


Abstract
The chemokine receptor CCR2 is a G protein–coupled receptor that is activated primarily by the endogenous CC chemokine ligand 2 (CCL2). Many different small-molecule antagonists have been developed to inhibit this receptor, as it is involved in a variety of diseases characterized by chronic inflammation. Unfortunately, all these antagonists lack clinical efficacy, and therefore a better understanding of their mechanism of action is warranted. In this study, we examined the pharmacological properties of small-molecule CCR2 antagonists in radioligand binding and functional assays. Six structurally different antagonists were selected for this study, all of which displaced the endogenous agonist 125I-CCL2 from CCR2 with nanomolar affinity. Two of these antagonists, INCB3344 [N-(2-(((3S,4S)-1-((1r,4S)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide] and CCR2-RA, were radiolabeled to study the binding site in greater detail. We discovered that [3H]INCB3344 and [3H]CCR2-RA bind to distinct binding sites at CCR2, the latter being the first allosteric radioligand for CCR2. Besides the binding properties of the antagonists, we examined CCR2 inhibition in multiple functional assays, including a novel label-free whole-cell assay. INCB3344 competitively inhibited CCL2-induced G protein activation, whereas CCR2-RA showed a noncompetitive or allosteric mode of inhibition. These findings demonstrated that the CCR2 antagonists examined in this study can be classified into two groups with different binding sites and thereby different modes of inhibition. We have provided further insights in CCR2 antagonism, and these insights are important for the development of novel CCR2 inhibitors.
Introduction
The CC chemokine receptor 2 (CCR2) is a G protein–coupled receptor that can be activated by the endogenous CC chemokine ligand CCL2, CCL7, CCL8, CCL11, and CCL13. CCR2 is expressed on monocytes, dendritic cells, activated T lymphocytes, and basophils and plays an important role in the immune system (Luster, 1998; Fantuzzi et al., 1999; Jimenez et al., 2010). These immune cells migrate to increasing concentrations of chemokines at sites of inflammation as part of the immune response, also known as chemotaxis. Besides this important role in physiology, increased levels of CCR2 and its ligands can induce severe tissue damage, resulting in a variety of diseases, such as multiple sclerosis (Mahad and Ransohoff, 2003), atherosclerosis (Boring et al., 1998), rheumatoid arthritis (Quinones et al., 2005), and neuropathic pain (White et al., 2005), which makes CCR2 an attractive target for the pharmaceutical industry.
Chemokines are thought to bind to their receptors in a two-step manner. First, they interact with the extracellular side of the receptor, after which the N terminus of the chemokine can enter the interhelical binding pocket in the transmembrane (TM) domain to activate the receptor (Monteclaro and Charo, 1997; Pease et al., 1998). This binding pocket of chemokine receptors has been divided into a major binding pocket (TM helices 3, 4, 5, 6, and 7) and a minor binding pocket (TM helices 1, 2, 3, and 7) (Surgand et al., 2006). Small-molecule antagonists (∼600 Da) are at least 10-fold smaller than the endogenous chemokines (∼8600 Da), and therefore at best their binding sites can only partly overlap. For CCR2, several mutagenesis studies have provided evidence for the binding of small-molecule antagonists in the major and minor pocket (Mirzadegan et al., 2000; Berkhout et al., 2003; Hall et al., 2009). These ligands often contain a positively charged basic nitrogen that interacts with the conserved negatively charged glutamic acid residue (E291) in TM7, which is directly located between the major and minor binding pocket (Rosenkilde and Schwartz, 2006). In addition, several other CCR2 antagonists have been developed that do not possess such a basic nitrogen, and their binding site remains to be elucidated (Struthers and Pasternak, 2010). As there is growing evidence of multiple ligand binding sites for other chemokine receptors, we sought to determine whether CCR2 contains several binding sites as well.
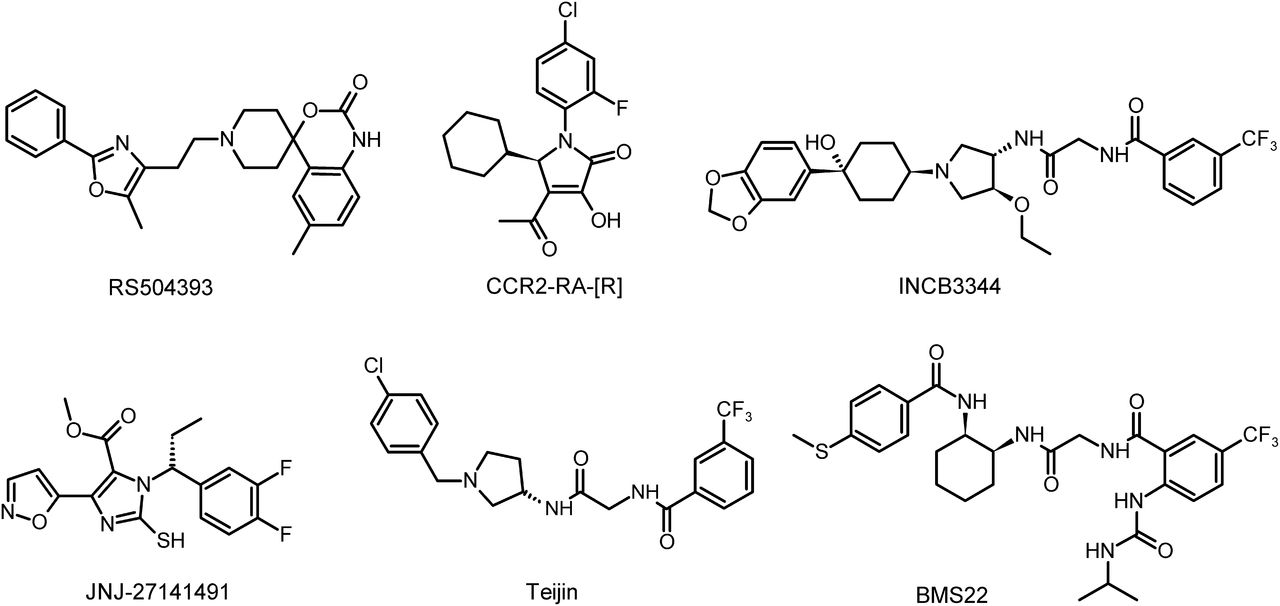
To gain a better understanding of the interaction of CCR2 and its ligands, we examined the binding sites and pharmacological properties of six chemically distinct CCR2 antagonists. These antagonists are RS504393 (6-methyl-1′-[2-(5-methyl-2-phenyl-4-oxazolyl)ethyl]-spiro[4H-3,1-benzoxazine-4,4′-piperidin]-2(1H)-one) (Mirzadegan et al., 2000), BMS22 (2-[(isopropylaminocarbonyl)amino]-N-[2-[[cis-2-[[4-(methylthio)benzoyl]amino]cyclohexyl]amino]-2-oxoethyl]-5-(trifluoromethyl)benzamide) (Cherney et al., 2008), Teijin compound 1 (N-[2-[[(3R)-1-[(4-chlorophenyl)methyl]-3-pyrrolidinyl]amino]-2-oxoethyl]-3-(trifluoromethyl)benzamide) (Moree et al., 2008), INCB3344 [N-(2-(((3S,4S)-1-((1r,4S)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide] (Brodmerkel et al., 2005), the (R) isomer CCR2-RA-[R] (Bhangoo et al., 2007), and JNJ-27141491 [(S)-3-[3,4-difluorophenyl)-propyl]-5-isoxazol-5-yl-2-thioxo-2,3-dihydro-1H-imidazole-4-carboxyl acid methyl ester] (Buntinx et al., 2008) (Fig. 1). In this study, for the first time, we provide evidence for two distinct binding sites of small-molecule antagonists at the CCR2 receptor. We also discuss the possible biased antagonism of some of the compounds. This work contributes to a better understanding of the nature of the interactions of diverse ligands at the CCR2 receptor.

Chemical structures of reference CCR2 small-molecule antagonists.
Materials and Methods
CCL2 was purchased from PeproTech (Rocky Hill, NJ), and the CCR2 antagonists BMS22 (Cherney et al., 2008), RS504393 (Mirzadegan et al., 2000), and Teijin compound 1 (Moree et al., 2008) were obtained from Tocris Bioscience (Bristol, UK). INCB3344, JNJ-27141491, and CCR2-RA-[R] were synthesized in-house as described previously (Xue et al., 2004; Brodmerkel et al., 2005; Zou et al., 2007; Doyon et al., 2008). [3H]INCB3344 (specific activity 32 Ci mmol−1) was custom-labeled by ViTrax (Placentia, CA), by means of direct titration of the parent ligand. Notably, INCB3344 was labeled as a racemic mixture of the two isomers N-(2-(((3S,4S)-1-((1r,4S)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide and N-(2-(((3R,4R)-1-((1r,4R)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide, but only the first isomer had sufficient affinity to label CCR2 in our experiments (see later discussion herein). The racemic radioligand [3H]CCR2-RA (specific activity 63 Ci mmol−1) was custom-labeled by ViTrax, for which a dehydrogenated precursor of CCR2-RA was provided. CCR2-RA was labeled as a racemic mixture of the two isomers (R)-4-acetyl-1-(4-chloro-2-fluorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one and (S)-4-acetyl-1-(4-chloro-2-fluorophenyl)-5-cyclohexyl-3-hydroxy-1,5-dihydro-2H-pyrrol-2-one. 125I-CCL2 (2200 Ci/mmol) and guanosine 5′-O-(3-[35S]thio)triphosphate ([35S]GTPγS) (1250 Ci/mmol) were purchased from PerkinElmer (Waltham, MA). Bovine serum albumin (fraction V) was purchased from Sigma-Aldrich (St. Louis, MO). Bicinchoninic acid (BCA) and BCA protein assay reagent were obtained from Pierce Chemical Company (Rockford, IL). Tango CCR2-bla U2OS cells stably expressing human CCR2 (U2OS-CCR2) were obtained from Invitrogen (Carlsbad, CA). All other chemicals were obtained from standard commercial sources.
Cell Cultures.
U2OS-CCR2 cells were cultured in McCoy’s 5a medium supplemented with 10% fetal calf serum, 2 mM glutamine, 0.1 mM nonessential amino acids, 25 mM HEPES, 1 mM sodium pyruvate, 100 IU/ml penicillin, 100 µg/ml streptomycin, 100 µg/ml G418, 50 µg/ml hygromycin, and 125 µg/ml zeocin in a humidified atmosphere at 37°C and 5% CO2. Cells were subcultured twice weekly at a ratio of 1:6 on 10-cm ø or 15-cm ø plates by trypsinization.
Cell Membrane Preparation.
Cells were detached from 15-cm ø plates by scraping into 5 ml of phosphate-buffered saline and subsequently centrifuged for 5 minutes at 3000g. The pellets were resuspended in ice-cold 50 mM Tris-HCl buffer and 5 mM MgCl2, pH 7.4, and homogenized with an Ultra Turrax homogenizer (IKA-Werke GmbH & Co. KG, Staufen, Germany). Membranes and the cytosolic fraction were separated by centrifugation at 31,000g in an Optima LE-80 K ultracentrifuge (Beckman Coulter, Inc., Fullerton, CA) at 4°C for 20 minutes. The pellet was resuspended in 10 ml of Tris-HCl buffer, and the homogenization and centrifugation steps were repeated. Finally, the membrane pellet was resuspended in 50 mM Tris-HCl buffer and 5 mM MgCl2, pH 7.4, and aliquots of 250 µl were stored at −80°C. Membrane protein concentrations were measured using a BCA protein determination (Smith et al., 1985).
125I-CCL2 Binding Assays.
Binding assays were performed in a 100-µl reaction volume containing 50 mM Tris-HCl buffer (pH 7.4), 5 mM MgCl2, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonic acid (CHAPS) and 15 μg of membrane protein at 37°C. For homologous competition experiments, increasing concentrations of CCL2 were incubated with 0.1 nM or 0.05 nM 125I-CCL2 for 150 minutes. At this concentration, total radioligand binding did not exceed 10% of the amount added to prevent ligand depletion. Nonspecific binding was determined with 10 μM INCB3344. Displacement assays were performed with 0.1 nM 125I-CCL2 using 10 concentrations of competing ligand for 150 minutes of incubation. Association experiments were performed with 0.1 nM 125I-CCL2 at different time intervals of incubation for 3 hours. Nonspecific binding was determined for every time point with 10 µM INCB3344. For dissociation experiments, the membranes were first incubated with 0.1 nM 125I-CCL2 for 2 hours. Dissociation was initiated by the addition of 10 µM INCB3344 at different time points. For all experiments, incubations were terminated by dilution with ice-cold 50 mM Tris-HCl buffer supplemented with 0.05% CHAPS and 0.5 M NaCl. Separation of bound from free radioligand was performed by rapid filtration through a 96-well GF/B filter plate precoated with 0.25% polyethyleneimine using a PerkinElmer Filtermate-harvester (PerkinElmer, Groningen, The Netherlands). Filters were washed 10 times with ice-cold wash buffer, and 25 μl of Microscint scintillation cocktail (PerkinElmer) was added to each well; the filter-bound radioactivity was determined by scintillation spectrometry using the P-E 1450 Microbeta Wallac Trilux scintillation counter (PerkinElmer).
[3H]INCB3344 Binding Assays.
Binding assays were performed in 100-µl reaction volume containing 50 mM Tris-HCl buffer (pH 7.4), 5 mM MgCl2, 0.1% CHAPS, and 10 µg of membrane protein at 25°C. Saturation experiments were carried out using nine different concentrations of radioligand from 0.5 to 45 nM for 120 minutes of incubation. Nonspecific binding was determined at three concentrations of radioligand with 10 μM BMS22. Displacement assays were carried out with 1.8 nM [3H]INCB3344 using 10 concentrations of competing ligand for 120 minutes of incubation. Association experiments were performed with 1.8 nM [3H]INCB3344 at different time intervals of incubation for 150 minutes. For dissociation experiments, the membranes were first incubated with 1.8 nM [3H]INCB3344 for 90 minutes. Dissociation was initiated by adding 10 µM of BMS22 at different time points. In all cases, total radioligand binding did not exceed 10% of the amount added to prevent ligand depletion. For all experiments, incubations were terminated by dilution with ice-cold 50 mM Tris-HCl buffer supplemented with 0.05% CHAPS. Separation of bound from free radioligand was performed as described in the section entitled “125I-CCL2 Binding Assays” using uncoated 96-well GF/B filter plates.
[3H]CCR2-RA Binding Assays.
Assay conditions were similar to those described for [3H]INCB3344 binding assays. Saturation experiments were carried out using 12 different concentrations of 0.1–75 nM radioligand for 120 minutes of incubation. Nonspecific binding was determined at three concentrations of radioligand with 10 μM JNJ-27141491. Displacement assays were carried out with 3 nM [3H]CCR2-RA using six concentrations of competing ligand for 120 minutes of incubation. Association experiments were performed with 3 nM [3H]CCR2-RA at different time intervals of incubation for 180 minutes. For dissociation experiments, the membranes were first incubated with 3 nM [3H]CCR2-RA for 90 minutes. Dissociation was initiated by the addition of 10 µM of JNJ-27141491 at different time points. In all cases, total radioligand binding did not exceed 10% of the amount added to prevent ligand depletion. All experiments were terminated as described in “[3H]INCB3344 Binding Assays.”
Tango β-Arrestin Recruitment Assay.
The assay was performed using the Tango CCR2-bla U2OS β-arrestin recruitment assay kit (Invitrogen), following the kit protocol. Tango CCR2-bla U2OS (U2OS-CCR2) cells were cultured in medium supplemented with 10% dialyzed fetal calf serum (Invitrogen) instead of normal serum for two passages prior to the assay. Briefly, cells were seeded at a density of 40,000 cells per well in Freestyle Expression Medium (Invitrogen) into a black-wall clear-bottom 96-well plate (PerkinElmer). Cells were stimulated with increasing concentrations of CCL2 and incubated for 16 hours at 37°C and 5% CO2. For antagonist assays, the cells were exposed to increasing concentrations of compound for 30 minutes before stimulation with an EC80 concentration (5 nM) of CCL2 for 16 hours at 37°C and 5% CO2. The final dimethylsulfoxide concentration was 0.1% for all assay points. After agonist exposure, the cells were loaded with 16 µl LiveBLAzer FRET B/G substrate (Invitrogen) for 2 hours at room temperature. After excitation at 400 nm, fluorescence emission values at 460 and 535 nm were measured in an EnVision multilabel plate reader (PerkinElmer). The ratio of the emission at 460 and 535 nm was calculated for each well.
Label-Free Whole-Cell Impedance Assay.
The xCELLigence RTCA system (Roche Applied Science, Mannheim, Germany) was used to perform whole-cell assays (Yu et al., 2006; Xi et al., 2008).
Tango CCR2-bla U2OS (U2OS-CCR2) cells were cultured in medium supplemented with 10% dialyzed fetal calf serum (Invitrogen) instead of normal serum for two passages before the assay. Initially, 50 µl of culture medium was added to wells in E-plates 96 to obtain background readings, followed by the addition of 50 µl of cell suspension containing 20,000 cells per well. The E-plate containing the cells was left at room temperature for 30 minutes before insertion into the xCELLigence station in the incubator at 37°C and 5% CO2. The cell index (CI) was monitored overnight every 15 minutes, during which time the cells grew to near confluence. After 16–18 hours, the cells were stimulated with increasing concentrations of CCL2. For the antagonist assays, cells were first preincubated for 30 minutes with increasing concentrations of antagonist or vehicle control that was added in 5 µl of compound solution (final concentration of 0.25% dimethylsulfoxide). Subsequently, cells were stimulated with an EC80 concentration (3 nM) of CCL2. Directly after stimulation, the measurement frequency was increased to 15-second intervals, followed by 30-second, 1-minute, and 5-minute intervals. For data analysis, the CI values were normalized to CI values before ligand addition, and baseline was corrected with CI traces obtained from vehicle control-treated cells.
[35S]GTPγS Binding Assay.
Ten micrograms of membranes were diluted in 100 µl of assay buffer containing 50 mM Tris-HCl buffer (pH 7.4), 5 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 0.05% bovine serum albumin, 10 µM GDP, and 10 µg of saponin per assay point. To determine the IC50 values of antagonists, the membranes were preincubated with varying concentrations of antagonist for 30 minutes at 25°C. Then CCL2 (10 nM) was added, followed by another incubation of 30 minutes; finally, the mixture was incubated for 90 minutes after the addition of [35S]GTPγS (0.3 nM). To determine the EC50 value of CCL2, the membranes were preincubated with varying concentrations of CCL2 in the absence (control) or presence of fixed concentrations of antagonist for 30 minutes at 25°C. Then [35S]GTPγS (0.3 nM) was added, and the mixture was incubated for 90 minutes. For all experiments, the incubation was terminated by dilution with ice-cold 50 mM Tris-HCl buffer supplemented with 5 mM MgCl2. Separation of bound from free radioligand was performed as described under “125I-CCL2 Binding Assays” using uncoated 96-well GF/B filter plates.
Data Analysis.
All experiments were analyzed using the nonlinear regression curve fitting program Prism 5 (GraphPad, San Diego, CA). The KD values of [3H]INCB3344 and [3H]CCR2-RA were obtained by computer analysis of saturation curves according to the equation bound = (Bmax*[L])/([L] + KD), where Bmax is the maximal number of binding sites (pmol/mg) and KD is the concentration of radioligand required to reach half-maximal binding. The KD value of 125I-CCL2 was calculated from homologous competition experiments using the Cheng-Prusoff equation, assuming that unlabeled and labeled CCL2 had identical affinities (Cheng and Prusoff, 1973). The dissociation rate constant (koff) was obtained by computer analysis of the exponential decay of radioligand binding to the receptor. Association rate constants (kon) were calculated according to the equation kon = (kobs − koff)/[L], where kobs , the observed rate constant to approach equilibrium, was obtained by computer analysis of the exponential association, and [L] is the amount of radioligand used for the association experiments. All experiments were fit according to monophasic equations except for dissociation of [3H]CCR2-RA, which occurred in a biphasic manner. From radioligand displacement data, Ki values were calculated from IC50 values using the Cheng-Prusoff equation (Cheng and Prusoff, 1973). [35S]GTPγS, β-arrestin recruitment and xCELLigence curves were analyzed by nonlinear regression to obtain IC50 or EC50 values, where xCELLigence peak responses were obtained within 5 minutes after the addition of compound using the RTCA software 1.2 (ACEA Biosciences, Inc., San Diego, CA). Data shown are the mean ± S.E.M. of at least three separate experiments performed in duplicate. Statistical analysis was performed with a two-tailed unpaired Student’s t test. Comparison of the means of multiple data sets was performed by one-way analysis of variance, followed by a Tukey’s multiple comparison test.
Results
Radioligand Binding Assays
Characterization of 125I-CCL2.
In this study, we performed radioligand binding assays for CCR2 with the labeled endogenous agonist 125I-CCL2 as a tracer ligand. To determine the affinity of 125I-CCL2 for CCR2, homologous displacement assays were performed on membranes of U2OS cells stably expressing CCR2 (U2OS-CCR2). This resulted in a KD of 0.068 ± 0.014 nM and a Bmax of 0.31 ± 0.03 pmol/mg (Fig. 2A; Table 1). Kinetic association and dissociation experiments were performed to determine the rate constants kon and koff (Fig. 2B). Both the association and dissociation of 125I-CCL2 were best fit by monophasic curves, yielding a kon of 0.29 ± 0.06 nM−1 min−1 and a koff of 0.033 ± 0.003 min−1. The calculated kinetic KD value (koff/kon) of 0.12 nM thus agreed fairly well with the affinity obtained in the homologous competition assay.

Characterization of 125I-CCL2 binding to membranes of U2OS cells stably expressing CCR2. (A) Homologous competition assay of 0.1 and 0.05 nM 125I-CCL2 in the presence of increasing concentrations of unlabeled CCL2. Nonspecific binding was determined in the presence of 10 µM INCB3344. (B) Association and dissociation kinetics of 0.1 nM 125I-CCL2 binding to CCR2 at 37°C. Dissociation was initiated by the addition of 10 μM INCB3344. Association and dissociation rate constants were 0.29 ± 0.06 nM−1 min−1 and 0.033 ± 0.003 min−1, respectively. Data were best fitted using a one-phase association and one-phase exponential decay function. For all experiments, a representative graph of one experiment performed in duplicate is shown (see Table 1 for KD, Bmax values).
Equilibrium binding parameters of 125I-CCL2, [3H]INCB3344, and [3H]CCR2-RA, determined on U2OS membranes expressing CCR2
Data are presented as mean ± S.E.M. of three experiments performed in duplicate.
Displacement of 125I-CCL2 from CCR2.
A panel of reference CCR2 antagonists (Fig. 1) was selected for analysis in radioligand displacement assays. U2OS-CCR2 membranes were incubated with 125I-CCL2 and increasing concentrations of antagonist (Fig. 3A). All antagonists fully displaced 125I-CCL2 from the CCR2 receptor, with IC50 values reported in Table 2. INCB3344 had the highest affinity for CCR2 (IC50 = 5.4 nM), followed by BMS22 (IC50 = 27 nM), CCR2-RA-[R] (IC50 = 103 nM), RS504393 (IC50 = 132 nM), JNJ-27141491 (IC50 = 172 nM), and Teijin compound 1 (IC50 = 220 nM).

Displacement of (A) 125I-CCL2 binding, (B) [3H]INCB3344 binding, and (C) [3H]CCR2-RA binding to U2OS membranes stably expressing CCR2 by increasing concentrations of CCL2 and six reference CCR2 antagonists. Results are presented as percentage of bound radioligand for one representative experiment performed in duplicate (see Table 2 for affinity values).
Displacement of 125I-CCL2, [3H]INCB3344, and [3H]CCR2-RA from U2OS membranes expressing CCR2
Data are presented as mean ± S.E.M. of at least three experiments performed in duplicate.
The antagonists INCB3344, BMS22, RS504393, and Teijin share structural similarities, whereas JNJ-27141491 and CCR2-RA-[R] form a different set of molecules (Fig. 1). We decided to tritium-label the two high-affinity racemic compounds INCB3344 and CCR2-RA within the two classes, for which we used two unsaturated precursor molecules that we synthesized in-house. We anticipated that these two molecules would help us to characterize in more detailed the binding sites of low-molecular-weight antagonists targeting CCR2.
Characterization of [3H]INCB3344.
Tritium-labeled INCB3344 is a racemic mixture of two isomers. In 125I-CCL2 displacement experiments with the isolated unlabeled isomers, we had found that the (3S,4S) isomer had an IC50 of 3.8 ± 0.5 nM, whereas the affinity of the other isomer was at least 1000-fold lower (IC50 = 4.3 ± 0.9 µM). Since we used low nanomolar concentrations (1.8 nM) of [3H]INCB3344 in the binding studies, we concluded that CCR2 was solely labeled by the high affinity (3S,4S) isomer.
Saturation binding experiments yielded a KD of 0.90 ± 0.03 nM with a Bmax of 7.1 ± 0.2 pmol/mg (Fig. 4; Table 1). Equilibrium binding of [3H]INCB3344 to U2OS-CCR2 membranes was reached within 1 hour at 25°C as assessed with kinetic association experiments (Fig. 4B). This radioligand was specifically bound to CCR2 as no binding was detected in U2OS membranes that do not express CCR2 (data not shown). Dissociation of [3H]INCB3344 was initiated by 10 μM BMS22 and resulted in a dissociation half-life of 53 minutes at 25°C (Fig. 4B). Both kinetic studies were best fit by monophasic curves, which confirmed that CCR2 was labeled by only one of the isomers. From these data, we calculated the association and dissociation rate constants, 0.054 ± 0.002 nM−1 min−1 and 0.013 ± 0.002 min−1, respectively, which resulted in a kinetic KD of 0.23 nM, in fair agreement with the equilibrium value of 0.90 nM.
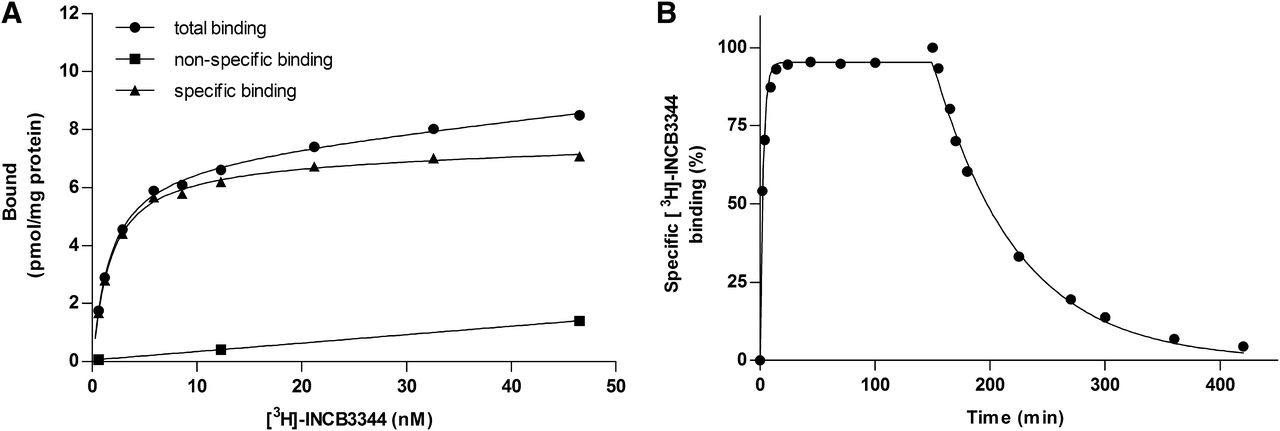
Characterization of [3H]INCB3344 binding to membranes of U2OS cells stably expressing CCR2. (A) Saturation binding of [3H]INCB3344 to CCR2. Different concentrations of [3H]INCB3344 were incubated in the presence (nonspecific binding) or absence (total binding) of 10 μM BMS22. Specific binding was determined by subtracting the nonspecific binding from the total binding. (B) Association and dissociation kinetics of 1.8 nM [3H]INCB3344 to CCR2 at 25°C. Dissociation was initiated by the addition of 10 μM BMS22. Association and dissociation rate constants were 0.054 ± 0.002 nM−1 min−1 and 0.013 ± 0.002 min−1, respectively. Data were best fitted using a one-phase association and one-phase exponential decay function. For all experiments, a representative graph of one experiment performed in duplicate is shown (see Table 1 for KD, Bmax values).
Displacement of [3H]INCB3344 from CCR2.
Homologous displacement by INCB3344 yielded a Ki of 1.2 ± 0.2 nM (Fig. 3B; Table 2), which corresponded to the KD that was obtained from saturation binding experiments. CCL2 displaced only 21% of bound [3H]INCB3344 from the receptor. The antagonists BMS22, RS504393, and Teijin were able to displace fully [3H]INCB3344 from CCR2 with nanomolar affinities, whereas JNJ-27141491 and CCR2-RA-[R] did not displace [3H]INCB3344, which indicates that they bind at different sites at the CCR2 receptor. At a concentration of 1 μM, JNJ-27141491 and CCR2-RA-[R] rather significantly increased [3H]INCB3344 binding, with 12 and 9% (Student’s t test P < 0.01; Table 2), respectively.
Characterization of [3H]CCR2-RA.
CCR2-RA was also tritium-labeled as a racemic mixture of two isomers. In 125I-CCL2 displacement experiments, the unlabeled (R) and (S) isomers had IC50 values of 103 ± 18 and 216 ± 21 nM, respectively. We therefore cannot exclude that CCR2 was labeled to some extent by the lower-affinity isomer, too, although we took care to use a low concentration of 3 nM in the displacement assays. Saturation binding experiments yielded a KD of 5.8 ± 0.2 nM with a Bmax of 9.7 ± 0.2 pmol/mg (Fig. 5A; Table 1). Equilibrium binding of [3H]CCR2-RA to U2OS-CCR2 membranes was reached within 2 hours at 25°C as assessed with kinetic association experiments after a monophasic fit (Fig. 5B). This radioligand was specifically bound to CCR2 as no binding was detected in U2OS membranes that do not express CCR2 (data not shown). Dissociation of [3H]CCR2-RA was initiated by 10 μM of JNJ-27141491 and resulted in a biphasic dissociation pattern with koff1 and koff2 of 0.24 ± 0.02 min−1 and 0.029 ± 0.005 min−1, respectively (Fig. 5C).

Characterization of [3H]CCR2-RA binding to membranes of U2OS cells stably expressing CCR2. (A) Saturation binding of [3H]CCR2-RA to CCR2. Different concentrations of [3H]CCR2-RA were incubated in the presence (nonspecific binding) or absence (total binding) of 10 μM JNJ-27141491. Specific binding was determined by subtracting the nonspecific binding from the total binding. (B) Association kinetics of 3 nM [3H]CCR2-RA to CCR2 at 25°C. Data were best fitted using a one-phase association function resulting in a kobs of 0.089 ± 0.005 min−1. (C) Dissociation kinetics of 3 nM [3H]CCR2-RA to CCR2 at 25°C. Dissociation was initiated by the addition of 10 μM JNJ-27141491. Data were best fitted using a two-phase exponential decay function, resulting in a koff1 of 0.24 ± 0.02 min−1 and a koff2 of 0.029 ± 0.005 min−1. For all experiments, a representative graph of one experiment performed in duplicate is shown (see Table 1 for KD, Bmax values).
Displacement of [3H]CCR2-RA from CCR2.
CCL2 displaced only 23% of bound [3H]CCR2-RA from the receptor (Fig. 3C; Table 2). The antagonists JNJ-27141491 and the unlabeled (R) isomer CCR2-RA-[R] were able to displace fully [3H]CCR2-RA from CCR2 with nanomolar affinities, whereas antagonists INCB3344, BMS22, RS504393, and Teijin did not displace [3H]CCR2-RA, suggesting that they bind at a different site at the CCR2 receptor. In contrast, INCB3344, BMS22, RS504393, and Teijin significantly increased [3H]CCR2-RA binding with 69, 64, 57, and 59% at a concentration of 1 µM, respectively (Student’s t test P < 0.05; Table 2).
Functional Assays
In addition to radioligand binding experiments, we performed a number of functional assays, both G protein–dependent and –independent.
Inhibition of β-Arrestin Recruitment to CCR2.
We first performed β-arrestin recruitment assays (G protein–independent) to assess the inhibitory potency of all six antagonists. CCL2 induced β-arrestin recruitment to CCR2 with an EC50 of 1.4 ± 0.4 nM (n = 4) (Fig. 6A). All antagonists were able to inhibit β-arrestin recruitment induced by 5 nM CCL2 (Fig. 6B; Table 3). Interestingly, JNJ-27141491 and CCR2-RA-[R] were more potent inhibitors of β-arrestin recruitment than were Teijin and RS504393, whereas these antagonists showed equal affinities in the 125I-CCL2 displacement assay.

(A) CCL2-induced β-arrestin recruitment to U2OS cells stably expressing CCR2. (B) Inhibition of CCL2-induced β-arrestin recruitment to CCR2. Cells were incubated with a concentration of CCL2 that evoked 80% of the maximum response (5 nM). Increasing concentrations of the antagonists were added to determine their IC50 value. For all experiments, basal activity was set at 0%, and the maximum response at 100%. Results are presented for one representative experiment performed in duplicate (see Table 3 for IC50 values).
Inhibition of CCL2-induced cellular responses measured in [35S]GTPγS membrane binding assays as well as β-arrestin recruitment and xCELLigence label-free whole-cell assays
Data are presented as mean ± S.E.M. of three experiments performed in duplicate.
Analysis of CCR2 Inhibition in an Impedance-Based, Label-Free Whole-Cell Assay.
The xCELLigence RTCA system was used to study the activation and inhibition of CCR2 in a label-free whole-cell assay. Typically, the addition of CCL2 resulted in an immediate dose-dependent increase in the CI to a peak level within 5 minutes, followed by a second peak after approximately 20 minutes, which then returned to baseline after 1 hour of incubation (Fig. 7A). Concentration-effect curves were obtained by analysis of the peak level that appeared within 5 minutes after stimulation and resulted in an EC50 value of 1.1 ± 0.1 nM for CCL2 (n = 5) (Fig. 7B). Preincubation with all antagonists resulted in inhibition of the CCL2-induced response (Fig. 7C; Table 3). The antagonists were equally potent as inhibitors of CCL2-induced impedance effects as they were able to inhibit β-arrestin recruitment, except for Teijin and CCR2-RA-[R], which were less potent as inhibitors of the impedance response with IC50 values of 292 ± 66 nM and 64 ± 14 nM, respectively.

Impedance measurements in the xCELLigence label-free assay for U2OS cells stably expressing CCR2. (A) Representative graph of baseline-corrected normalized CI after the addition of different concentrations of CCL2. (B) Concentration-response curve for CCL2 derived from peak-height analysis of CI changes for the 5-minute interval after application. (C) Inhibition of CCL2-induced impedance measurements. Cells were incubated with a concentration of CCL2 that evoked 80% of the maximum response (3 nM). Increasing concentrations of the antagonists were added 30 minutes before agonist stimulation to determine their IC50 value, derived from peak-height analysis of CI changes for the 5-minute interval after the application of CCL2. Results are presented for one representative experiment performed in duplicate (see Table 3 for IC50 values).
[35S]GTPγS Binding to CCR2.
We also performed a G protein–dependent functional assay. For this purpose, we used a [35S]GTPγS binding assay on U2OS-CCR2 membranes, where we measured G protein activation by CCL2 in the absence or presence of different antagonists (Fig. 8). CCL2 stimulated [35S]GTPγS binding with an EC50 value of 5.7 ± 0.9 nM (n = 8; see also, Fig. 8, B and C). All antagonists were able to inhibit CCL2-induced G protein activation (Fig. 8A; Table 3). Notably, RS504393 and JNJ-27141491 were more potent inhibitors of G protein activation than of β-arrestin recruitment or the impedance response evoked by CCL2, their IC50 values being 19 ± 7 nM and 3.9 ± 1.0 nM, respectively.

[35S]GTPγS binding to membranes of U2OS cells stably expressing CCR2. (A) Inhibition of CCL2-induced [35S]GTPγS binding by six small-molecule antagonists. Membranes were incubated with a concentration of CCL2 that evoked 80% of the maximum response (10 nM). Increasing concentrations of the antagonists were added to determine their IC50 value. (B andC) Concentration-effect curves of CCL2-induced [35S]GTPγS binding. Increasing concentrations of CCL2 were added simultaneously with indicated concentrations of INCB3344 (B) or CCR2-RA-[R] (C). For all experiments, the basal activity was set at 0% and the maximum response at 100%. Results are presented as mean percentage ± S.E.M. of three experiments performed in duplicate (see Table 4 for pEC50 and Emax values).
Based on the displacement data obtained with [3H]INCB3344 and [3H]CCR2-RA, INCB3344 and CCR2-RA-[R] were selected as representative antagonists for the two putative distinct binding sites. To examine their mechanism of inhibition in more detail, we analyzed the shift of CCL2-induced [35S]GTPγS binding in the presence of fixed concentrations of antagonist. The maximal effect of CCL2 in the presence of CCR2-RA-[R] was significantly reduced, whereas CCL2’s potency was slightly, but similarly, decreased at all three antagonist concentrations (Fig. 8C; Table 4). In contrast, the addition of INCB3344 markedly decreased the EC50 value of CCL2, whereas the efficacy of CCL2 was unaffected (Fig. 8B; Table 4). A Schild plot analysis of the INCB3344 data yielded a KB value of 0.4 nM, which is in close agreement with the KD value of 0.9 nM determined in the saturation binding assay. Overall, these data indicate that INCB3344 behaved as a competitive antagonist, whereas CCR2-RA-[R] clearly showed noncompetitive antagonism for CCR2 with respect to CCL2, as indicated by a decrease in CCL2’s efficacy in the presence of increasing concentrations of CCR2-RA-[R].
G protein activation by CCL2 measured by [35S]GTPγS binding
Potency and maximum effect of CCL2 in the absence or presence of different concentrations of CCR2-RA-[R]. Data are presented as mean ± S.E.M. of at least three experiments
Discussion
Previous studies reported CCR2 and its endogenous ligand CCL2 as major players in a variety of diseases (Boring et al., 1998; Mahad and Ransohoff, 2003; White et al., 2005). Given this potential role as a drug target, the pharmaceutical industry has developed many CCR2 antagonists (Struthers and Pasternak, 2010), although without much clinical success. Given the variety in chemical scaffolds, it is surprising that little attention, if any, has been paid to the binding mode of these ligands to the receptor. Therefore, the present study was designed to compare the binding site and mechanism of inhibition for a selection of these antagonists.
To study the binding of these antagonists to CCR2, we used the radioligand 125I-CCL2 and the custom-labeled small-molecule radioligands [3H]INCB3344 and [3H]CCR2-RA. We found an affinity constant of 0.068 nM for 125I-CCL2, which corresponds to reported data in literature (KD 0.05 nM) (Samson et al., 1997; Springael et al., 2006). In addition, the dissociation rate constant of 125I-CCL2 from U2OS-CCR2 membranes of 0.033 min−1 is in agreement with the previously reported koff of 0.036 min−1 (Springael et al., 2006). [3H]INCB3344 was previously published and characterized as a radioligand for CCR2 (Shin et al., 2009). In those studies a KD value of 5 nM was reported for human embryonic kidney 293 cells stably expressing CCR2. Our experiments with U2OS-CCR2 cells resulted in a 5-fold lower KD value of 0.9 nM. This difference could be the result of the different cell type, different assay buffer, or the longer incubation time used in our assay.
In this study, [3H]CCR2-RA is reported as the first allosteric radioligand for CCR2. It was characterized as a high-affinity radioligand (KD = 5.8 nM) that reversibly binds to the receptor. The biphasic dissociation of [3H]CCR2-RA from CCR2 could be a result of the racemic nature of the radioligand. The Bmax values for [3H]CCR2-RA and [3H]INCB3344 were 31- and 23-fold higher than for 125I-CCL2. A possible explanation might be that some of the receptors present in vesicular structures in the membrane preparations are accessible to membrane-permeable molecules, such as CCR2-RA and INCB3344, but not to CCL2. In addition, allosteric interactions described within chemokine receptor oligomers might modify the apparent number of binding sites for some radioligands (El-Asmar et al., 2005; Sohy et al., 2007). Finally, antagonists like CCR2-RA and INCB3344 supposedly bind to the G protein–coupled state as well as the uncoupled state of the receptor, whereas the agonist CCL2 presumably binds only to the G protein–coupled state (i.e., labeling a smaller population of receptors). This was confirmed by experiments in the presence of GTP, which uncouples the receptor from its G protein, since these resulted in a complete loss of binding of 125I-CCL2, whereas binding of [3H]INCB3344 and [3H]CCR2-RA was not affected (data not shown). Similar extensive differences in Bmax values between radiolabeled agonist and antagonist have previously been reported for the CXCR2 chemokine receptor (de Kruijf et al., 2009). In any case, the three radioligands do not label the same receptor populations, which may therefore contribute to the different IC50 values derived from competition experiments performed with either CCL2 or a small molecule as a radioligand.
INCB3344 and CCR2-RA-[R] were able to displace fully 125I-CCL2 receptor binding, whereas CCL2 was capable only of displacing 21% of [3H]INCB3344 binding and 19% of [3H]CCR2-RA binding. This finding could be explained by the heterogeneity of binding sites and the G protein–uncoupling of a fraction of CCR2. In addition, the incomplete displacement could suggest that CCL2, INCB3344, and CCR2-RA bind to different sites at CCR2. Given the size of INCB3344 (578 Da) and CCR2-RA (352 Da), these ligands can at best only partly overlap with the binding site of the large peptide CCL2 (8600 Da).
The three different radioligands were used to study and compare six structurally different CCR2 antagonists. All antagonists displaced 125I-CCL2 from the receptor (Fig. 3A; Table 2) with affinities similar to previously reported data (Mirzadegan et al., 2000; Brodmerkel et al., 2005; Zou et al., 2007; Buntinx et al., 2008; Cherney et al., 2008; Moree et al., 2008). BMS22, RS504393, and Teijin were also able to displace [3H]INCB3344 binding, which indicated that these four ligands share a common binding site. On the contrary, JNJ-27141491 and CCR2-RA-[R] did not displace [3H]INCB3344 from CCR2 (Fig. 3B). Analogous results were found in the [3H]CCR2-RA displacement assay, where INCB3344, BMS22, RS504393, and Teijin did not displace [3H]CCR2-RA from CCR2 (Fig. 3C). Therefore, we conclude that there are at least two different binding sites for small-molecule antagonists at CCR2. Notably, at high concentrations, JNJ-27141491 and CCR2-RA-[R] significantly increased [3H]INCB3344 binding to CCR2, whereas INCB3344, BMS22, RS504393, and Teijin significantly increased [3H]CCR2-RA binding to CCR2 (Table 2). This behavior is indicative for allosteric enhancement, best explained by the two compounds stabilizing the same conformation of the receptor by binding at two topographically different sites (Lazareno and Birdsall, 1995).
For RS504393, Teijin and BMS22 mutagenesis studies have shown that these molecules bind to the major or minor binding pocket of CCR2. RS504393 and Teijin interact with the highly conserved glutamic acid residue E291, most likely via their basic nitrogen atom (Mirzadegan et al., 2000; Hall et al., 2009). For BMS22, the adjacent T292 was found to be important for binding (Cherney et al., 2008), indicating that it shares the same binding pocket as RS504393 and Teijin. Notably, for INCB3344, no such data have been reported yet, and here we established that it binds to the same site as RS504393, Teijin, and BMS22. The presence of a basic nitrogen in the pyrrolidine ring of INCB3344 suggests a similar interaction with E291.
The structures of JNJ-27141491 and CCR2-RA-[R] contain different chemical features compared with the other antagonists. JNJ-27141491 and CCR2-RA-[R] lack a basic nitrogen, have a lower molecular weight, and are acidic. Their exact binding site or sites at CCR2 remain to be determined. For several other chemokine receptors, the presence of an allosteric binding site has been reported (Scholten et al., 2012). Whereas some antagonists interact with both the major and minor binding pocket in the transmembrane region, others bind exclusively to either one of these sites. Given the large size of this binding pocket, the two different binding sites that we have identified for CCR2 could both be located in this transmembrane region. In addition, an allosteric binding site on the intracellular side of the receptor in the C-terminal domain has been identified for the chemokine receptors CXCR2, CCR4, and CCR5 (Andrews et al., 2008; Nicholls et al., 2008; Salchow et al., 2010). This binding site resides close to the site of G protein–coupling to the receptor, and therefore it is assumed that activation of the G protein is prevented in the presence of an antagonist at this site. The intracellular antagonists of CXCR2 contain an acidic center (Salchow et al., 2010), which is also present in JNJ-27141491 and CCR2-RA-[R]. In addition, CCR2 and CCR5 are closely related based on sequence similarity. Hence, the presence of such an intracellular binding site for CCR2 is not unlikely.
By means of functional assays, we confirmed that the six compounds described in this article are indeed CCR2 antagonists, at the G protein level, in β-arrestin recruitment and in a novel label-free impedance-based functional assay (Table 3). Analysis of these differential functional responses allowed us to explore whether these ligands show biased antagonism, which has been described for allosteric ligands of other G protein–coupled receptors (Kenakin and Miller, 2010; Magnan et al., 2013). Notably, it has been reported that some CCR2 antagonists are capable of discriminating between different functional states of the receptor (Kredel et al., 2011). In our hands, RS504393 and JNJ-27141491 were slightly more potent inhibitors of G protein activation, whereas Teijin was most potent in the β-arrestin recruitment assay. Except for these small but significant differences, all antagonists were equally potent among the different functional assays, and as such there is little indication of biased antagonism in our assays. As the effect in functional assays is dependent on the off-rate of the antagonists, which are at present unknown, we realize that it is difficult to compare the three functional assays as their incubation times varies from minutes to hours.
We next determined the mechanism of inhibition in a [35S]GTPγS assay for INCB3344 and CCR2-RA-[R] as representative compounds binding to different binding sites. INCB3344 behaved as a competitive antagonist, whereas CCR2-RA-[R] showed noncompetitive antagonism for CCR2 with respect to CCL2 (Fig. 7). Agonist stimulation after preincubation with an antagonist can result in submaximal receptor stimulation if the antagonist is not sufficiently dissociated to liberate the entire population at the time at which the maximal response is measured (Vauquelin et al., 2002). Therefore, we coincubated increasing concentrations of CCL2 in the presence of fixed amounts of antagonist to rule out insurmountable antagonism resulting from slow dissociation kinetics. The mechanism of INCB3344 inhibition was previously addressed in a calcium flux assay in human monocytes and a competition binding assay using 125I-CCL2 (Shin et al., 2009). Both experiments showed a competitive mode of inhibition with respect to CCL2. These results are in good agreement with our competitive profile of INCB3344 in the [35S]GTPγS assay.
For CCR2-RA-[R], no detailed pharmacological data were previously published except for its inhibition of pain behavior in an in vivo model with nerve-injured rats (Bhangoo et al., 2007). We now provide evidence for a noncompetitive mode of inhibition of CCR2. Based on the results of our binding studies, we assume that CCR2-RA-[R] and JNJ-27141491 bind to a similar site at CCR2 or at least bind to a site that is distinct from the INCB3344′s binding pocket. Since JNJ-27141491 was previously described as a noncompetitive antagonist of CCR2, it is likely that CCR2-RA-[R] and JNJ-27141491 bind and inhibit CCR2 via a similar mechanism (Buntinx et al., 2008).
In summary, we have demonstrated that the CCR2 antagonists examined in this study can be classified into at least two groups with a different binding site and thereby a different mode of inhibition. Hence, we have provided further insights into CCR2 antagonism, which may be relevant for the development of novel CCR2 inhibitors.
Authorship Contributions
Participated in research design: Zweemer, Smit, Stamos, Saunders, IJzerman, Heitman.
Conducted experiments: Zweemer, Nederpelt, Doornbos, Hafith, de Vries, Vrieling.
Contributed new reagents or analytic tools: Abt, Gross.
Performed data analysis: Zweemer, Nederpelt, Doornbos, Hafith, de Vries, Vrieling.
Wrote or contributed to the writing of the manuscript: Zweemer, Smit, IJzerman, Heitman
Footnotes
- Received April 16, 2013.
- Accepted July 22, 2013.
This study was performed and financially supported within the framework of Top Institute Pharma, project number D1-301.
Abbreviations
- BCA
- bicinchoninic acid
- BMS22
- 2-[(isopropylaminocarbonyl)amino]-N-[2-[[cis-2-[[4-(methylthio)benzoyl]amino]cyclohexyl]amino]-2-oxoethyl]-5-(trifluoromethyl)benzamide
- CCL2
- CC chemokine ligand 2
- CCR2
- CC chemokine receptor 2
- CCR2-RA
- CC chemokine receptor 2 antagonist
- CHAPS
- 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonic acid
- CI
- cell index
- [35S]GTPγS
- guanosine 5′-O-(3-[35S]thio)triphosphate
- INCB3344
- N-(2-(((3S,4S)-1-((1r,4S)-4-(benzo[d][1,3]dioxol-5-yl)-4-hydroxycyclohexyl)-4-ethoxypyrrolidin-3-yl)amino)-2-oxoethyl)-3-(trifluoromethyl)benzamide
- JNJ-27141491
- (S)-3-[3,4-difluorophenyl)-propyl]-5-isoxazol-5-yl-2-thioxo-2,3-dihydro-1H-imidazole-4-carboxyl acid methyl ester
- kobs
- the observed rate constant to approach equilibrium
- koff
- dissociation rate constant
- kon
- association rate constant
- RS504393
- 6-methyl-1′-[2-(5-methyl-2-phenyl-4-oxazolyl)ethyl]-spiro[4H-3,1-benzoxazine-4,4′-piperidin]-2(1H)-one
- Teijin compound 1
- N-[2-[[(3R)-1-[(4-chlorophenyl)methyl]-3-pyrrolidinyl]amino]-2-oxoethyl]-3-(trifluoromethyl)benzamide
- TM
- transmembrane
- U2OS-CCR2
- human osteosarcoma cells stably expressing CCR2
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}