


Abstract
Despite the discovery of a diverse range of novel agonists and allosteric modulators of the M4 muscarinic acetylcholine (ACh) receptor (mAChR), little is known about how such ligands activate the receptor. We used site-directed mutagenesis of conserved residues in transmembrane 3 (TMIII), a key region involved in G protein-coupled receptor activation, to probe the binding and function of prototypical orthosteric mAChR agonists, allosteric modulators, and “atypical” agonists. We found that most mutations did not affect the binding of the allosteric modulators, with the exception of W1083.28A and L1093.29A (which may contribute directly to the interface between allosteric and orthosteric sites) and mutation D1123.32N (which may cause a global disruption of a hydrogen bond network). Although numerous mutations affected signaling, we did not identify amino acids that were important for the functional activity of any one class of agonist (orthosteric, allosteric, or atypical) to the exclusion of any others, suggesting that TMIII is key for the transmission of stimulus irrespective of the agonist. We also identified two key residues, Trp1083.28 and Asp1123.32, that are essential for the transmission of binding cooperativity between 3-amino-5-chloro-6-methoxy-4-methyl-thieno[2,3-b]pyridine- 2-carboxylic acid cyclopropylamide (LY2033298) and ACh. Finally, we found that LY2033298 was able to rescue functionally impaired signaling of ACh at the majority of mutants tested in a manner that was inversely correlated with the ACh signaling efficacy, indicating that a key part of the mechanism of the positive cooperativity mediated by LY2033298 on the endogenous agonist involves a global drive of the receptor toward an active conformation.
Introduction
A vast array of ligands, ranging from photons and ions to large glycoproteins, interact with cell-surface GPCRs; these mediate a plethora of physiological functions and comprise one of the largest families of drug targets in the genome (Hopkins and Groom, 2002). Given such importance, an understanding of the structural basis underlying ligand binding and activation of GPCRs is vital. Although a handful of high-resolution GPCR crystal structures have been solved in recent years, the process remains challenging and the current structures are mostly of inactive state receptors (Lodowski et al., 2009). As a consequence, substantial insights into molecular events governing ligand action at GPCRs continue to be gained through structure-function analyses based on alternative approaches, such as mutagenesis.
The mAChRs are prototypical members of the biogenic amine family A GPCRs, and have long-served as a model system for understanding the structural basis of small-molecule action at these receptors (Hulme et al., 2003). In recent years, there has been a particular resurgence of interest in mechanisms of mAChR activation due to the discovery of novel selective agonists and allosteric modulators of these receptors, especially for the M1 and M4 mAChR subtypes (Conn et al., 2009), which represent very attractive targets for the treatment of cognitive deficits associated with diseases such as schizophrenia (Chan et al., 2008; Jones et al., 2008). In contrast to the M1 mAChR, however, relatively few mutational analyses have been performed on the M4 mAChR, and thus the structural basis of the subtype-selectivity of novel allosteric modulators and selective agonists is not well established.
The conserved orthosteric site that binds the endogenous agonist, ACh, is located in the top third of the transmembrane helical bundle of the mAChRs and is believed to use contacts provided by inward-facing residues in TM domains III to VII (Hulme et al., 2003). In particular, TMIII contains a number of residues that have been implicated in both the binding and activating mechanisms of the mAChRs by participating in a “global” activation switch that involves a separation of the cytoplasmic region of TMVI away from TMIII and TMVII (Altenbach et al., 1996; Farrens et al., 1996; Gether et al., 1997; Jensen et al., 2001; Hubbell et al., 2003), and movement of the extracellular region of TMVI toward TMIII and TMVII (Elling et al., 1999; Schwartz et al., 2006). Although we recently identified residues in the extracellular portions of the M4 mAChR that also contribute to ligand-specific activation of the receptor (Nawaratne et al., 2010), the contribution of key TMIII residues in this receptor to the activation mediated by orthosteric and allosteric mAChR ligands remains undetermined.
The current study therefore aimed to provide insight into the role of TMIII on the binding and activation mechanisms used by different classes of M4 mAChR ligands (Supplementary Fig. 1). These include the prototypical orthosteric agonists, ACh and pilocarpine, the negative allosteric modulator, heptane-1,7-bis-(dimethyl-3′-phthalimidopropyl) ammonium bromide (C7/3-phth), the novel allosteric agonist and modulator, 3-amino-5-chloro-6-methoxy-4-methyl-thieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide (LY2033298), and the functionally selective agonists, 4-I-[3-chlorophenyl]carbamoyloxy)-2-butynyltrimethylammnonium chloride (McN-A-343) and N-desmethylclozapine (NDMC). Although the basis of the functional selectivity of the latter two compounds remains undetermined, it has been attributed in the past to them interacting with an allosteric site (Sur et al., 2003; May et al., 2007a); in the case of McN-A-343, we have shown that its interaction with the M2 mAChR, at least, is via a “bitopic” mode that involves concomitant binding to both the orthosteric and an allosteric site (Valant et al., 2008). It is currently unknown whether such a bitopic mechanism is operative at the M4 mAChR, and thus both McN-A-343 and NDMC are referred to herein as “atypical” agonists. The availability of this diverse repertoire of small-molecule orthosteric, allosteric, and atypical ligands affords, for the first time, an unprecedented opportunity to delineate the role of TMIII in the processes of M4 mAChR binding, activation, and transmission of cooperativity between two topographically distinct binding domains.
Materials and Methods
Materials.
Chinese hamster ovary (CHO) FlpIn cells were from Invitrogen (Carlsbad, CA). Hygromycin B was purchased from Roche Diagnostics (Indianapolis, IN). Dulbecco's modified Eagle's medium and fetal bovine serum (FBS) were from Invitrogen (Carlsbad, CA) and JRH Biosciences (Lenexa, KS), respectively. Primers used for the generation of mutant receptors were purchased from GeneWorks (Hindmarsh, SA, Australia). The AlphaScreen SureFire phospho-ERK1/2 reagents were kindly donated by Drs. Michael Crouch and Ron Osmond (TGR Biosciences, Thebarton, SA, Australia). AlphaScreen streptavidin donor beads and anti-IgG (protein A) acceptor beads, [3H]quinuclidinyl benzilate ([3H]QNB; specific activity, 52 Ci/mmol), and [3H]N-methylscopolamine ([3H]NMS; specific activity, 72 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). LY2033298 was synthesized in-house at Eli Lilly (Indianapolis, IN), whereas C7/3-phth was synthesized in-house by Dr. Celine Valant at the Monash Institute of Pharmaceutical Sciences. All other chemicals were from Sigma-Aldrich (St. Louis, MO).
Receptor Mutagenesis and Generation of Cell Lines.
DNA encoding the human M4 mAChR with a triple HA tag at its amino terminus was purchased from Missouri University of Science and Technology (http://www.cdna.org) and cloned into the Gateway destination vector, pEF5/frt/v5/dest, as described previously (Nawaratne et al., 2008). This construct was used to generate M4 mAChR sequences with the desired amino acid substitutions using QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA) with primers shown in Supplementary Table 1. DNA constructs were transfected into FlpIn CHO cells (Invitrogen) for stable expression according to manufacturer's instructions. Cells were maintained in high-glucose Dulbecco's modified Eagle's medium containing 10% FBS, 16 mM HEPES, and 400 μg/ml hygromycin B.
Radioligand Binding Assays.
Cell membranes were prepared as described previously (Nawaratne et al., 2008; Leach et al., 2010). For equilibrium binding, cell membranes (50 μg) were incubated in a final volume of 1 ml of binding buffer [20 mM HEPES, 100 mM NaCl, 10 mM MgCl2, and 100 μM guanosine-5′-(βγ-imino)triphosphate, pH 7.4] for 2 h at 37°C with [3H]QNB as described previously (Nawaratne et al., 2008; Leach et al., 2010); [3H]QNB was chosen for the equilibrium binding experiments because it retained appreciable affinity for most of the mutations studied, in contrast to an alternative (and commonly used) prototypical orthosteric antagonist, [3H]NMS. Radioligand dissociation was determined by equilibrating cell membranes (50 μg/1 ml; 1 h, 37°C) with 0.2 nM (wild-type M4 mAChR) or 1 nM (W1083.28A1 and L1093.29A) [3H]NMS in binding buffer before the addition of 10 μl of atropine (10 μM) in the absence or presence of modulator using a reverse-time protocol. Receptor-bound radioligand was separated from free radioligand using rapid filtration with a Brandel harvester and radioactivity determined by liquid scintillation counting.
ERK1/2 Phosphorylation Assays.
Cells were seeded at 40,000 cells/well into a transparent 96-well plate and grown overnight. Initial time course experiments were used to determine the time required to stimulate maximum ERK1/2 phosphorylation by each agonist, and subsequent concentration-response experiments were conducted by stimulating cells with agonist for 4 min. For interaction studies, in which ACh-stimulated ERK1/2 phosphorylation was measured in the presence of LY2033298, the orthosteric agonist was added to cells immediately after the addition of LY2033298. Phosphorylated ERK1/2 was detected using an AlphaScreen assay as described previously (Nawaratne et al., 2008).
Enzyme-Linked Immunosorbent Assay.
Cells were seeded at 75,000 cells/well into a transparent 48-well plate and grown overnight. Cells were washed three times with phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.5 mM KH2PO4) and fixed with 4% paraformaldehyde for 15 min at 4°C before two more washes with PBS. Cells were blocked with 5% bovine serum albumin for 45 min and incubated with rabbit anti-HA antibody (Abcam Inc., Cambridge, MA) for 1 h at 37°C. Cells were washed three times with PBS and blocked for a further 15 min with 5% bovine serum albumin. Cells were subsequently incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling Technology, Danvers, MA) in PBS for 1 h at 37°C before three washes with PBS. The signal was developed using Sigma OPD tablets, and the reaction was stopped by the addition of 3 M HCl. Absorbance was read at 492 nM using an Envision plate reader (PerkinElmer Life and Analytical Sciences).
Data Analysis.
Radioligand dissociation kinetic experiments were fitted to a monoexponential decay equation (Motulsky and Christopoulos, 2004), and rate constants determined in the presence of LY2033298 were normalized to those determined in its absence. Competition binding curves between [3H]QNB and unlabeled orthosteric ligands were fitted to a one-site binding model (Motulsky and Christopoulos, 2004). Inhibition experiments that used the allosteric modulators, LY2033298 or C7/3-phth, were fitted to the following allosteric ternary complex model (Ehlert, 1988):
 where Y is the specific radioligand binding, Bmax is the total number of receptors, [A], [B], and [I] are the concentrations of radioligand, allosteric modulator, and unlabeled orthosteric ligand, respectively, KA, KB, and KI are the equilibrium dissociation constants of the radioligand, allosteric modulator, and unlabeled orthosteric ligand, respectively, and α′ and α are the cooperativity factors between allosteric modulator and the radioligand or unlabeled orthosteric ligand, respectively. For binding experiments performed with C7/3-phth versus [3H]QNB, [I] was set to 0 (i.e., no ACh was present in the experiment and the interaction was between C7/3-phth and [3H]QNB only).
where Y is the specific radioligand binding, Bmax is the total number of receptors, [A], [B], and [I] are the concentrations of radioligand, allosteric modulator, and unlabeled orthosteric ligand, respectively, KA, KB, and KI are the equilibrium dissociation constants of the radioligand, allosteric modulator, and unlabeled orthosteric ligand, respectively, and α′ and α are the cooperativity factors between allosteric modulator and the radioligand or unlabeled orthosteric ligand, respectively. For binding experiments performed with C7/3-phth versus [3H]QNB, [I] was set to 0 (i.e., no ACh was present in the experiment and the interaction was between C7/3-phth and [3H]QNB only).
All ERK1/2 phosphorylation assays were analyzed using an operational model of allosterism and agonism according to eq. 2 (Leach et al., 2007, 2010):
 where Em is the maximum possible tissue response, [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively, KA and KB are the equilibrium dissociation constant of the orthosteric and allosteric ligands, respectively, τA and τB are operational measures of orthosteric and allosteric ligand efficacy, respectively, α is the binding cooperativity parameter between the orthosteric and allosteric ligand, and β denotes the allosteric effect of the modulator on the efficacy of the orthosteric agonist. When no allosteric modulator was present, the concentration of [B] was set to 0, and the model becomes identical with the original operational model of agonism described by Black and Leff (1983). Thus, in all instances, agonism is expressed in operational model terms. For the analysis to converge when an allosteric modulator was present, the binding cooperativity with ACh, α, was fixed to that determined separately in radioligand binding assays. In all instances, the equilibrium dissociation constant of each agonist was also fixed to that determined from the binding assays. Finally, to account for effects of the expression level of each of the different mutant receptors on the observed efficacy of each agonist, the Bmax values determined from saturation binding were used to normalize the logτ values derived from the operational model analysis to what they would be if the mutant were expressed at the same level as the wild-type receptor (Gregory et al., 2010); this corrected efficacy value is denoted as logτC.
where Em is the maximum possible tissue response, [A] and [B] are the concentrations of orthosteric and allosteric ligands, respectively, KA and KB are the equilibrium dissociation constant of the orthosteric and allosteric ligands, respectively, τA and τB are operational measures of orthosteric and allosteric ligand efficacy, respectively, α is the binding cooperativity parameter between the orthosteric and allosteric ligand, and β denotes the allosteric effect of the modulator on the efficacy of the orthosteric agonist. When no allosteric modulator was present, the concentration of [B] was set to 0, and the model becomes identical with the original operational model of agonism described by Black and Leff (1983). Thus, in all instances, agonism is expressed in operational model terms. For the analysis to converge when an allosteric modulator was present, the binding cooperativity with ACh, α, was fixed to that determined separately in radioligand binding assays. In all instances, the equilibrium dissociation constant of each agonist was also fixed to that determined from the binding assays. Finally, to account for effects of the expression level of each of the different mutant receptors on the observed efficacy of each agonist, the Bmax values determined from saturation binding were used to normalize the logτ values derived from the operational model analysis to what they would be if the mutant were expressed at the same level as the wild-type receptor (Gregory et al., 2010); this corrected efficacy value is denoted as logτC.
All affinity, potency, and cooperativity values were estimated as logarithms (Christopoulos, 1998), and statistical comparisons between values for agonists at each different mutant receptor were by one-way analysis of variance using a Dunnett's multiple comparison post test to determine significant differences between mutant receptors and the wild-type M4 mAChR. A value of P < 0.05 was considered statistically significant.
Results
Rationale for the Choice of Amino Acid Substitutions.
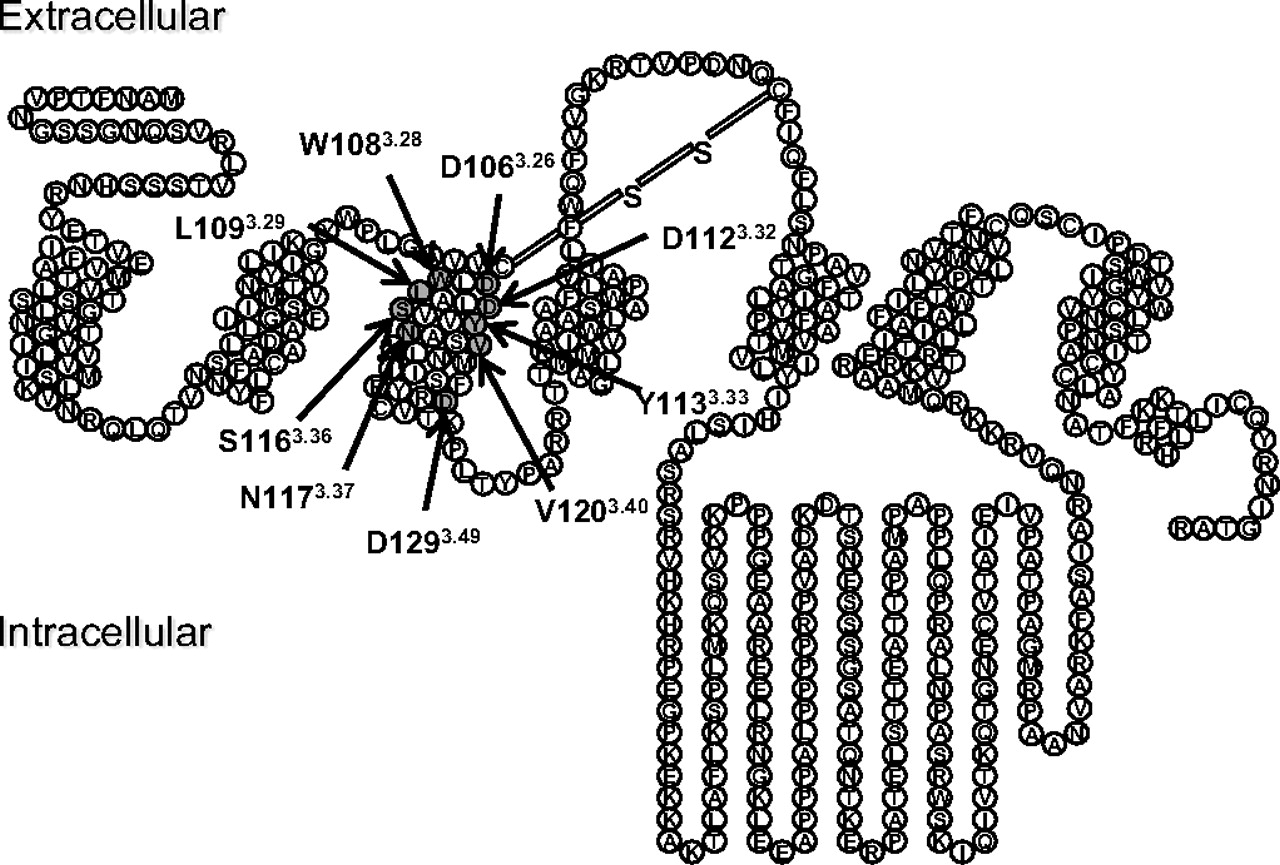
A snake diagram of the M4 mAChR is shown in Fig. 1, highlighting the amino acid residues that were mutated in the current study. We have focused on residues conserved across all five mAChR subtypes located within TMIII because this region is vitally important for both the binding of orthosteric ligands and the activation of the receptor (Hulme et al., 2001, 2003). Very little is known, however, about the role of these residues in the actions of allosteric ligands.

Snake diagram of the M4 mAChR. The amino acid residues mutated in this study are highlighted in dark gray.
Effects of TMIII Substitutions on the Affinity of Orthosteric and Atypical Ligands.
In all assays, the triple HA-tagged wild-type human M4 mAChR (referred to herein as WT) was compared with the untagged receptor to ensure that the pharmacology of the receptor was not altered by the tag. Although the presence of the HA tag caused a significant reduction in the number of [3H]QNB binding sites detected using saturation binding assays (untagged, 1.35 ± 0.18 pmol/mg protein; HA-tagged, 0.16 ± 0.03pmol/mg protein; n = 3–6), no significant differences were observed between the binding affinity of any of the ligands tested at the tagged and untagged receptor (data not shown). Thus, all subsequent receptor constructs were prepared with the triple HA tag.
As determined by [3H]QNB saturation binding analysis, the exchange of Asp1063.26 for alanine or Asp1293.49 for asparagine led to a significant reduction in receptor expression compared with the WT, whereas substitution of Val1203.40 for alanine resulted in a significant increase in receptor expression levels (Table 1). No [3H]QNB binding was detected after the exchange of Asp1123.32 or Tyr1133.33 for alanine, although this probably reflects a dramatic inhibitory effect on orthosteric ligand affinity rather than receptor expression because a separate enzyme-linked immunosorbent assay indicated that these latter two constructs were indeed expressed in our FlpIn CHO cells (Supplementary Fig. 2). No other amino acid substitutions had any significant effects on the expression level of the M4 mAChR (Table 1).
Equilibrium binding parameter estimates for ligands at M4 mAChR constructs
Values represent the mean ± S.E.M. from at least three separate experiments performed in triplicate. Bmax is the maximum density of binding sites. pKA is the negative logarithm of the radioligand equilibrium dissociation constant. pKB is the negative logarithm of the unlabeled ligand equilibrium dissociation constant.
In addition to substitution of Asp1123.32 or Tyr1133.33 with alanine, substantial effects were noted on ligand affinity for other receptor mutants. In particular, substituting Asp1123.32 with asparagine to eliminate the potential ionic interaction with the common ammonium cation of prototypical orthosteric mAChR ligands resulted in a large reduction in the binding affinity of both orthosteric and atypical ligands (Table 1 and Fig. 3). Elongation of the amino acid side chain while retaining the charge of Asp1123.32, by substitution with glutamic acid, had a more selective effect, causing a large reduction in ACh affinity, a modest reduction in pilocarpine affinity, but no effect on the binding of [3H]QNB, McN-A-343, or NDMC (Table 1 and Figs. 2 and 3). A considerable effect on ACh affinity was also observed after substitution of Leu1093.29 for alanine, and smaller effects were observed for other ligands as well (Table 1 and Figs. 2 and 3). Substitution of Ser1163.36 or Asn1173.37 for alanine had significant inhibitory effects on the binding affinity of ACh but minimal to no effect on the affinity of any of the other ligands. In general, these findings are consistent with the important role these residues play in defining part of the orthosteric pocket for the endogenous agonist.

Mutations in TMIII differentially alter the equilibrium dissociation constant of orthosteric M4 ligands. Bars represent the change in pKB of each ligand at the mutant receptor relative to the wild-type receptor, determined from equilibrium radioligand binding assays. Data are mean ± S.E.M. from at least three experiments performed in triplicate. N/D, no detectable binding; *, significant difference to wild type, p < 0.05, one-way analysis of variance, Dunnett's post test.

Mutations in TMIII have differential effects on the equilibrium dissociation constant of allosteric and atypical M4 ligands. Bars represent the change in pKB of each ligand at the mutant receptor relative to the wild-type receptor, determined either from equilibrium radioligand binding assays or from nonequilibrium radioligand dissociation assays. Data are mean ± S.E.M. from at least three experiments performed in triplicate. N/D, no detectable binding; *, significant difference to wild-type, p < 0.05, one-way analysis of variance, Dunnett's post test.
In agreement with recent findings at the M2 mAChR (Gregory et al., 2010), the W1083.28A mutant M4 mAChR had opposing effects on ACh affinity (reduction) relative to that of the atypical ligand, NDMC (increase), suggesting that this residue is a key discriminator between the binding poses adopted by these ligands. A different profile was noted upon substitution of Val1203.40 for alanine, which caused a modest decrease in the affinity of [3H]QNB and NDMC but a modest increase in the affinity of ACh and pilocarpine. No significant differences were noted in ligand affinities at the D1293.49E mutant, but removal of the charge (D1293.49N) caused a modest increase in ACh affinity.
Effects of TMIII Substitutions on the Affinity of Allosteric Ligands.
The interaction between the allosteric modulator C7/3-phth and the orthosteric antagonist [3H]QNB is characterized by substantial negative cooperativity, and thus, effects of amino acid substitutions on the binding of the modulator could be determined by application of a simple allosteric ternary complex model to the data. In contrast, the interaction between the allosteric ligand LY2033298 and [3H]QNB is neutrally cooperative (i.e., α′ = 1; eq. 1) (Nawaratne et al., 2008; Leach et al., 2010), and thus, the effects of receptor mutations on the binding of the latter modulator were determined via an analysis of the interaction between ACh and [3H]QNB in the presence of LY2033298 (Nawaratne et al., 2010).
Perhaps not unexpectedly, the majority of the mutations in TMIII did not have any significant effects on the affinity of the two allosteric modulators (Table 2 and Fig. 3), with a few notable exceptions. Specifically, allosteric ligand affinity was reduced at both the W1083.28A and L1093.29A mutant M4 mAChRs (Table 2 and Figs. 3 and 4), suggesting that these two residues represent an interface between the prototypical orthosteric binding site and a more extracellular allosteric site; a previous study of the M1 mAChR had identified a similar inhibitory effect of the W3.28A on the binding of the allosteric modulator gallamine (Matsui et al., 1995). Because of the indirect nature of the experiments used to determine mutational effects on LY2033298, we also used a second experimental paradigm for determining the potency of this modulator at the two mutants. Specifically, we took advantage of the ability of LY2033298 to allosterically retard the dissociation rate of the orthosteric antagonist [3H]NMS (Leach et al., 2010), which enabled us to measure the LY2033298 concentration range over which this occurred at different receptor constructs (Fig. 5A). Because the cooperativity between LY2033298 and [3H]NMS is also close to neutral at the M4 mAChR (Leach et al., 2010) (Fig. 5B), the potency (IC50) determined in dissociation kinetic assays will be approximately equal to the KB value of LY2033298 at the allosteric site (Lazareno and Birdsall, 1995); [3H]NMS was used instead of [3H]QNB for these experiments because the former antagonist has a much faster rate of dissociation than the latter, thus allowing the determination of dissociation kinetics over a reasonable experimental time frame. Accordingly, there was a clear reduction in the potency of LY2033298 to slow [3H]NMS dissociation at both the W1083.28A (pIC50 4.24 ± 0.07, n = 3) and L1093.29A (pIC50 4.28 ± 0.14, n = 3) mutant M4 mAChRs compared with the WT (pIC50 4.70 ± 0.07, n = 3) (Fig. 5), confirming the results of the equilibrium binding data and indicating that the affinity of LY2033298 for the allosteric site was indeed reduced.
Allosteric modulator equilibrium binding parameters
Values represent the mean ± S.E.M. from at least three separate experiments performed in triplicate. pKB is the negative logarithm of the allosteric modulator equilibrium dissociation constant. Logα′ (α′) is the logarithm of the cooperativity factor for the interaction between C7/3-phth and [3H]QNB; antilogarithms are shown in parentheses. Logα (α) is the logarithm of the cooperativity factor for the interaction between LY2033298 and ACh; antilogarithms are shown in parentheses.

Radioligand binding interaction studies reveal mutations that alter the cooperativity and/or binding affinity of LY2033298. The competition between a KA equivalent concentration of [3H]QNB and a fixed concentration of ACh, which inhibited approximately 20% [3H]QNB binding (100 μM at Trp1083.28, 300 μM at L1093.29A, 10 mM at D1123.32N, and 1 mM at D1123.32E) was determined in the presence of increasing concentrations of LY2033298 at the indicated mAChR constructs. The curves drawn through the points represent the best global fit of an allosteric ternary complex model (eq. 1) to each pair of datasets (ACh competition of [3H]QNB binding and the IC20 concentration of ACh in the presence of LY2033298), with the cooperativity between LY2033298 and [3H]QNB (α′) fixed to a value of 1. Data points represent the S.E.M. of at least three experiments performed in triplicate.

[3H]NMS dissociation kinetic studies confirm that the W1083.28A and L1093.29A mutations reduce the binding affinity of LY2033298. Concentration-effect relationships for LY2033298 on the dissociation rate (A) or equilibrium binding (B) of [3H]NMS at the wild-type, W1083.28A, or L1093.29A mutants. Data represent mean ± S.E.M. from three experiments performed in duplicate.
A second surprising finding was that removal of the charge on Asp1123.32 through substitution with asparagine had a profound inhibitory effect on the affinity of C7/3phth whereas slightly enhancing the affinity of LY2033298 (Table 2, Fig. 3, and Supplementary Fig. 3). Given the important role of this residue as a contact point for ACh and other orthosteric ligands, it is likely that the differential effect of mutation of Asp1123.32 on allosteric ligand affinity represents an indirect conformational effect.
Residues in TMIII of the M4 mAChR Are Involved in Receptor Activation by All Classes of Ligand.
We used ERK1/2 phosphorylation as a measure of agonist-stimulated receptor activity because this is a convergent pathway downstream of both G protein-dependent and -independent mechanisms. With the exception of C7/3-phth, all ligands displayed some agonistic activity at this pathway. We identified a number of residues that contributed to the signaling of orthosteric, allosteric and atypical agonists, as quantified by the operational model logτC values (Fig. 6 and Table 3). Mutations D1063.26A, L1093.29A, D1123.32E, D1123.32N, S1163.36A, and N1173.37A were detrimental to the efficacy of all agonists tested. We did not identify any residues that contributed solely to the efficacy of one particular class of agonist (e.g., orthosteric, allosteric, or atypical), but substitution of Val1203.40 with alanine caused a significant decrease in the efficacy of ACh, LY2033298, and McN-A-343, whereas the efficacy of pilocarpine and NDMC was enhanced by this mutation. Likewise, exchange of Asp1293.49 for asparagine had no significant effect on the efficacy of ACh or LY2033298, yet the efficacy of McN-A-343, pilocarpine, and NDMC was increased.

Agonist efficacy is differentially modified by TMIII mutations. Bars represent the difference in logτC of each agonist, derived from an operational model of agonism (eq. 2), relative to the wild-type receptor value for that agonist. Data represent the mean ± S.E.M. of at least three experiments performed in triplicate. No response, no detectable response; *, significant difference from the wild-type receptor, p < 0.05, one-way analysis of variance, Dunnett's post test.
Coupling efficiency (logτC) parameters of ligands at M4 mAChR constructs
Values represent the mean ± S.E.M. from at least three separate experiments performed in triplicate. LogτC (τC) is the logarithm of the corrected operational efficacy parameter, τC, determined via nonlinear regression of the concentration-response data to an operational model of agonism; antilogarithms are shown in parentheses.
Residues in TMIII Contribute to the Transmission of Cooperativity between the Orthosteric and Allosteric Sites.
The ability of an allosteric modulator to alter the function of an orthosteric ligand can be manifested as either changes in the binding affinity and/or changes in the signaling efficacy of the orthosteric ligand (May et al., 2007b). Thus, we determined the extent to which different amino acid residues in TMIII were involved in the transmission of binding cooperativity (α′) between C7/3-phth and [3H]QNB and both binding cooperativity (α) and efficacy modulation (β) between LY2033298 and ACh; allosteric effects of C7/3-phth on ACh could not be investigated because of the very high negative cooperativity between this modulator and the agonist (data not shown).
Two mutations, namely W1083.28A and L1093.29A, had substantial effects on the binding cooperativity between each of the modulators and their respective orthosteric interactants (Table 2 and Fig. 4). With regard to W1083.28A, it was of note that both the negative cooperativity between C7/3-phth and [3H]QNB and the positive cooperativity between LY2033298 and ACh were blunted in each instance (Table 2), suggesting that this residue is not only important for modulator binding affinity but also for the optimal transmission of cooperative effects. In contrast, L1093.29A had opposing effects on cooperativity (i.e., increasing the positive cooperativity between LY2033298 and ACh whereas reducing the negative cooperativity between C7/3-phth and [3H]QNB). D1063.26A also had a selective (blunting) effect on the negative cooperativity between C7/3-phth and [3H]QNB. The greatest measurable effect on the binding cooperativity between LY2033298 and ACh was observed at the D1123.32E and D1123.32N mutants, in which a large reduction in the ability of LY2033298 to potentiate ACh binding affinity was observed (Fig. 4 and Table 2). Although the cooperativity between C7/3-phth and [3H]QNB at the D1123.32N-mutant could not be determined accurately because of the pronounced reduction of modulator affinity at this construct (Supplementary Fig. 3), the data were consistent with the retention of substantial negative cooperativity at the mutant because a significant reduction in the binding of [3H]QNB was noted in the presence of high C7/3-phth concentrations (Supplementary Fig. 3), suggesting that the principal effect of the mutation is on modulator affinity rather than cooperativity.
To determine the effects of amino acid substitutions on the ability of LY2033298 to modulate the signaling efficacy of ACh, we performed functional interaction studies using ERK1/2 phosphorylation as a measure of agonist-mediated receptor activation and fitted the data to an operational model of allosterism and agonism (eq. 2) (Leach et al., 2007, 2010). An internal check of the robustness of the analysis was the excellent correlation between logτC estimates obtained for ACh or LY2033298 in experiments when the ligands were tested on their own as agonists (Table 3) compared with when they were coadministered together in the interaction studies (Table 4 and Fig. 7); for the ACh comparison, r2 = 0.89, and for the LY2033298 comparison, r2 = 0.95.
Operational allosteric ternary complex model parameters for the functional interaction between ACh and LY2033298
Parameter values are the mean ± S.E.M. from at least three experiments performed in triplicate and analyzed according to eq. 2. Logβ (β) is the logarithm of the allosteric effect on orthosteric agonist efficacy, β. LogτC (τC) is the logarithm of the corrected operational efficacy parameter, τC.

LY2033298 rescues ACh efficacy at inactivating mutations. Peak levels of pERK1/2 were assessed as described under Materials and Methods and normalized to the response elicited by 10% FBS. The curves drawn through the points for data at the WT, Trp1083.28, Leu1093.29, and D1123.32E constructs represent the best global fit of an operational model of allosterism (eq. 2) to each family of datasets, with the affinity of each agonist fixed to the pKB value determined in separate binding assays (Table 1). Curves drawn through the points for data at the D1123.32N construct represent the fit of each curve to a simple Hill equation. Data points represent the mean ± S.E.M. of at least three experiments performed in triplicate.
With the exception of the D1123.32A, D1123.32N, and Y1133.33A mutations, in which little or no response was observed to ACh in both the absence or presence of LY2033298, the key finding from the functional interaction experiments was that the allosteric modulator was able to rescue ACh function to varying extents at all other mutant receptors, with significant increases in the strength of the allosteric modulation of ACh efficacy (β) noted at the L1093.29A, D1123.32E, and S1163.36A mutant receptors (Table 4 and Fig. 7). Indeed, there was a significant inverse correlation (r2 = 0.71) between the observed signaling efficacy of ACh and the ability of LY2033298 to potentiate ACh-mediated signaling, in which lower ACh logτC values (more impaired orthosteric agonist signaling) were associated with higher logβ values for the interaction (Fig. 8). This finding indicates that a key part of the mechanism of the positive cooperativity mediated by LY2033298 on the endogenous agonist involves a global drive of the receptor toward an active conformation, irrespective of the nature of mutational impairment in TMIII residues.

The ability of LY2033298 to potentiate ACh activity correlates well with the efficacy of ACh at each mutant. ACh logτC and logβ values between ACh and LY2033298 were determined from fitting pERK1/2 interaction data at the WT and mutant receptors to an operational model of allosterism (eq. 2), as described under Materials and Methods. The line drawn through the points represents the best fit of a linear regression analysis to the data, where r2 represents the correlation between logτC and logβ.
Discussion
The mAChRs are an important model system for understanding structure-function relationships at family A GPCRs because of the growing number of ligands with novel modes of action being identified for these receptors. In addition to Asp1293.49, located near the cytosolic end of TMIII as part of the highly conserved (E)/DRY activation motif, the current study focused predominantly on amino acid residues that contribute to the top (extracellular-facing) half of TMIII of the M4 mAChR, which remains less characterized than other mAChR subtypes despite emerging as an exciting drug target for cognitive disorders such as schizophrenia (Chan et al., 2008). The fact that mutations in this region of the receptor had substantial effects on orthosteric, allosteric, and atypical mAChR ligands highlights the pivotal role that TMIII plays in mechanisms of activation and transmission of allosteric effects at the M4 mAChR. Given the high degree of conservation of these residues across the mAChR and biogenic GPCR families, it is likely that some of the mechanisms inferred from our study will be operative at other GPCRs.
As expected, the majority of TMIII mutations did not substantially affect the binding affinity of the allosteric modulators, C7/3-phth and LY2033298, with the exception of W1083.28A, L1093.29A, and D1123.32N. Trp1083.28 and Leu1093.29 are predicted to lie near the junction of TMIII and the extracellular entrance to the orthosteric binding pocket, and it is thus reasonable to hypothesize that these residues may contribute to lining the “bottom” of a more extracellularly located allosteric binding domain. This hypothesis is also supported by our recent finding that C7/3-phth and LY2033298 interact in an apparently competitive manner at the M4 mAChR (Leach et al., 2010), suggesting that they share overlapping binding regions on the receptor. The effect of removing the charge on Asp1123.32 was unexpected because of the fact that this residue is the key counter ion that interacts with the ammonium headgroup of ACh (Curtis et al., 1989; Kurtenbach et al., 1990; Spalding et al., 1994). However, Asp1123.32 is believed to participate in a hydrogen bond network with key residues near the top TMVII, a region that has been implicated previously in contributing to the binding pocket of prototypical modulators such as C7/3-phth (Matsui et al., 1995; Voigtländer et al., 2003; Huang et al., 2005; Prilla et al., 2006; May et al., 2007a), and disruption of this network may account for the deleterious effects on C7/3-phth binding.
In contrast, the prototypical orthosteric ligands and atypical agonists used in this study were generally characterized by reduced binding affinity at a variety of the TMIII mutants with a few notable exceptions. The first was McN-A-343, which exhibited no significant difference in binding affinity at any mutant except the key D1123.32N-M4 mAChR construct. This finding is consistent with prior studies of the M2 mAChR, in which McN-A-343 was unaffected by key orthosteric site mutations, in agreement with our hypothesis that this compound adopts a bitopic binding mode that extends up from Asp3.32 toward the extracellular loop regions (Valant et al., 2008; Gregory et al., 2010). The second exception was the modest increases in affinity noted for NDMC at the W1083.28A mutant, for ACh and pilocarpine at the V1203.40A mutant, and for ACh at the D1293.49N mutant, respectively. The first of these observations is consistent with what has been noted for NDMC at the equivalent mutation introduced into the M2 mAChR (Gregory et al., 2010) but is the opposite to the small decrease in its binding affinity at the equivalent mutation of the M1 mAChR (Lebon et al., 2009), suggesting that NDMC adopts a slightly different binding pose at the even-numbered mAChRs compared with the M1 mAChR.
It was also interesting to note that substitution of Ser1163.36 and Asn1173.37 to alanine caused a large reduction in the binding affinity of ACh but not in the affinity of any of the other ligands tested, indicating that ACh adopts a unique binding pose in comparison to the other agonists. It is unclear whether these residues are direct contact points for ACh, because although Ser1163.36 is predicted to face directly into the core of the orthosteric binding pocket (Han et al., 2005), Asn1173.37 is located below the predicted orthosteric binding site. Therefore, Asn1173.37 may serve to stabilize an important receptor conformation that is essential for ACh binding but not for any of the other ligands tested.
With regard to mutational effects on ligand signaling efficacy, it was of particular note that we did not identify any amino acids that were important for the functional activity of one particular class of agonist (orthosteric, allosteric, or atypical) to the exclusion of any others, suggesting that the location of the agonist binding site does not necessarily govern a unique activation mechanism after agonist binding and that there are some global receptor conformations that are favored by all agonists. Furthermore, with the exception of ligand-specific effects of the V1203.40A and D1293.49N mutations, the rest of the mutations introduced into TMIII were generally deleterious to receptor activation. The substitution of Val1203.40 with alanine caused a significant increase in the signaling efficacy of pilocarpine, suggesting that even prototypical orthosteric ligands can sense different conformations compared with the endogenous ligand for the receptor. In contrast, the removal of the charge on Asp1293.49 caused an increase in the efficacy of all agonists tested, with the interesting exceptions of the endogenous orthosteric ligand ACh and the allosteric agonist LY2033298. The general view of mechanisms involving Asp3.49 in family A GPCR activation is that it forms an important hydrogen bond interaction with Arg3.50, which itself interacts with Glu6.30 in the formation of a key “ionic lock”; disruption of this lock is proposed to be part of the activation mechanism for many GPCRs (Schwartz et al., 2006). This could explain why compounds such as pilocarpine, McN-A-343, and NDMC displayed enhanced signaling at this mutant receptor, assuming that the mutation placed the receptor in a partially active state with respect to ERK1/2 phosphorylation. However, prior studies at the M1 and M5 mAChRs have found minimal effects of the D3.49N mutation on the efficacy of ACh itself (Lu et al., 1997; Burstein et al., 1998), which is also in agreement with our current study at the M4 mAChR, suggesting that this mechanism need not be universal. It is noteworthy that the signaling efficacy of the allosteric agonist LY2033298 was also completely insensitive to the D3.49N mutation. Overall, these findings are concordant with recent experiments on the β2 adrenergic receptor, which showed that diverse agonists disrupt a different combination of molecular interactions responsible for stabilizing that receptor's inactive state (Yao et al., 2006).
Given that a key aim of our study was to also quantify the effects of TMIII mutations on the cooperativity between cobound orthosteric and allosteric ligands on the M4 mAChR, we reveal for the first time two key residues required for the transmission of binding cooperativity between LY2033298 and ACh. Trp1083.28 and Asp1123.32 were essential for the ability of LY2033298 to potentiate the binding affinity of ACh. Intriguingly, however, even though the binding cooperativity was almost abolished at the D1123.32E mutant, the allosteric effect on signaling between ACh and LY203329 was retained. In fact, at the L1093.29A, D1123.32E, and S1163.36A mutants, an increase in efficacy modulation between LY2033298 and ACh was observed. This may be expected where ACh-mediated receptor signaling events are significantly impaired by an amino acid substitution if LY2033298 can restore them to functional receptors. It also highlights that an important yet often underappreciated mechanism of allosteric modulation is a “resetting” of energy barriers governing global transitions between receptor states by an allosteric modulator that subsequently facilitates (or hinders) the interaction with an orthosteric ligand.
In conclusion, the present study has identified conserved amino acid residues in the key TMIII domain that play a role in the binding, signaling and transmission of cooperativity in a diverse range of orthosteric, allosteric and atypical mAChR ligands. We have shown that although common activation switches are used by all classes of M4 mAChR agonist, some subtle differences exist between the intramolecular interactions that are altered after the binding of different agonists.
Authorship Contributions
Participated in research design: Leach, Davey, Sexton, and Christopoulos.
Conducted experiments: Leach and Davey.
Contributed new reagents or analytic tools: Felder.
Performed data analysis: Leach, Davey, and Christopoulos.
Wrote or contributed to the writing of the manuscript: Leach, Davey, Felder, Sexton, and Christopoulos.
Other: Christopoulos and Sexton acquired funding for the research.
Acknowledgments
We thank Dr. Celine Valant for the synthesis of C7/3-phth and Drs. Michael Crouch and Ron Osmond (TGR Biosciences) for the generous donation of AlphaScreen SureFire phospho-ERK1/2 reagents.
Footnotes
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.This work was supported by the National Health and Medical Research Council (NHMRC) of Australia [Grant 519461]. A.C. is a Senior and P.S. a Principal Research Fellow of the NHMRC.
A.C. is a consultant for Johnson and Johnson and Alchemia. C.C.F. is an employee of Eli Lilly and Co.
↵1 Ballesteros and Weinstein numbers are provided (in superscript) to indicate relative position of residues within the transmembrane domain.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.111.070938.
-
ABBREVIATIONS:
- GPCR
- G protein-coupled receptor
- ACh
- acetylcholine
- C7/3-phth
- heptane-1,7-bis-(dimethyl-3′-phthalimidopropyl) ammonium bromide
- CHO
- Chinese hamster ovary
- FBS
- fetal bovine serum
- LY2033298
- 3-amino-5-chloro-6-methoxy-4-methyl-thieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide
- mAChR
- muscarinic acetylcholine receptor
- McN-A-343
- 4-I-[3-chlorophenyl]carbamoyloxy)-2-butynyltrimethylammnonium chloride
- NDMC
- N-desmethylclozapine
- [3H]NMS
- [3H]N-methylscopolamine
- [3H]QNB
- [3H]quinuclidinyl benzilate
- TM
- transmembrane domain
- ERK1/2
- extracellular signal-regulated kinase 1/2
- PBS
- phosphate-buffered saline
- WT
- wild type
- HA
- hemagglutinin.

- Received January 2, 2011.
- Accepted February 7, 2011.
- Copyright © 2011 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}