


Abstract
We have previously identified Ser293 in transmembrane segment 5 as a determinant for selective KCa2.1 channel activation by GW542573X (4-(2-methoxyphenylcarbamoyloxymethyl)-piperidine-1-carboxylic acid tert-butyl ester). Now we show that Ser293 mediates both activation and inhibition of KCa2.1: CM-TPMF (N-{7-[1-(4-chloro-2-methylphenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine) and B-TPMF (N-{7-[1-(4-tert-butyl-phenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine), two newly identified and structurally related [1,2,4]triazolo[1,5-a]pyrimidines, act either as activators or as inhibitors of the human KCa2.1 channel. Whereas (−)-CM-TPMF activates KCa2.1 with an EC50 value of 24 nM, (−)-B-TPMF inhibits the channel with an IC50 value of 31 nM. In contrast, their (+)-enantiomers are 40 to 100 times less active. Both (−)-CM-TPMF and (−)-B-TPMF are subtype-selective, with 10- to 20-fold discrimination toward other KCa2 channels and the KCa3 channel. Coapplication experiments reveal competitive-like functional interactions between the effects of (−)-CM-TPMF and (−)-B-TPMF. Despite belonging to a different chemical class than GW542573X, the KCa2.1 selectivity of (−)-CM-TPMF and (−)-B-TPMF depend critically on Ser293 as revealed by loss- and gain-of-function mutations. We conclude that compounds occupying the TPMF site may either positively or negatively influence the gating process depending on their substitution patterns. It is noteworthy that (−)-CM-TPMF is 10 times more potent on KCa2.1 than NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime), an unselective but hitherto the most potent KCa3/KCa2 channel activator. (−)-B-TPMF is the first small-molecule inhibitor with significant selectivity among the KCa2 channel subtypes. In contrast to peptide blockers such as apamin and scyllatoxin, which preferentially affect KCa2.2, (−)-B-TPMF exhibits KCa2.1 selectivity. These high-affinity compounds, which exert opposite effects on KCa2.1 gating, may help define physiological or pathophysiological roles of this channel.
Introduction
Small and intermediate conductance Ca2+-activated K+ channels (the KCNN family: KCa2.1, KCa2.2, KCa2.3, and KCa3.1) contain six transmembrane segments (S1–S6) arranged as a tetramer around a central pore. Each subunit associates with one calmodulin (CaM) molecule, acting as a β-subunit at the C-terminal CaM binding domain (CaMBD) (Xia et al., 1998; Khanna et al., 1999). Ca2+ binding to CaM rearranges the CaM/CaMBD region and the S6/S5 domains, thereby opening the channel. The nature of the gate remains elusive, but cysteine scanning experiments have confined its possible location to residues in the inner pore vestibule close to the selectivity filter. In contrast to Kv channels, KCNN channels are assumed to exhibit “deep-pore gating” (Bruening-Wright et al., 2002, 2007; Klein et al., 2007; Garneau et al., 2009), which is important for the understanding of the molecular pharmacology of positive and negative KCNN channel gating modifiers.
The KCa2 channel subtypes are widely and distinctly expressed in the CNS, controlling somatic excitability, pacemaker firing rates, and synaptic plasticity (Bond et al., 2005; Pedarzani and Stocker, 2008). Clinically used drugs with KCa2-activating properties are the centrally acting muscle relaxants zoxazolamine and chlorzoxazone, which are used for treating spasticity, and riluzole, which is registered for amyotrophic lateral sclerosis. The KCa2-facilitating properties (probably KCa2.2) of both chlorzoxazone and riluzole have recently been suggested to account for their beneficial effects in rodent ataxia models (Janahmadi et al., 2009; Alviña and Khodakhah, 2010) and in patients (Ristori et al., 2010). KCa2 activation (probably KCa2.3) also arguably underlies reduced craving and dampening of excessive alcohol intake observed with chlorzoxazone in rats (Hopf et al., 2011). Important hallmarks for a therapeutic KCa2-facilitating mechanism of these drugs are that 1-EBIO (1-ethyl-2-benzimidazolinone), the prototype KCNN channel activator, exerts a similar antiataxic action when applied locally into the cerebellum of tottering mice (Walter et al., 2006) and antiaddictive effect against alcohol when applied into rat nucleus accumbens core region (Hopf et al., 2010). In contrast, inhibition of KCa2 channels has not been demonstrated to account for clinical efficacy of any drug. The antidepressant fluoxetine is a weak blocker of cloned KCa channels (Terstappen et al., 2003) and KCa2.3 channel block may mediate an antidepressant mechanism (Galeotti et al., 1999; Jacobsen et al., 2008). However, it is doubtful whether this mechanism for fluoxetine plays a role compared with serotonin transporter inhibition.
Validation of specific KCa channel subtypes as targets for CNS drug discovery is hampered by lack of selective and potent molecules that pass the blood-brain barrier. Classic KCa2 channel blockers such as UCL1684 [6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)-diquinolinacyclodecaphane] mimic the inhibition by the KCa2 selective peptide apamin and are unselective among the KCa2 subtypes. Like apamin, they further contain basic and charged moieties that limit their passage into the CNS. 1-EBIO and the KCa2-activating drugs are nonselective KCa2 enhancers and act more potently on KCa3.1. Unfortunately, this is also the case for the more potent positive modulators DCEBIO, NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime), 4,5-dichloro-1,3-diethyl-2,3-dihydro-1H-1,3-benzodiazol-2-one (NS4591), and SKA-31, even though these have emerged from dedicated chemical optimization programs (Wulff and Zhorov, 2008).
Subtype-selective positive gating modulators and nonselective negative gating modulators have recently been identified. These have been instrumental in defining new sites and new modes of action for gating modulation of KCa2 channels. The negative gating modulator NS8593 [(R)-N-(benzimidazol-2-yl)-tetrahydro-1-naphtylamine] causes a reduction in the apparent Ca2+-sensitivity for channel activation (Strøbæk et al., 2006; Sørensen et al., 2008). This effect depends on Ser507 and Ala532 (KCa2.3 numeration) positioned in the S5 pore helix and S6 transmembrane segment just below the selectivity filter and was therefore interpreted as NS8593 interaction with deep-pore gating structures (Jenkins et al., 2011). These amino acids are conserved among the KCa2 channels, thus explaining the lack of KCa2 subtype selectivity of NS8593 and close analogs (Sørensen et al., 2008). The KCa2.1 selective activator GW542573X, which acts dually by directly opening the channel and by increasing the apparent Ca2+ sensitivity (Hougaard et al., 2009), depends on Ser293 in the S5 transmembrane helix, a KCa2.1-specific amino acid. These mixed effects differ significantly from the mode of action of nonselective activators but also from the KCa2.2/3 selective modulator cyclohexyl-[2-(3,5-dimethylpyrazol-1-yl)-6-methylpyrimidin-4-yl]-amine, which increases the apparent Ca2+ sensitivity via interaction with the C-terminal (Pedarzani et al., 2001; Hougaard et al., 2007; Li et al., 2009).
Here we describe two novel and closely related [1,2,4]triazolo[1,5-a]pyrimidines, B-TPMF [(N-{7-[1-(4-tert-butyl-phenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine] and CM-TPMF [N-{7-[1-(4-chloro-2-methylphenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine], that both exhibit high-potency (∼30 nM) and subtype-selective interactions with KCa2.1 via Ser293. These compounds show opposite functions with (−)-CM-TPMF acting as an activator and (−)-B-TPMF as an inhibitor.
Materials and Methods
Chemistry.
B-TPMF and CM-TPMF were purchased as racemates from Key Organics Ltd. (Camelford, Cornwall, UK). The compounds were resolved into their enantiomeric pairs by preparative chiral chromatography, using for B-TPMF a Chiracel OD-H column (solvent 40% ethanol in hexane; flow rate, 1.0 ml/min) and for CM-TPMF a Chiralpak AD-H column (solvent 40% ethanol in hexane; flow rate, 1.0 ml/min). Determination of chiral purity was performed on an analytical Waters Thar Investigator instrument equipped with a photodiode array detector: for B-TPMF using a Phenomenex (Torrance, CA) Lux2 column, 250 × 4.6 mm, 5-μm particle size (solvent super critical CO2 with ethanol as cosolvent and isocratic elution with 45% cosolvent and a flow rate of 3.2 ml/min); for CM-TPMF using a Phenomenex Lux1 Column, 250 × 4.6 mm, 5-μm particle size (solvent super critical CO2 with methanol as cosolvent and isocratic elution with 40% cosolvent and a flow rate of 3.2 ml/min). Chiral purity was determined by integration of the photodiode array (total absorbance) trace. Optical rotations were measured on a polarimeter (PerkinElmer Life and Analytical Sciences, Waltham, MA). Absolute configurations were not determined. NMR spectra were recorded on a Bruker UltraShield Plus 400 MHz (Avance II) instrument. Characterization of (−)-B-TPMF (C19H24N6O2): specific optical rotation [α]D25 = −291° (CHCl3); enantiomeric excess >99%; HR-MS(ES+) m/z 369.20362 ([M + 1]+, 100%), calculated, 369.20328; 1H NMR (CDCl3) δ 1.26 (s, 9H), 1.78 (d, 3H, J = 6.6 Hz), 3.92 (s, 3H), 5.89 (q, 1H, J = 6.5 Hz), 6.75 (m, 2H), 7.10 (m, 2H), 7.25 (m, 2H), 7.82 (d, 1H, J = 10.4 Hz), 7.98 (d, 1H, J = 10.4 Hz), and 8.60 (d, 1H J = 4.7 Hz). Characterization of (−)-CM-TPMF (C16H17ClN6O2): Specific optical rotation [α]D25 = −275° (CHCl3); enantiomeric excess = 96.4%; HR-MS(ES+) m/z 361.11775 ([M + 1]+, 100%), calculated 361.11738; 1H NMR (CDCl3) δ 1.80 (d, 3H, J = 6.5 Hz), 2.34 (s, 3H), 3.93 (s, 3H), 5.89 (q, 1H, J = 6.4 Hz), 6.44 (d, 1H, J = 8.8 Hz), 6.95–7.04 (m, 2H), 7.17 (m, 1H), 7.81(d, 1H, J = 10.4 Hz), 7.97 (d, 1H, J = 10.2), and 8.61 (d, 1H, J = 4.7 Hz).
Molecular Biology.
The generation of point-mutated hKCa2.3L476S has been described previously (Hougaard et al., 2009), and hKCa2.1S293L was generated by mutagenesis on hKCa2.1. In brief, the plasmid encoding hKCa2.1 was uracilated by the Escherichia coli RZ1032 and used as template in a mutagenesis reaction using the oligonucleotide GTGCTGCTGGTCTTCtcgATaTCCctCTGGATCATCGCAGC, T7 DNA polymerase, and T4 DNA ligase. The mutation was verified by sequencing.
Cell Cultures.
For patch clamp experiments with hKCa2.1, hKCa2.2, hKCa2.3, and hKCa3.1, HEK293 cell lines stably expressing these channels were used (Hougaard et al., 2009). Point-mutated channels (hKCa2.1S293L and hKCa2.3L476S) were transiently transfected into HEK293 cells using Lipofectamine (Invitrogen, Carlsbad, CA) and standard transfection methods. Electrophysiological measurements were performed 2 to 3 days after transfection. Cells were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich, Brøndby, Denmark) enriched with 10% fetal calf serum (Invitrogen) at 37°C and 5% CO2. At approximately 75% confluence, the cells were washed once with phosphate-buffered saline, harvested by trypsin/EDTA (Sigma-Aldrich) treatment, and transferred to Petri dishes containing coverslips (diameter 3.5 mm; VWR International, Herlev, Denmark).
Electrophysiology.
Membrane currents were recorded using the whole-cell or the inside-out configuration of the patch-clamp technique. Cells seeded on cover slips were transferred to a 15-μl recording chamber and continuously superfused at 1 ml/min. An integrated Ag/AgCl pellet electrode served as reference. Experiments were conducted at room temperature. Patch pipettes (approximately 2 MΩ) were pulled from borosilicate tubes with an outside diameter of 1.32 mm (Vitrex Medical, Herlev, Denmark) using a horizontal electrode puller (Zeitz Instruments, Augsburg, Germany). An electronically controlled micromanipulator (Eppendorf, Radiometer, Denmark) was used for the positioning of pipettes, and the experiments were controlled by an EPC-9 amplifier (HEKA, Lambrecht, Germany). Data were filtered at 3 kHz. Currents were elicited by applying a 200-ms linear voltage ramp from −80 to +80 mV every 5 s from a holding potential of 0 mV. In whole-cell experiments, the cell capacitance and series resistance (Rs below 8 MΩ, 80% compensation) were updated before each voltage ramp.
In all experiments a solution with a high K+ concentration was applied to the extracellular side of the membrane: 154 mM KCl, 2 mM CaCl2, 1 mM MgCl2 and 10 mM HEPES, pH adjusted to 7.4 with 1 M KOH. The intracellular solutions contained 154 mM KCl, 10 mM HEPES, 10 mM EGTA, or a combination of EGTA and NTA (10 mM in total). Concentrations of MgCl2 and CaCl2 required to obtain the desired free concentrations (Mg2+ always 1 mM, Ca2+ 0.01–10 μM) were calculated (EqCal, Cambridge, UK) and added. In the nominally Ca2+-free intracellular solution, no Ca2+ was added. The intracellular solutions were adjusted to pH 7.2 with 1 M KOH.
Calculations and Statistics.
In inside-out experiments, EC50 and IC50 as well as Hill (nH) values for compounds and Ca2+ were estimated from equilibrium concentration-response relationships by fitting to the Hill equation. In whole-cell experiments, EC50 values for current activation are not readily determined because of poor clamping conditions at higher degrees of channel activation. Instead, the concentrations of compound giving 100% current increase (called the SC100 value; see Table 1) were estimated. In some experiments, Kd values were calculated from single concentration applications by fitting to the kinetics of inhibition (Strøbæk et al., 2006). All results are presented as the mean ± S.E.M., with the number of experiments indicated. Significance testing was performed by Student's t test for unpaired samples.
Enantiomeric effects of CM-TPMF and B-TPMF at hKCa channels in whole-cell experiments
Results
The chemical structures of the two new compounds, B-TPMF and CM-TPMF, as well as GW542573X, are shown in Fig. 1. The effects of the isolated enantiomers of B-TPMF and CM-TPMF (see Chemistry above) were determined in whole-cell voltage-clamp experiments by applying 10 nM concentrations of the compounds to HEK293 cells expressing human KCa2.1 channels. Representative current traces are shown in Fig. 2A, and the current recorded at −75 mV is plotted versus time in Fig. 2B. The (−)-enantiomer of B-TPMF inhibited the KCa2.1 current, whereas the (+)-enantiomer had no effect. In contrast, (−)-CM-TPMF, induced a robust activation, again with the (+)-enantiomer exerting no effect. The time course of this experiment (Fig. 2B) underscores the reversibility of these actions, and the I-V curves show preservation of the normal inward rectification in presence of all compounds. Figure 2C summarizes this series of experiments and specifically documents that 10 nM (−)-B-TPMF significantly inhibits the KCa2.1 current, whereas (−)-CM-TPMF enhances it (p < 0.001, Student's t test for unpaired samples). The corresponding (+)-enantiomers of both compounds, by contrast, are inactive at this concentration. Whole-cell experiments were also performed to quantify the enantiomer selectivity (experiments not illustrated; data in Table 1): Kd values for (+)-B-TPMF and (−)-B-TPMF were 1.6 ± 0.5 μM (n = 5) and 15 ± 7 nM (n = 5), respectively. Likewise, (+)-CM-TPMF activated KCa2.1 with an SC100 of 200 ± 44 nM (n = 5) compared with 5 ± 0.2 nM (n = 6) for (−)-CM-TPMF. Thus, for B-TPMF and CM-TPMF, the activity on KCa2.1 channels resides primarily in the (−)-enantiomers, with enantiomer selectivities of ∼100- and ∼40 fold.

Chemical structures of the negative KCa2.1 channel modulator B-TPMF and the positive modulators CM-TPMF and GW542573X.

(−)-CM-TPMF acts as an activator and (−)-B-TPMF acts as an inhibitor of KCa2.1 channels. A, whole-cell current recorded from HEK293 cells stably expressing KCa2.1. I-V relationships were obtained at symmetrical [K+] and with the free [Ca2+] in the pipette solution buffered to 0.3 μM. I-V relationships were measured upon application of 200-ms voltage ramps (−80 to +80 mV), elicited every 5 s from a holding potential of 0 mV in the absence (Ctrl) or in the presence of 10 nM (+)-B-TPMF, (−)-B-TPMF, (+)-CM-TPMF, and (−)-CM-TPMF, as indicated. B, whole-cell KCa2.1 current at −75 mV obtained from the voltage ramps (A) as a function of time. The cell was exposed to 10 nM (−)- and (+)-forms of CM-TPMF and B-TPMF as indicated by the bars. C, average currents (mean ± S.E.M.) in the presence of (−)- and (+)-forms of CM-TPMF and B-TPMF (10 nM). Current measured in the presence of compound is shown relative to the current level before compound addition (control; n = 4–11). (−)-B-TPMF was more potent than (+)-B-TPMF (p < 0.001, Student's t test) in inhibiting KCa2.1 current, whereas (−)-CM-TPMF was more potent than (+)-CM-TPMF in potentiating the current (p < 0.001, Student's t test).
To investigate the subtype selectivity of the more active (−)-enantiomers, experiments were conducted on HEK293 cells stably expressing hKCa2.1, hKCa2.2, or hKCa2.3 channels (Fig. 3). Because (−)-B-TPMF and (−)-CM-TPMF act equally well from both sides of the membrane, the inside-out configuration of the patch-clamp technique was chosen to achieve a well defined [Ca2+]i. At a concentration of 30 nM, (−)-B-TPMF inhibited the KCa2.1 current by approximately 50%; conversely, (−)-CM-TPMF induced a 3-fold increase. At the same concentration, only minor effects were detectable on KCa2.2 and KCa2.3 (Fig. 3A), indicating that both compounds exhibit selectivity for KCa2.1 over the closely related isoforms. Full concentration-response relationships were obtained for the three KCa2 subtypes, and results for both compounds are summarized in Fig. 3, B and C. The data generated for (−)-B-TPMF-induced inhibition conformed well to the Hill equation with an IC50 value for KCa2.1 of approximately 30 nM and a complete inhibition of the current at 1 μM, whereas KCa2.2 and KCa2.3 both were inhibited with IC50 values around 1 μM (see Table 2, and Fig. 3 legend). Hence (−)-B-TPMF is a selective inhibitor of KCa2.1, which displays approximately 30-fold selectivity over the other KCa2 channels. The concentration-response relationships obtained from applications of (−)-CM-TPMF to marginally activated KCa2 channels (0.2 μM Ca2+; degree of activation, ∼15%) yielded an EC50 value of 24 nM for KCa2.1 and a maximal degree of activation of approximately 80% compared with a [Ca2+]i of 10 μM. KCa2.2 and KCa2.3 were activated with EC50 values of 290 and 250 nM, showing that (−)-CM-TPMF is 10-fold more selective for KCa2.1. Similar selectivity ratios were obtained against KCa3.1, except for (−)-CM-TPMF, which acted as a (very weak) activator (Table 1). Table 3 also summarizes the effects of (−)-CM-TPMF and (−)-B-TPMF on more distantly related ion channels. Only marginal inhibitory effects were observed for both compounds at 10 μM on members of other K+ channel gene families (KCa1.1, Kv7.2+Kv7.3, and Kv11.1) as well as Nav1.2 channels. A broader selectivity profile was obtained for the two racemates by testing at concentrations of 10 μM in the 69-receptor LeadProfilerScreen (internal reference number PT# 1107076) at MDSPharma (now Ricerca Biosciences LLC), Taiwan. Neither of the racemates reached the predefined significance criteria of >50% effect in any assay.
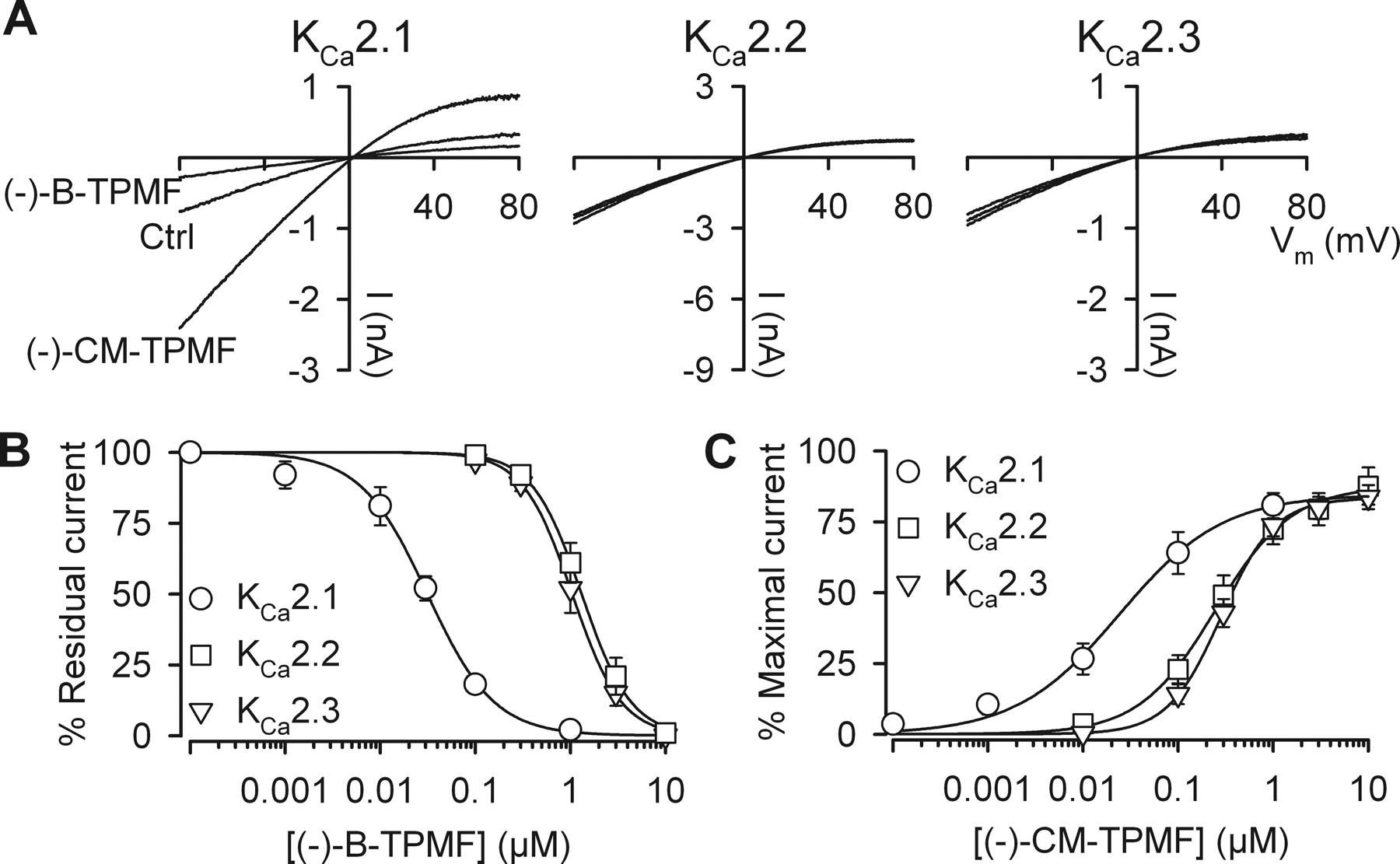
(−)-B-TPMF is a KCa2.1 subtype-selective inhibitor and (−)-CM-TPMF a KCa2.1-selective activator. A, I-V relationships measured from inside-out patches obtained from HEK293 cells stably expressing hKCa2.1, hKCa2.2, or hKCa2.3. Each I-V plot shows a control trace, a trace obtained in the presence of 30 nM (−)-B-TPMF, and a trace in the presence of 30 nM (−)-CM-TPMF. The free [Ca2+] in the bath/intracellular solution was 0.3 μM (resulting in approximately 30% of the maximal current level) to allow for both positive and negative modulation. B, concentration-response relationships of (−)-B-TPMF on the three KCa2 subtypes. Residual current is depicted as a function of the (−)-B-TPMF concentration. Currents were measured from inside-out patches at 0.5 μM free Ca2+ (approximately 90% of the maximal current level), and each data point is the mean ± S.E.M. of five to seven experiments. The solid lines are the fit of data to the Hill equation yielding the following IC50 values and Hill coefficients: KCa2.1, 0.031 μM and −1.2; KCa2.2, 1.05 μM and −1.7; KCa2.3, 1.32 μM and −1.7. C, concentration-response relationships of (−)-CM-TPMF on KCa2.1, KCa2.2, and KCa2.3. Currents were measured from inside-out patches at 0.2 μM free Ca2+ (approximately 15% activation) and shown relative to the current level obtained at 10 μM Ca2+ in the absence of compound. Each data point is the mean ± S.E.M. of six to eight experiments, and the solid lines are the fit of data to the Hill equation yielding the following EC50 values and Hill coefficients KCa2.1, 0.024 μM and 0.8; KCa2.2, 0.29 μM and 1.5; KCa2.3; 0.25 μM and 1.1. Efficacy with respect to saturating Ca2+ was in the range of 83 to 87%.
Effects of (−)-CM-TPMF and (−)-B-TPMF at hKCa2 channels in inside out experiments
Effects of (−)-CM-TPMF and (−)-B-TPMF at other ion channels
We next investigated the modes of action of (−)-CM-TPMF and (−)-B-TPMF. Classic enhancers of KCa2 channel activity increase the apparent Ca2+-sensitivity and are unable to activate the channels in the absence of intracellular Ca2+. In contrast, GW542573X (see Fig. 1), a recently described KCa2.1-selective compound, acts by a dual mechanism, increasing the apparent Ca2+ sensitivity and inducing a small Ca2+-independent channel activation. Figure 4A shows the time course of an inside-out experiment in which the [Ca2+]i was varied (0, 0.2, and 10 μM) and the ability of 100 nM (−)-CM-TPMF to activate the KCa2.1 current at each Ca2+ concentration was investigated. Figure 4B shows the corresponding I-V relationships. Addition of (−)-CM-TPMF clearly enhanced the KCa2.1 current in the nominal absence of Ca2+ and shifted the I-V relationship from a small, nonspecific linear leak current to an inwardly rectifying current typical for KCa2 channels recorded under symmetrical K+ conditions. At an intermediate Ca2+ concentration of 200 nM, where the KCa2.1 channels are partially Ca2+-activated, as well as at saturating Ca2+ concentrations of 10 μM, where the channels are maximally activated, application of (−)-CM-TPMF induced an increase in the current level. At all [Ca2+]i, the current measured in the presence of (−)-CM-TPMF maintained the characteristic inward rectification. Figure 4C shows the Ca2+-response relationship of KCa2.1 in the absence of compound (Ctrl) as well as in the presence of 0.1 or 1 μM (−)-CM-TPMF. It is noteworthy that (−)-CM-TPMF exerts a dual action on the KCa2.1 channel consisting of a leftward shift in the Ca2+-response curve (i.e., positive modulation) as well as a substantial direct activating effect, which is independent of Ca2+ (close to 40% at low Ca2+ and in the presence of 1 μM (−)-CM-TPMF). Thus, from a mode-of-action perspective, (−)-CM-TPMF belongs to the GW542573X functional class of KCa2.1 activators. Viewed from a quantitative perspective, however, (−)-CM-TPMF is considerably more potent (>100 fold) than GW542573X, and the fraction of Ca2+-independent activation induced by the compound is much larger (∼40% versus 5%).

(−)-CM-TPMF is a KCa2 channel opener and a positive modulator. A, KCa2.1 current at −75 mV, measured from an inside-out patch and depicted as a function of time. The patch was exposed to 0.1 μM (−)-CM-TPMF either in the absence of Ca2+ (0), at 0.2—or at 10 μM Ca2+. B, I-V relationships at 0, 0.2 - or 10 μM Ca2+ in the absence (Ctrl) or presence of 0.1 μM (−)-CM-TPMF as indicated. C, Ca2+-response curves for KCa2.1 measured from inside-out patches either in the absence (Ctrl) or in the presence of (−)-CM-TPMF (0.1 or 1 μM). Currents from individual patches were normalized with respect to the effect of 10 μM Ca2+. Data points are the mean ± S.E.M. of at least six experiments. The lines are the fit of data to the Hill equation yielding the following EC50 values and Hill coefficients: Ctrl, 0.32 μM and 4.4; 0.1 μM (−)-CM-TPMF, 0.15 μM and 2.9; 1 μM (−)-CM-TPMF, 0.13 μM and 2.8. Note the substantial Ca2+-independent opener effect of (−)-CM-TPMF at very low Ca2+ concentrations [14 ± 3 and 32 ± 5% of maximal activity at 0.1 and 1 μM (−)-CM-TPMF] and the higher efficacy at saturating Ca2+ (I/Imax >1) in the presence of (−)-CM-TPMF.
KCa2 channels are inhibited by peptides such as apamin and scyllatoxin as well as by small molecules carrying positive charges mimicking the charges on apamin. These molecules bind from the outside, and despite the fact that an allosteric mode of action has been demonstrated for apamin (Lamy et al., 2010), their inhibition is independent of the degree of channel activation by Ca2+. In contrast, negative gating modulators such as NS8593 interfere with the gating mechanism of KCa2 channels, causing reduced apparent affinity for Ca2+. A characteristic feature of this mode of action is potent inhibition at low [Ca2+]i and reduced effect at higher Ca2+ concentrations. The following experiment was designed to elucidate whether the inhibition by (−)-B-TPMF is dependent on the [Ca2+]i. Figure 5A shows the time course of an inside-out experiment in which the ability of 30 nM (−)-B-TPMF to inhibit the KCa2.1 was evaluated at two different Ca2+ concentrations: 0.3 μM, equal to approximately 30% of maximal channel activity, and 10 μM Ca2+, corresponding to maximal activity. (−)-B-TPMF inhibited approximately 75% of the current at the lower Ca2+ concentration, whereas it inhibited only approximately 20% at 10 μM Ca2+. In Fig. 5B, the degree of inhibition by 30 nM (−)-B-TPMF at three Ca2+ concentrations (0.3-, 0.5-, and 10 μM) is quantified. The effect is clearly dependent on the intracellular Ca2+ concentration/channel activity, and (−)-B-TPMF accordingly fulfills the criteria for being a negative gating modulator.

(−)-B-TPMF inhibits the KCa2.1 current in a Ca2+-dependent manner. A, KCa2.1 current at −75 mV plotted versus time. Current was measured from an inside-out patch exposed to 30 nM (−)-B-TPMF at 0.3 or 10 μM Ca2+. As control for the background/leak current level, periods with 0 free Ca2+ were included. B, percentage current inhibition induced by (−)-B-TPMF (30 nM) at the different Ca2+ concentrations. Data points are the mean ± S.E.M. of five to six experiments at each Ca2+ concentration.
Because of the very similar structures of (−)-B-TPMF and (−)-CM-TPMF, we envisioned that they might interact with a common binding site on KCa2.1. To test this hypothesis, we conducted a series of patch clamp experiments to elucidate possible competitive-like functional interactions. In the first series of experiments, we investigated whether KCa2.1 activated by different concentrations (1 and 10 μM) of (−)-CM-TPMF would affect experimentally determined IC50 values for (−)-B-TPMF. These concentrations of (−)-CM-TPMF were chosen because they both induce near-maximal activation of KCa2.1 (see Fig. 3C) at a permissive fixed [Ca2+]i of 200 nM. Figure 6A shows the I-V relations obtained in the presence of 1 μM (left) and 10 μM (right) of (−)-CM-TPMF. When 1 μM (−)-B-TPMF was coapplied, an approximate 60% reduction in the current activated by 1 μM (−)-CM-TPMF was obtained, whereas the same concentration of (−)-B-TPMF inhibited only 15% of the current activated by 10 μM (−)-CM-TPMF. Inhibition curves for (−)-B-TPMF were generated, and Fig. 6B clearly shows that increasing the concentration of (−)-CM-TPMF from 1 to 10 μM resulted in an increase in the IC50 value of (−)-B-TPMF from 0.43 to 2.96 μM. Although no rigorous comparison is possible, it is noteworthy that the IC50 value for (−)-B-TPMF at 200 nM [Ca2+]i in the absence of (−)-CM-TPMF is expected to be in the low nanomolar range (Fig. 5). In the second series of experiments, we attempted to prove that the increase in IC50 values in the presence of (−)-CM-TPMF is dependent of the presence of this compound per se rather than being an effect of the high open-state probability prevailing under the experimental conditions in Fig. 6. Therefore, we compared the inhibition by (−)-B-TPMF at high [Ca2+]i (10 μM) with the inhibition at semimaximal [Ca2+]i (0.5 μM) in combination with 3 μM (−)-CM-TPMF, resulting in similar degrees of activation (Fig. 7). The time course of the experiment (Fig. 7A) shows that at 10 μM Ca2+, (−)-B-TPMF (0.1 μM) inhibits the current reversibly by 75%. However, after wash-out and reactivation with 0.5 μM Ca2+ + 3 μM (−)-CM-TPMF, no effect of 0.1 μM (−)-B-TPMF was observed, and even a 100 times higher concentration was needed to get the same degree of inhibition as when the channel was similarly activated by Ca2+ alone. Note also the strongly reduced rate of inhibition compared with the faster rate obtained at 0.1 μM (−)-B-TPMF at the first addition. We conclude from these experiments that (−)-B-TPMF and (−)-CM-TPMF interact functionally on the KCa2.1 channel in a manner likely to reflect direct competition on a common binding site.

Maximal (−)-CM-TPMF activation concentration-dependently decreases the apparent affinity of the inhibitor (−)-B.TPMF. A, I-V relationships measured from inside-out patches obtained from HEK293 cells stably expressing hKCa2.1 at a free [Ca2+] of 0.2 μM. I-V curves were obtained in the presence of 1 or 10 μM (−)-CM-TPMF alone or in combination with 1 μM (−)-B-TPMF. B, concentration-response relationship of (−)-B-TPMF in the presence of 1 or 10 μM (−)-CM-TPMF. Residual current is depicted as a function of the (−)-B-TPMF concentration. Each data point is the mean ± S.E.M. of four to nine experiments, and the solid lines are the fit of data to the Hill equation, resulting in the following IC50 values and Hill coefficients: 0.43 μM and −0.9 (at 1 μM (−)-CM-TPMF) and 2.96 μM and −1.4 (in the presence of 10 μM (−)-CM-TPMF).

The reduced apparent affinity of (−)-B-TPMF is due to (−)-CM-TPMF competition, not the degree of channel activation. A, KCa2.1 current measured at −75 mV as a function of time. The inside of the patch was exposed to a [Ca2+] of 0, 0.5, or 10 μM as indicated. (−)-B-TPMF was added once steady current activation was obtained by 10 μM Ca2+ alone or by the combination of 0.5 μM Ca2+ and 3 μM (−)-CM-TPMF. B, I-V relationships measured at the time points indicated by letters in A. Data are from a single experiment representative of four independent experiments.
The action of both positive and negative modulators of KCa2 channels depend on very specific amino acid residues. 1-EBIO and cyclohexyl-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methylpyrimidin-4-yl]-amine interact with the intracellular C-terminal CaMBD, whereas NS8593 depends on transmembrane amino acids. The KCa2.1-selective activator GW542573X acts via a single amino acid, Ser293, located in the S5 transmembrane region of KCa2.1. Because of the similar modes of action of GW542573X and (−)-CM-TPMF, we conducted a series of experiments to investigate the possible involvement of Ser293 in the actions of both (−)-CM-TPMF and (−)-B-TPMF. Two point-mutated KCa2 channels were used, one (KCa2.1S293L) in which Ser293 in KCa2.1 is substituted with the equivalent leucine (Leu321 in KCa2.2 and Leu476 in KCa2.3) and one with the reverse mutation (Leu476 in KCa2.3 changed to serine, KCa2.3L476S; see Fig. 8A for approximate position in S5 and amino acid alignment of the S5 region of KCa2.1 and KCa2.3). Both channel constructs were activated by [Ca2+]i within the normal range with EC50 values of 0.44 ± 0.05 μM (KCa2.1S293L, n = 3) and 0.47 ± 0.06 μM (KCa2.3L476S, n = 4). At 30 nM, a compound concentration that significantly modulated KCa2.1 but had limited effect on KCa2.3 (see Fig. 3), both (−)-CM-TPMF and (−)-B-TPMF failed to affect KCa2.1S293L (Fig. 8B) but, on the other hand, significantly modulated the KCa2.3L476S current level (Fig. 8C). Full concentration-response relationships were generated for (−)-B-TPMF (Fig. 8D) and (−)-CM-TPMF (Fig. 8E) on KCa2.1S293L and KCa2.3L476S in excised inside-out patches. For comparison, the best fit curves for KCa2.1 and KCa2.3, obtained from Fig. 3, are included as dotted lines. As seen from the figure, data obtained on KCa2.1S293L was similar to those generated on KCa2.3, thus demonstrating full loss of sensitivity, both with respect to inhibition by (−)-B-TPMF and activation by (−)-CM-TPMF. Likewise, data obtained on KCa2.3L476S was virtually identical to those on KCa2.1, demonstrating full gain of sensitivity of the KCa2.3L476S construct for both compounds.

The activity of (−)-B-TPMF and (−)-CM-TPMF is critically dependent on Ser293 in KCa2.1. A, schematic illustrating the approximate S5 location of the single amino acid that is mutated in the channel constructs. In KCa2.1, serine is replaced with leucine, which is the equivalent amino acid in KCa2.3, and in KCa2.3 leucine is replaced with serine. Amino acid alignment of the S5 region of KCa2.1 and KCa2.3 is shown on the right. B and C, I-V relationships measured from inside-out patches obtained from HEK293 cells transiently transfected with KCa2.1S293L (B) or KCa2.3L476S (C). Each plot shows a trace representing the absence of compound (Ctrl), a trace representing the presence of 30 nM (−)-B-TPMF, and a trace representing the presence of 30 nM (−)-CM-TPMF. The free [Ca2+] in the bath/intracellular solution was 0.3 μM to allow for both positive and negative modulation of the current level. D, concentration-response relationship of (−)-B-TPMF on KCa2.1S293L and KCa2.3L476S. Residual current in the presence of (−)-B-TPMF is depicted as a function of the (−)-B-TPMF concentration. Currents were measured from inside-out patches at 0.5 μM free Ca2+ and each data point is the mean ± S.E.M. of five or six experiments. The solid lines are the fit of data to the Hill equation yielding the following IC50 values and Hill coefficients: hKCa2.1S293L, 0.81 μM and −1.2; KCa2.3L476S, 0.05 μM and −1.6. For illustrative purposes, data obtained on wild type KCa2.1 and KCa2.3 are included as dotted lines. E, concentration-response relationship of (−)-CM-TPMF on KCa2.1S293L and KCa2.3L476S. Currents were measured at 0.2 μM free Ca2+ and depicted relative to the current level obtained at 10 μM Ca2+ and in the absence of compound. Data points are the mean ± S.E.M. of four to five experiments, and the solid lines are the fit of data to the Hill equation: hKCa2.1S293L, 0.7 μM and 1.3; KCa2.3L476S, 0.03 μM and 1.2. Efficacy with respect to maximal Ca2+ was 89% (hKCa2.1S293L) and 85% (KCa2.3L476S). Again, for illustrative purposes, data on KCa2.1 and KCa2.3 are included as dotted lines.
Discussion
We have described two structurally related compounds as new pharmacological modulators of Ca2+-activated K+ channels belonging to the KCNN gene family. Both are [1,2,4]triazolo[1,5-a]pyrimidines, a chemical series hitherto not described as having ion channel-modulating properties. Both (−)-CM-TPMF and (−)-B-TPMF potently modulated the KCa2.1 subtype but had considerably weaker activity on KCa2.2, KCa2.3, and KCa3.1. (−)-CM-TPMF is a mixed opener/positive gating modulator, whereas the analog (−)-B-TPMF is a negative gating modulator. Coapplication experiments clearly revealed competition-like interactions between (−)-CM-TPMF and (−)-B-TPMF, and the high-affinity actions of both compounds were shown to depend critically on the Ser293 positioned in the S5 segment, as demonstrated by loss-of-sensitivity (KCa2.1S293L) as well as gain-of-sensitivity (KCa2.3L476S) mutations. CM-TPMF and B-TPMF are remarkably similar structurally, and the pharmacology leading to either positive or negative gating modulation is determined entirely by the substituents on the terminal phenyl ring, a 4-tert-butyl group in B-TPMF or 4-chloro-2-methyl substitution in CM-TPMF. We are currently further exploring the structural components leading to either activation or inhibition and the general structure-activity relationship associated with chemical modification within this compound class (Sørensen et al., 2010a,b,c). It is noteworthy that for each of these compounds, the chirality introduced by the carbon atom in the linker separating the phenyl and the bicyclic heteroaromatic groups led to pairs consisting of a highly potent (−)-enantiomer and a 40- to 100-fold less active (+)-isomer. Overall, we conclude that KCa2 channels—in particular the KCa2.1 isoform—possess a common high-affinity binding-site for (−)-[1,2,4]triazolo[1,5-a]pyrimidines, here exemplified by (−)-CM-TPMF and (−)-B-TPMF, that pivotally influences the gating process in a facilitating or dampening way depending on minor changes in the substitution pattern of the interacting molecules. This is the first demonstration of a common site for both positive and negative pharmacological modulation of gating in KCa channels, a phenomenon that is well established for the dihydropyridine pharmacology of L-type Ca2+ channels [i.e., nifedipine versus S-(−)-1,4-dihydro-2,6-dimethyl-5-nitro-4-(2-[trifluoromethyl]phenyl)-3-pyridine carboxylic acid methyl ester (Bay-k-8644) (Greenberg et al., 1984)] as well as for the benzodiazepine pharmacology of GABAA receptors [i.e., diazepam versus methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate (Sieghart, 1994)].
It is interesting to compare the chemical structures of CM-TPMF/B-TPMF with the previously described (Hougaard et al., 2009) nonchiral and lower-potency KCa2.1 activator GW542573X (Fig. 1), which also depends on Ser293 and has a qualitatively identical opener/positive modulator mode of action as described here for (−)-CM-TPMF. Both structural classes are relatively flexible, small molecules of similar sizes and molecular weights; both classes contain a terminal phenyl ring, although with different substitution pattern; and both GW542573X and the [1,2,4]triazolo[1,5-a]pyrimidines have a single potential hydrogen bond donating group and seven to eight hydrogen bond acceptors. Still, it is evident that GW542573X represents a structurally very different compound class containing two carbamate moieties of which one contains a terminal tert-butyl group also present in B-TPMF. Despite this apparent point of similarity, it should be noted that GW542573X is an activator, whereas a closer structure activity analysis comprising further structural analogs (not shown) revealed that the tert-butyl group is crucial for the effect of B-TPMF as an inhibitor. Overall, based on the structural differences described above and also the fact that the small structural difference arising from chirality has such detrimental effects on the pharmacology in CM-TPMF and B-TPMF, we consider it unlikely that the [1,2,4]triazolo[1,5-a]pyrimidines described herein would have exactly identical interaction points as GW542573X and share an identical binding site. Despite the parallels between GW542573X and (−)-CM-TPMF in terms of mode of action and interaction with Ser293, we therefore cannot exclude that it actually interacts with a site neighboring the [1,2,4]triazolo[1,5-a]pyrimidines and just shares the Ser293 and the positive coupling to the gate with (−)-CM-TPMF as downstream functional effects. From this also follows uncertainty about whether Ser293 per se actually constitutes part of the physical binding site for the [1,2,4]triazolo[1,5-a]pyrimidines.
A series of experiments exploring the apparent Ca2+ affinity of rKCa2.2 through mutational analysis has demonstrated the complexity and the extended distribution of amino acids participating in or influencing the gating process in rKCa2.2 channels (Li and Aldrich, 2011). Charged residues in S6 were found to form a ring of positivity near the inner pore mouth that causes intrinsic rectification but also influences the gating via an electrostatic mechanism, in particular by keeping the open-state probability very low at zero Ca2+. Charge reversal mutations of these amino acids strongly increase the open state probability at zero Ca2+ and very significantly left-shift the Ca2+ activation curve. Without in any way suggesting a common causality, these findings are phenomenologically very similar to the effects of (−)-CM-TPMF and GW542573X and serve to illustrate the point that amino acids in the transmembrane segments, far away from the CaM/CaMBD in the C terminus, are pivotally important for gating and determining apparent Ca2+ affinity of KCa2 channels.
The physiological role of the KCa2.1 subtype remains obscure in contrast to the KCa2.2 and KCa2.3 subtypes, which, through their contribution to action potential afterhyperpolarizations and functional coupling with the NMDA receptor, are involved in controlling firing precision in pacemaking neurons and determining synaptic plasticity (Bond et al., 2005). KCa2.1 is predominantly expressed in the cortical/limbic structures of the brain, a distribution overlapping to a large extent with the expression of KCa2.2, but is essentially absent from the basal ganglia and monoaminergic neurons, where KCa2.3 is the dominating subtype. It is noteworthy that KCa2.1 is also the KCNN subtype with the most specific CNS expression and seems confined to neurons (Rimini et al., 2000). In contrast, the other KCNN members are also broadly expressed in the periphery on both neurons and somatic cell types, and KCa2.3 is also present on glia cells (Armstrong et al., 2005). A serious obstacle toward exploration of the physiology of the KCa2.1 channel is the inability of the rat (and mouse) isoforms to express recombinantly in oocytes as well as mammalian cells. Furthermore, the phenotype of the KCa2.1 knockout mouse is still largely unknown, although it has been reported that the mAHP in CA1 is unaltered in these mice (Bond et al., 2004). An additional complication is the lack of subtype-selective tools targeting KCa2.1. In contrast to GW542573X, which is not very potent (EC50 ∼ 7 μM), both (−)-CM-TPMF and (−)-B-TPMF are high-potency KCa2.1 modulators (∼30 nM) that display acceptable selectivity over KCa2.2 and KCa2.3 and good to excellent selectivity over other ion channels and receptors. In particular, we foresee that the “package” of the two very close analogs with opposite effects on KCa2.1 may constitute a useful combination for many experimental situations: a detailed look at the concentration-response curves in Fig. 3 shows that (−)-CM-TPMF and (−)-B-TPMF can be applied up to 200 nM (causing 90% inhibition and 40% enhancement of KCa2.1) without significant functional activity on the other subtypes. (A note of caution: because of the Ca2+-dependent modulatory actions of both compounds, the corresponding percentages for natively expressed channels may deviate slightly). The reversibility properties of both compounds (Fig. 2) are also promising with respect to their use in a sequential manner on the same cell, at least in isolated neuronal preparations. We tentatively suggest that the opposite effects of (−)-CM-TPMF and (−)-B-TPMF on biological responses obtained from complex in vitro or in vivo biological systems—possibly combined with lack of effects of their respective (+)-enantiomers—would be a strong indication of the participation of the KCa2.1 isoform in that particular process.
Authorship Contributions
Participated in research design: Hougaard, Jensen, Strøbæk, and Christophersen.
Conducted experiments: Hougaard and Hammami.
Contributed new reagents or analytic tools: Eriksen, Sørensen, and Jensen.
Performed data analysis: Hougaard and Hammami.
Wrote or contributed to the writing of the manuscript: Hougaard, Eriksen, Sørensen, Jensen, Strøbæk, and Christophersen.
Acknowledgments
A very special thank you to Sofia Hammami for dedicated and highly significant contribution to the electrophysiological work conducted during a 4-week sabbatical at NeuroSearch A/S in January 2011. We also greatly appreciate the expert technical assistance provided by Lene Gylle Larsen (molecular biology), Susanne Kalf Hansen, Anne Stryhn Meincke, Vibeke Meyland-Smith, and Jette Sonne (electrophysiology and cell culturing), as well as Torben Skov (determination of chiral purity) and Tove Thomsen (optical rotation measurements). Drs. Heike Wulff and Gordon Munro are acknowledged for critical reading of the manuscript.
Footnotes
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
ABBREVIATIONS:
- KCNN
- small and intermediate conductance Ca2+-activated K+ channel
- CaM
- calmodulin
- CaMBD
- CaM binding domain
- CNS
- central nervous system
- 1-EBIO
- 1-ethyl-2-benzimidazolinone
- UCL1684
- 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)-diquinolinacyclodecaphane
- DCEBIO
- 5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one
- NS4591
- 4,5-dichloro-1,3-diethyl-2,3-dihydro-1H-1,3-benzodiazol-2-one
- SKA-31
- naphtho[1,2-d]thiazol-2-ylamine
- NS8593
- (R)-N-(benzimidazol-2-yl)-tetrahydro-1-naphtylamine
- NS309
- 6,7-dichloro-1H-indole-2,3-dione 3-oxime
- GW542573X
- 4-(2-methoxyphenylcarbamoyloxymethyl)-piperidine-1-carboxylic acid tert-butyl ester
- B-TPMF
- (N-{7-[1-(4-tert-butyl-phenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine
- CM-TPMF
- N-{7-[1-(4-chloro-2-methylphenoxy)ethyl]-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl}-N′-methoxy-formamidine
- 1-EBIO
- 1-ethyl-2-benzimidazolinone
- HEK
- human embryonic kidney
- I-V
- current-voltage
- Bay-k-8644
- S-(−)-1,4-dihydro-2,6-dimethyl-5-nitro-4-(2-[trifluoromethyl]phenyl)-3-pyridine carboxylic acid methyl ester
- KCa2
- small conductance Ca2+-activated K+ channel (SK channel)
- KCa3.1
- intermediate conductance Ca2+-activated K+ channel (IK channel).
- Received June 22, 2011.
- Accepted November 1, 2011.
- Copyright © 2012 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}