of GW3965 (Fig. 6B). We noted that little receptor occupancy of the promoter, especially for LXRα, was detected in the absence of ligands. The lack of more obvious basal occupancy in ChIP assay may be due to the experimental conditions and the limitation of sensitivity. The expression of the transfected receptors was confirmed by Western blot analysis (Fig. 6C).

Effects of CAR and LXR agonists on receptor recruitment to target gene promoters in vivo. A and B, Flag-LXRα and HA-CAR expression vectors were hydrodynamically transfected into the mouse liver. Transfected mice were treated with GW3965 and/or TOPOBOP for 8 h before sacrificing and ChIP assay using anti-Flag and anti-HA antibodies. PCRs in A and B encompass the Srebp-1c/DR4 LXRE and Cyp2b10/PBRE, respectively. ChIP with normal IgG was included as negative controls. Lanes represent individual mice. C, the expression of Flag-LXRα and HA-CAR proteins in transfected livers was confirmed by Western blot analysis. The β-actin blot was included as a protein loading control.
Discussion
In this study, we have uncovered a mutual suppression between LXRα and CAR that links these two seemingly distinct pathways of lipogenesis and xenobiotic response. Based on our results and as summarized in Fig. 7, we propose a model of LXRα-CAR cross-talk, in which the activation of LXR suppresses CAR-mediated xenobiotic response, leading to sensitization of animals to xenotoxicants. In contrast, activation of CAR may suppress the LXR-mediated lipogenesis.

A model of functional cross-talk between LXR and CAR in regulating lipogenesis and xenobiotic responses. The mutual suppression may have linked LXR-mediated endobiotic and CAR-mediated xenobiotic metabolism in the liver and intestine.
We showed that LXRα exhibited constitutive activity by interacting with coactivators in the absence of an exogenously added agonist. The competition for coactivators has been proposed to be a mechanism for the mutual inhibition between CAR and the estrogen receptor (Min et al., 2002), CAR and PXR (Saini et al., 2005), CAR and hepatic nuclear factor-4 (Miao et al., 2006), and LXR and retinoid-related orphan receptor-α (Wada et al., 2008). In the current study, we showed that although CAR can compete with LXRα for coactivators, the constitutive activity and recruitment of coactivators did not seem to be required for CAR to suppress the activity of LXRα. However, CARΔ8, a CAR mutant that bears the disruption of the AF-2 region and thus fails to bind to coactivators (Choi et al., 1997; Min et al., 2002), was efficient to suppress the constitutive and ligand-inducible activity of LXRα (Fig. 5G). Moreover, down-regulation of SRC1 by small interfering RNA also did not enhance the inhibitory effect of CAR on LXRα (data not shown). Although the CARΔ8 results cannot eliminate the possibility that the LXR suppression of CAR involves coactivator competition, our results suggest that competition for coactivators is unlikely to be the primary mechanism for the mutual suppression between LXRα and CAR. The mechanism for the inhibitory effect of CARΔ8 on LXR remains to be clearly defined. Because CARΔ8 is transcriptionally inactive (Fig. 5E), our results suggested that it is unlikely that unknown CAR target gene(s) are responsible for the inhibitory effect of CAR. Most, if not all, nuclear receptors have two activation functions (AFs), the C-terminal AF-2 and the N-terminal AF-1. Although AF-2 is not required, as suggested by the CARΔ8 results, it remains to be determined whether AF-1 of CAR is necessary for the inhibitory effect of CAR on LXR. The AF-1 of PPARα has been reported to be important for the bidirectional inhibitory cross-talk between PPARα and signal transducer and activator of transcription 5b (Shipley and Waxman, 2004).
The loss of inhibitory effect of CARΔ37 is particularly intriguing. CARΔ37 failed to heterodimerize with RXR and bind to DNA (Choi et al., 1997). However, because CAR/RXR heterodimers cannot bind to LXRE, and a forced expression of RXR failed to abolish the inhibitory effect of CAR, we cannot conclude that the loss of RXR binding is responsible for the lack of inhibition by CARΔ37. The lack of RXR rescue was in contrast to the reported mutual suppression between LXRα and PPARα, in which the inhibitory effect of PPARα on LXRα was completely abolished by a forced expression of RXR (Yoshikawa et al., 2003). Among other potential mechanisms, several reports suggested that CAR or LXR can share or compete for DNA binding sites with other nuclear receptors (Xie et al., 2000a; Handschin et al., 2002). Both LXR and CAR have been reported to bind to DR-4-type nuclear receptor binding sites; however, our electrophoretic mobility shift assay results showed that LXR cannot bind to PBRE, and CAR had little affinity toward Srebp-1c/DR-4 (data not shown). Interestingly and despite the lack of share of DNA binding motifs, our ChIP results showed that cotreatment of a CAR agonist compromised the LXR agonist-responsive recruitment of LXRα to Srebp-1c, whereas an LXR agonist inhibited the CAR agonist responsive recruitment of CAR to Cyp2b10 (Fig. 6).
The high basal activity of LXRα is an interesting observation. The biological significance of the constitutive activity of CAR is obvious. As a xenobiotic receptor, CAR is essential in mammals' coping with obnoxious substances (Wei et al., 2000; Zhang et al., 2002). On the other hand, xenobiotic enzymes are mostly produced or induced as needed. As such, sustained over-activation of xenobiotic responses could be harmful, as evidenced by the sensitization to caffeine and acetaminophen toxicity in CAR-activated mice (Wei et al., 2000; Zhang et al., 2002). In this regard, the constitutive activity of LXRα and consequent suppression of CAR activity may have offered a mechanism of “checks and balances” to maintain a proper level of xenobiotic clearance. Reciprocally, lipogenesis is an essential function of the liver, in which LXRα plays an important role. However, overactivation of lipogenesis is potentially harmful, leading to both local and systemic metabolic disorders. It remains to be determined whether CAR represents a cellular factor that helps to keep the lipogenic activity of LXRα in check.
Our results have also shown that the balance between LXR and CAR activities can be shifted by activation of the receptors. This functional interplay between sterol receptor and xenobiotic receptor may have its implications in drug metabolism and lipogenesis. It is conceivable that CAR agonists may be used to limit the intensity and duration of LXR-mediated lipogenesis, thus alleviating the lipogenic side effect of LXR agonists. Indeed, we have reported recently that activation of CAR was beneficial in preventing obesity and relieving type 2 diabetes, in which the CAR-mediated suppression of hepatic lipogenesis played an important role (Gao et al., 2009). Reciprocally, because sustained activated of LXR may compromise drug metabolism, cautions to avoid drug accumulation and toxicity should be applied when LXRs are being explored as therapeutic targets.
Because LXRα and CAR have functions outside of lipogenesis and drug metabolism, the LXRα-CAR cross-talk might be implicated in other physiological and pathophysiological conditions. For example, cholesterol and bile acids homeostasis is tightly controlled by the functions of liver and intestine, in which both LXRα and CAR are highly expressed. Treatment of WT mice with LXR agonists increased high-density lipoprotein (HDL) cholesterol level (Jiang et al., 2003), whereas HDL cholesterol level was elevated in CAR(−/−) mice (Stedman et al., 2005). It is interesting to know whether the presumed increased activity of LXR in CAR(−/−) mice may have contributed to the elevated HDL cholesterol level in this genotype. In another example, the serum bile acid level after bile duct ligation in CAR(−/−) mice was significantly lower than that in WT mice (Stedman et al., 2005), whereas our previous study showed that LXR DKO mice had increased level of circulating bile acids upon bile duct ligation (Uppal et al., 2007). The opposite effect of loss of CAR and LXRs on serum bile acid level also suggested that the LXRα-CAR cross-talk might also play a role in the homeostasis of bile acids.
A recent report suggested that activation of CAR can suppress lipid metabolism by reducing the protein level of the active form of SREBP-1 (Roth et al., 2008a), which was reasoned to be due to the CAR-mediated induction of Insig-1, an anti-lipogenic protein that blocks proteolytic activation of SREBPs (McPherson and Gauthier, 2004). In a subsequent study, the same group showed that activation of SREBP-1 by insulin or cholesterol inhibited the activity of CAR, in which SREBP-1 may function as a non-DNA binding inhibitor that blocks the interaction of CAR with coactivators (Roth et al., 2008b). These results suggest another possible but not mutually exclusive mechanism by which LXR and CAR might cross-talk. The relative contribution of Insig-1 induction by CAR and LXR inhibition by CAR in the overall effect of CAR on lipogenesis remains to be determined.
In summary, the current study has revealed a mutual repression between LXRα and CAR that links hepatic lipogenesis and xenobiotic responses. The in vivo significance of this cross-talk was strongly supported by recent reports that treatment with the CAR agonist TCPOBOP inhibited hepatic steatosis in high-fat diet-treated wild-type mice and ob/ob mice (Dong et al., 2009; Gao et al., 2009; Maglich et al., 2009).
Acknowledgments
We thank Ying Mu for preliminary transfections and some of the GST pull-down assays. We thank Jie Gao for some of the animal drug-dosing experiments. We thank Yong Li for GST-CAR protein. We also thank Dr. David Mangelsdorf for providing the LXR DKO mice.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported in part by the National Institutes of Health National Institute of Environmental Health Sciences [Grant ES014626]; the National Institute of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK076962]; and the National Natural Science Foundation of China [Grant 30870926].
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.064618.
-
ABBREVIATIONS:
- LXR
- liver X receptor
- CAR
- constitutive androstane receptor
- LXRE
- liver X receptor-responsive element
- PXR
- pregnane X receptor
- PBRE
- phenobarbital response element
- Acc-1
- acetyl CoA carboxylase 1
- Fas
- fatty acid synthase
- Scd-1
- stearoyl CoA desaturase-1
- SRC1
- steroid receptor coactivator 1
- PCR
- polymerase chain reaction
- Srebp-1c
- sterol regulatory element-binding protein 1c
- TO1317 (TO901317)
- N-methyl-n-[4-(2,2,2-trifluoro-1-hydroxy-1-trifluoromethylethyl)-phenyl]-benzenesulfonamide
- TCPOBOP
- 1,4-bis[2-(3,5 dichloropyridyloxy)] benzene
- VP
- viral protein 16
- ChIP
- chromatin immunoprecipitation
- GW3965
- 3-[3-[[[2-chloro-3-(trifluoromethyl)phenyl]methyl](2,2-diphenylethyl)amino]propoxy]benzeneacetic acid hydrochloride
- GST
- glutathione transferase
- DMSO
- dimethyl sulfoxide
- β-gal
- β-galactoside
- tk
- thymidine kinase
- DKO
- double knockout
- DR-4
- direct repeat spaced by four nucleotides
- PPAR
- peroxisome proliferator-activated receptor
- RXR
- retinoid X receptor
- HA
- hemagglutinin
- HDL
- high-density lipoprotein
- AF
- activator function
- BRL49653
- rosiglitazone.

- Received March 10, 2010.
- Accepted June 30, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}