


Abstract
Ranolazine is an approved drug for chronic stable angina that acts by suppressing a noninactivating current conducted by the cardiac sodium channel [persistent sodium ion current (INa)]. Ranolazine has also been shown to inhibit the increased persistent INa carried by NaV1.1 channels encoding epilepsy- and migraine-associated mutations. Here, we investigate the antiepileptic properties of ranolazine exhibited through the reduction of hippocampal neuronal excitability. At therapeutically relevant concentrations, ranolazine reduced action potential firing frequency of hippocampal neurons in response to repetitive depolarizing current injections. Similarly, using a single current injection paradigm, ranolazine required a long depolarization (4 seconds) to produce significant inhibition of excitability, which was similar to that observed for the anticonvulsants phenytoin (slowly binds to the fast-inactivated state) and lacosamide (binds to the slow-inactivated state). Ranolazine enhanced the development of fast and slow inactivation assessed with conditioning prepulses of 100, 1000, or 10,000 milliseconds. Recovery of channels from inactivated states was also slowed in the presence of ranolazine. Interestingly, the use-dependent inhibition of hippocampal neurons was dependent on the duration of the voltage step, suggesting ranolazine does not selectively affect the open state and may also interact with inactivated states. NEURON (Yale University, New Haven, CT) computational simulations predict equal inhibition of action potential generation for binding to either fast-inactivated or slow-inactivated states. Binding of ranolazine to either preopen or open states did not affect the excitability of the simulation. Ranolazine was able to significantly reduce the epileptiform activity of the neuronal cultures, suggesting possible antiepileptic activity.
Introduction
Epilepsy is a common neurologic disorder characterized by abnormal neuronal synchronization (Rogawski and Loscher, 2004). In several epileptic syndromes, the local network synchrony spreads to include one or both hemispheres in a process termed secondary generalization. In the most severe form generalization produces a life-threatening, nonterminating seizure (generalized tonic-clonic seizure). Current antiepileptic therapeutics targeting neuronal voltage-gated sodium (NaV) channels selectively reduce Na+ current (INa) during periods of high frequency firing (Rogawski and Loscher, 2004). Drug binding is thought to stabilize the Na+ channel–inactivation process, which serves to dampen neuronal activity and network synchronization, while leaving normal neuronal functioning largely intact. Central nervous system voltage-gated Na+ channels are responsible for the generation and propagation of neuronal action potentials. Several genetic epilepsies are caused by mutation of sodium channel genes SCN1A and SCN2A, which encode the pore-forming α-subunits of the NaV1.1 and NaV1.2 isoforms, respectively (Catterall et al., 2008).
Ranolazine is an antianginal drug that has been shown to inhibit cardiac persistent INa at a therapeutic concentration of 2–8 μM (Antzelevitch et al., 2004; Chaitman, 2006). Furthermore, ranolazine has been shown to preferentially block the persistent INa evoked by mutations in NaV1.5 (Fredj et al., 2006; Rajamani et al., 2009), as well as toxin or mutation-induced persistent INa carried by muscle [NaV1.4 (Wang et al., 2008)] and peripheral nerve [NaV1.7 and NaV1.8 (Rajamani et al., 2008; Wang et al., 2008)] Na+ channels. More recently, it was demonstrated that at an achievable brain concentration (∼1 μM), ranolazine blocked the increased ramp and persistent INa carried by epilepsy and migraine-associated NaV1.1 mutations more potently than the observed inhibition of transient (peak) INa (Kahlig et al., 2010a). However the ability of ranolazine to exert antiepileptic actions has not been reported.
Neuronal network excitability is achieved through a balance between transient (peak) INa and persistent INa. Transient INa generates the upstroke of the action potential and supports signal propagation. Although elevated levels of persistent INa are associated with disease causing Na+-channel mutations, recent work has confirmed that a small fraction of total INa is normally conducted through a persistent mechanism. This depolarizing persistent INa facilitates the integration of synaptic inputs and supports repetitive firing capabilities. Moreover, the normal balance between transient and persistent INa is activity-dependent, with periods of high stimulation frequency (e.g., seizure) increasing the proportion of persistent INa.
The consensus mechanism of action of typical Na+ channel–targeting antiepileptic drugs, such as phenytoin, has been the reduction of high frequency firing by the inhibition of transient INa. Kuo and Bean (1994) refined this mechanism by demonstrating phenytoin inhibition of hippocampal Na+ channels by the slow binding to fast-inactivated conformations. Studies by other investigators have shown that phenytoin can also reduce the persistent INa normally expressed by several neuron types (Chao and Alzheimer, 1995; Lampl et al., 1998) [therapeutic concentration 4–8 μM (Sherwin et al., 1973; Richens, 1979)]. However, the contribution of persistent INa to seizure and antiepileptic activity remains to be clarified. Lacosamide is a novel antiepileptic drug that selectively stabilizes slow-inactivated states of brain NaV channels with minimal interactions with fast-inactivated conformations [therapeutic concentration 17–41 μM (Ben-Menachem et al., 2007)]. Previous reports have demonstrated an inhibition of neuronal excitability with corresponding reduction of transient INa. The independent mechanisms of action of phenytoin and lacosamide provide useful tools to investigate the effects of ranolazine on brain neuronal excitability.
In this study, we investigated the antiepileptic properties of ranolazine using hippocampal neuronal cultures. Ranolazine reduced the number and frequency of action potentials evoked by depolarizing current injections with a time course similar to phenytoin and lacosamide. Computational modeling showed that this inhibition of neuronal NaV channels by ranolazine can result from either binding to fast- or slow-inactivated states with minimal interactions with other channel conformations. In addition, ranolazine decreased the epileptiform activity induced by N-methyl d-aspartate (NMDA) activation (removal of extracellular Mg2+). Our findings provide an initial observation that suggests ranolazine could be effective in controlling high frequency firing during epileptic seizure.
Materials and Methods
Primary Neuronal Culture.
All animal procedures were performed in strict adherence to the policies and procedures approved by the Gilead Sciences Institutional Animal Care and Use Committee. Rat hippocampal neurons were cultured as previously described with a slight modification (Brewer et al., 1993). Briefly, hippocampi were isolated from brains of newborn Sprague-Dawley rats (postnatal day 1–3, sex undetermined), followed by dissociation using papain (1 mg/ml; Worthington Biochemical, Lakewood, NJ) and gentle trituration. Cells were diluted in primary cell culture medium (Neurobasal-A medium; Invitrogen, Carlsbad, CA), 2% B27 supplement (Invitrogen), 0.5 mM l-glutamine, 100 IU/ml penicillin, and 100 mg/ml streptomycin) at a density of 6.5 × 105 cells/ml. A volume of 1 ml was added to a 35-mm dish containing coverslips coated with poly-d-lysine and laminin (BD Biosciences, Sparks, MD), which resulted in approximately 700 cells/mm2. Forty-eight hours after seeding, the media was changed and cytosine β-d-arabinoside (10 μM) was added to prevent the proliferation of non-neuronal cells. Subsequently, media was changed every 3–5 days. Neurons were cultured for at least 10 days prior to experimentation. Neurons exhibiting pyramidal morphology were used for these studies. Unless otherwise noted, all reagents were purchased from Sigma-Aldrich (St. Louis, MO).
Stably Expressed Human Brain NaV1.2.
Human embryonic kidney (HEK) cells stably expressing the human brain (h) NaV1.2 channel were described previously (Wang et al., 2010). The cell line was created by simultaneous stable integration of piggyBac transposons encoding the cDNA for either SCN2A (G418 selection) or SCN1B-IRES2-SCN2B (puromycin selection), as described previously (Kahlig et al., 2010b). The cells were grown in Dulbecco’s modified Eagle’s medium High Glucose (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 100 IU/ml penicillin, 100 μg/ml streptomycin, 1 mg/ml G418, and 3 μg/ml puromycin (InvivoGen, San Diego, CA).
Electrophysiology.
All experiments were performed at room temperature using a MultiClamp 700B amplifier, Digidata 1400 digitizer, and pClamp 10 software (Molecular Devices, Sunnyvale, CA). Patch electrodes (2–3 MOhm) were fabricated from borosilicate glass capillary tubes (World Precision Instruments, Sarasota, FL) using a DMZ-Universal Puller (Zeitz Instruments GmbH, Martinsried, Germany). Results are presented as mean ± S.E.M. Unless otherwise noted, data were analyzed with GraphPad Prism 4 (GraphPad Software, San Diego, CA) and statistical comparisons were made using one-way analysis of variance followed by a Tukey post-hoc test in reference to the control drug-free condition. Steady-state channel availability curves were fit with Boltzmann functions to determine the voltage for half-maximal activation/inactivation (V1/2) and a slope factor (k). Recovery from inactivation was evaluated by fitting the peak current recovery with a two-exponent function: where τf and τs denote time constants (fast and slow components, respectively), Af and As represent the fast and slow fractional amplitudes. For use-dependence studies, currents in response to pulse 40 were normalized to the peak current recorded in response to the first pulse in each frequency train.
where τf and τs denote time constants (fast and slow components, respectively), Af and As represent the fast and slow fractional amplitudes. For use-dependence studies, currents in response to pulse 40 were normalized to the peak current recorded in response to the first pulse in each frequency train.
Whole-Cell Voltage-Clamp.
Whole-cell voltage-clamp recordings were performed as described previously (Kahlig et al., 2010a). For voltage-clamp experiments with neuronal cultures, the pipette solution consisted of (in mM): 140 CsCl, 2 MgCl2, 1 EGTA, 10 HEPES, with a pH of 7.3 and osmolarity of 310 mOsmol/kg. The bath solution contained (in mM): 140 NaCl, 2.5 KCl, 1 CaCl2, 1 MgCl2, 10 dextrose, 10 HEPES, with a pH of 7.4 and osmolarity of 290 mOsmol/kg. For voltage-clamp experiments with stably expressed hNaV1.2 in HEK cells, the pipette solution consisted of (in mM): 110 CsF, 10 NaF, 20 CsCl, 2 EGTA, 10 HEPES, with a pH of 7.35 and osmolarity of 300 mOsmol/kg. The bath solution contained (in mM): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 dextrose, 10 HEPES, with a pH of 7.35 and osmolarity of 310 mOsmol/kg. For all solutions, the osmolarity was adjusted with sucrose. Cells were allowed to stabilize (3 minutes for neurons and 10 minutes for stably expressed NaV1.2) after establishment of the whole-cell configuration before current was measured. For all voltage-clamp experiments, series resistance was compensated 90% to minimize voltage error. Leak currents were subtracted by using an online P/4 procedure and all currents were low-pass Bessel filtered at 4 kHz and digitized at 50 kHz. Specific voltage-clamp protocols were used as depicted in figure insets.
Whole-Cell Current-Clamp.
Whole-cell current-clamp recordings were performed as described previously (Mitterdorfer and Bean, 2002). The pipette solution consisted of (in mM): 140 K-gluconate, 2 MgCl2, 10 EGTA, 10 HEPES, 0.5 Mg-ATP with a pH of 7.3 and osmolarity of 300 mOsmol/kg. The bath solution contained (in mM): 140 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 dextrose, 10 HEPES, with a pH of 7.4 and osmolarity of 320 mOsmol/kg. For experiments investigating spontaneous activity in the culture (action potential generation and synaptic transmission), the MgCl2 in the bath solution was isosmotically replaced by sucrose. Cells were allowed to stabilize for 3 minutes after establishment of the whole-cell configuration before switching to current-clamp mode. For all experiments, pipette neutralization and bridge balance corrections were used to minimize voltage errors and pipette filtering. All voltage records were low-pass Bessel filtered at 2 kHz and digitized at 10 kHz. For experiments investigating evoked action potential generation, the neuron was stimulated for either 1 or 4 seconds using a depolarizing current injection of 1.5× threshold. Only neurons with a resting membrane potential more negative than –50 mV were used in this study and the resting membrane potential of the neuron was not modified. Ranolazine (10 μM) did not alter the resting membrane potential compared with the drug-free condition (–64.0 ± 1.5 mV versus –64.4 ± 1.4 mV for control and ranolazine, n = 20, respectively).
In Vitro Pharmacology.
A stock solution of 50 mM ranolazine (Gilead Sciences, Foster City, CA) was prepared in 0.1 M HCl. A fresh dilution of ranolazine in the bath solution was prepared every experimental day and the pH was readjusted as necessary. Direct application of the test solution to the clamped cell was achieved using the Perfusion Pencil system (AutoMate Scientific, Berkeley, CA). Direct cell superfusion was driven by gravity at a flow rate of 350 μl/min using a 250-μm tip. This system sequesters the cell within a stream and enables complete solution exchange within 1 second. The cell was superfused continuously starting immediately after establishing the whole-cell configuration. Control currents were measured in drug free solution. Drug containing solutions were superfused for 1.5 minutes prior to current recordings to allow equilibration of tonic drug block (tonic block). Tonic block of peak current was measured from this steady-state condition using a depolarizing voltage step at a frequency of 0.2 Hz. Use-dependent block of peak current was measured during pulse number 40 of a depolarizing pulse train at either 10 or 25 Hz. Concentration inhibition curves were fit with the Hill equation: where IC50 is the concentration that produces half inhibition and nH is the Hill coefficient factor. The upper and lower limits were set to 1 and 0.
where IC50 is the concentration that produces half inhibition and nH is the Hill coefficient factor. The upper and lower limits were set to 1 and 0.
Computational Modeling.
The computational model reported here for a brain NaV channel is based on our previously described model of human brain NaV1.1 (Kahlig et al., 2006). The model has been simplified and generalized to allow better quantitation of drug binding to fast-inactivated and slow-inactivated states (Fig. 6). Figure 6A illustrates the Markov model, which includes states for the conditions: closed (C), preopen (PO), open (O), fast-inactivated (FI), and slow-inactivated (SI). Transitions between states are reversible and described by continuous equations whose instantaneous solution depends on the membrane voltage. Microscopic reversibility was ensured by setting the rate constant for β6 equal to (β4*α6*β5)/(α4*α5). Rate equations for all transitions are reported in Table 1. The state O represents the only conducting state and the occupancy of O determines the sodium current by: where INa is the sodium current, GNa_bar is the maximum sodium current density, O is the fractional occupancy of the open state, V is the membrane voltage and ENa is the sodium reversal potential.
where INa is the sodium current, GNa_bar is the maximum sodium current density, O is the fractional occupancy of the open state, V is the membrane voltage and ENa is the sodium reversal potential.
Transition rates for NaV Markov model
To evaluate channel gating, an NaV model was inserted into a single-compartment model of length of 20.0 μm, diameter of 12.1 μm and membrane capacitance of 1μf/cm2. The resulting computational surface area is equivalent to the average surface area measured previously for HEK cells (7.6 × 10−6 cm2, n = 15) (Rhodes et al., 2005). Voltage protocols used to assess activation, steady-state fast inactivation, recovery from fast inactivation, entry into slow inactivation, voltage-dependence of slow inactivation, and recovery from slow inactivation are included as figure insets. The reported model accurately reproduces all recorded behaviors of heterologously expressed brain NaV channels (Supplemental Fig. 3) (Kahlig et al., 2006). For action potential simulations, we used the somatic compartment of the previously published model of a pyramidal neuron (Uebachs et al., 2010). Our NaV model (0.37 S/cm2) replaced all sodium channels in the original model. The potassium channel current densities were adjusted to account for the different behaviors between our Markov-style model and the original Hodgkin–Huxley (HH) style NaV models. The simulated values were (in S/cm2): 0.002 IKCT, 0.00025 IKAHP, 0.0032 IKM, 0.002 IKA, 0.002 IKDR, 0.00004 IKleak, 0.012 IKslow. The resting membrane potential was initialized at –75 mV and the model was allowed 5000-millisecond simulation time for parameter stabilization prior to depolarizing current injection.
Inhibition of peak INa or action potential generation was simulated use four independent schemes (Fig. 6D) to evaluate binding to the following states: preopen (PO), open (O), fast-inactivated (FI), or slow-inactivated (SI). The apparent binding rates for ranolazine are KON = 1 M–1ms−1 and KOFF = 5 × 10−5ms−1 (Fig. 6C, apparent Kd of 50 μM). The microscopic binding rates were estimated by Kapp = KI/(1 h), where Kapp is the apparent dissociation constant for the inactivated state, KI is the dissociation constant for the inactivated state, and h is the fraction of available channels (Kuo and Bean, 1994). This relationship assumes minimal binding to closed states and accounts for reduced availability of the high affinity binding site due to membrane hyperpolarization. The apparent binding rates were measured at a holding potential of –70 mV, which results in 66 ± 6% channel availability (Fig. 6C, n = 6, h = 0.66). The unbinding rate (KOFF) was kept constant and the microscopic KON was calculated as 2.9 M–1ms−1 (0.99 M–1ms−1/0.34) and simulations were performed using a microscopic KON of 3 M–1ms−1 (microscopic Kd ∼16.67 μM to the inactivated state). This 3-fold increase corresponds well with the 4.5-fold increase in the KON (apparent to microscopic) measured for phenytoin (Kuo and Bean, 1994). Additional simulations were performed testing KON values of 10 and 20 M–1ms−1 (Kd values of 5 and 2.5 μM, respectively) in an attempt to force binding to PO and O states.
Computational modeling was performed using NEURON (www.neuron.yale.edu; Hines and Carnevale, 2001). All simulations were performed using the default integration strategy (Backward Euler) with an implicit fixed time step of 25 microseconds to maintain temporal accuracy and efficiency. Simulations were implemented on a Dell Precision T3500 employing a Quad Core Intel Xeon dual-core 3.2 GHz processors running Windows 7.
Results
Ranolazine Inhibits Evoked Action Potential Generation.
Figure 1A shows representative experiments in which cultured hippocampal neuronal action potentials were evoked in response to a 1-second depolarizing current injection in the absence of drug (Fig. 1A, top, control drug-free condition [CTR]). Application of 10 μM ranolazine had a minimal effect on the instantaneous firing frequency. However, the average number of action potentials generated was significantly decreased from 18.5 ± 1.8 in CTR to 16.3 ± 1.2 in the presence of ranolazine (RAN) (Fig. 1B, P < 0.05). In separate experiments, the effect of ranolazine to reduce evoked action potentials was compared with phenytoin and lacosamide. Application of 3 μM phenytoin (DPH) (Figs. 1A, middle, DPH) or 30 μM lacosamide (LCM) (Fig. 1A, bottom, LCM) produced a similar inhibition pattern of evoked action potentials. This inhibition caused a significant reduction in average number of action potentials evoked in the presence of phenytoin (Fig. 1B middle, 10.7 ± 1.5 in CTR compared with 8.7 ± 1.5 in the presence of phenytoin, P < 0.05). The reduction did not reach significance for lacosamide-treated neurons, likely reflecting the delayed accumulation of slow-inactivated states (Fig. 1B, bottom).

Ranolazine reduces central neuron excitability. Hippocampal neuronal cultures were used to determine the effect of ranolazine on central neuron excitability. (A) Representative experiments showing action potentials evoked using depolarizing current injections (1 second) performed either before (CTR) or after the application of 10 μM RAN, 3 μM DPH, or 30 μM LCM. The average resting membrane potentials in the absence of drug were –62.0 ± 1.5 mV, –63.1 ± 2.1 mV, and –61.0 ± 6.6 mV for the RAN, DPH and LCM recordings, respectively. (B) The average number of action potentials (APs) evoked during ranolazine, phenytoin, and lacosamide treatments (n = 6, 3, and 3, respectively). Significant differences from the CTR condition are denoted by *P < 0.05. WASH represents drug wash out.
Ranolazine (10 μM) was able to induce cessation of evoked action-potential generation when the depolarizing pulse duration was 4 seconds (Fig. 2A, top). On average, the number of action potentials generated during CTR was 49.2 ± 3.3, compared with 28.0 ± 4.9 in the presence of 10 μM ranolazine (Fig. 2B, P < 0.1). Similarly, for both phenytoin and lacosamide, extending the depolarizing injection duration to 4 seconds resulted in firing cessation (Fig. 2A) due to accumulated inhibition of NaV activity. The number of action potentials generated during CTR was 46.3 ± 3.5, compared with 24.7 ± 5.9 in presence of 3 μM phenytoin (Fig. 2B, P < 0.05). For experiments with lacosamide, the number of action potentials generated during CTR was 56.0 ± 5.5, compared with 44.3 ± 5.4 in the presence of drug (Fig. 2B, P < 0.05).

Ranolazine induces cessation of action potential firing during the extended depolarization. The experiments in Fig. 1 were repeated using a longer 4-second depolarizing current injection. (A) Representative experiments showing action potentials (APs) evoked from cultured hippocampal neurons using depolarizing current injections (4 seconds) performed either before (CTR) or after the application of 10 μM RAN, 3 μM DPH, or 30 μM LCM. Representative experiments illustrating the cessation (arrow) of excitability observed during each drug treatment. The resting membrane potentials in the absence of drug were –65.1 ± 3.2 mV, –66.6 ± 5.1 mV, and –64.5 ± 6.0 mV for the RAN, DPH, and LCM recordings, respectively. (B) All three drugs significantly reduced the average number of evoked action potentials during the extended depolarization (n = 5, 3, and 3, respectively). Significant differences from the CTR condition are denoted by *P < 0.05; **P < 0.01.
The effect of ranolazine on action potential firing was next assessed during nine sequential depolarizing current injections (1 second, 0.667 Hz). Figure 3A shows representative evoked action potential trains measured during pulses 1 and 9 in the absence of drug or following sequential superfusion of either 3 μM or 10 μM ranolazine. The accumulation of NaV inhibition in the presence of 10 μM RAN was sufficient to cause firing cessation (denoted by an arrow). Figure 3B shows the slow kinetics of inhibition as evidenced by a gradual reduction in the instantaneous firing frequency compared with CTR for each pulse. The average number of action potentials evoked for each pulse was plotted in Fig. 3C. The inhibition increased during the pulse train suggesting a slowly developing block of NaV channels similar to that previously described for phenytoin and lacosamide (Errington et al., 2008). Compared with control, the average number of evoked action potentials during pulse 9 was significantly reduced for 10 μM ranolazine (20.3 ± 3.0 to 7.5 ± 1.4, respectively, P < 0.05). These data suggest that the slow kinetics of NaV block by ranolazine during an extended depolarizing pulse likely reflects either: 1) slow interaction of drug with a site that rapidly becomes available (fast-inactivated states) or 2) rapid interaction with a site that slowly becomes available (slow-inactivated states).
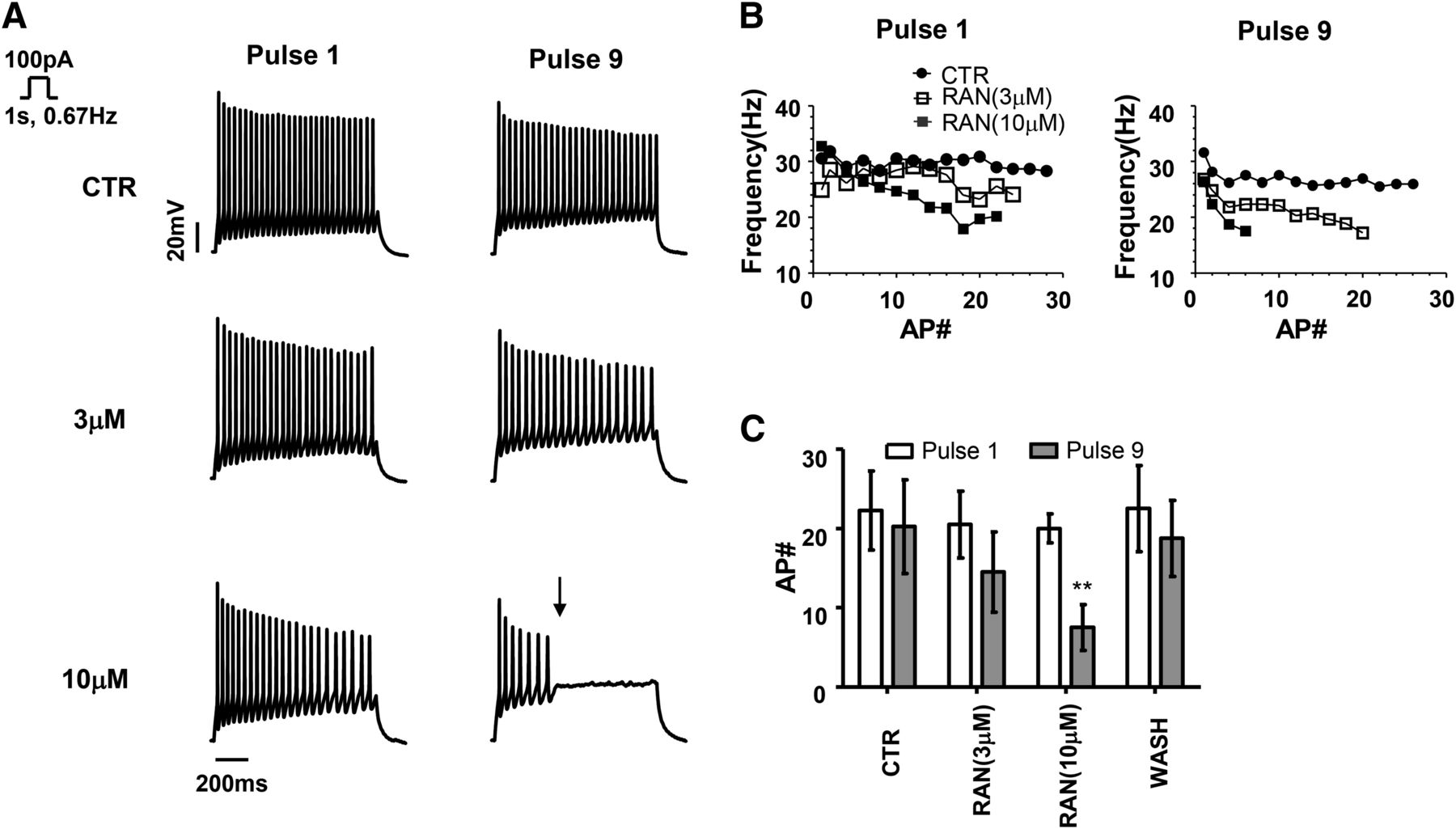
Repetitive depolarization potentiates the inhibition of neuronal excitability by ranolazine. (A) Representative experiments illustrating action potentials (APs) evoked from cultured hippocampal neurons using a repetitive depolarizing current injection (1 second, 0.67 Hz) either before (CTR, top) or after the application of 3 μM ranolazine (middle) or 10 μM ranolazine (bottom). For clarity, only data from Pulse 1 and Pulse 9 are shown and the arrow denotes cessation of action potential generation. The resting membrane potential in the absence of drug was –70.0 ± 3.4 mV. (B) The instantaneous frequency plot of each action potential train in A illustrates the concentration and stimulation dependence of the reduction in excitability. Odd numbered data points are omitted for clarity. (C) RAN (10 μM) reduced the average number of action potentials evoked in response to pulse 9 demonstrating a delayed inhibition of neuronal excitability (n = 4). Significant differences from the CTR condition are denoted by **P < 0.01.
Ranolazine Modulates Nav Fast Inactivation.
We next determined the effect of 10 μM ranolazine on hippocampal INa during voltage protocols designed to selectively engage fast inactivation. Fast inactivation was induced using 100-millisecond depolarizing voltage steps to various potentials in the absence of drug or following superfusion of 10 μM ranolazine (Supplemental Fig. 1). Ranolazine induced a negative shift in the V1/2 of steady-state inactivation (–5 mV; Table 2). The experiment was repeated with a longer inactivating prepulse (1000 milliseconds) designed to allow additional time for ranolazine to interact with the channel (Supplemental Fig. 2). The longer prepulse potentiated the shift in the V1/2 of steady-state inactivation (–8 mV; Table 2). There was a nonsignificant trend toward delayed recovery of hippocampal NaV channels from fast inactivation in the presence of ranolazine (Supplemental Fig. 1C; Table 2). Together, these data suggest ranolazine slowly stabilizes fast inactivation by interacting with NaV channel fast-inactivated states.
Biophysical parameters for fast inactivation
Ranolazine Modulates Nav Slow Inactivation.
We next evaluated the effect of 10 μM ranolazine on hippocampal INa during voltage protocols engaging both fast and slow inactivation. Inactivation was induced using long (10-second) depolarizing voltage steps to various potentials in the absence of drug or following superfusion of 10 μM ranolazine (Supplemental Fig. 1D). Ranolazine caused a significant negative shift in the V1/2 of steady-state slow inactivation compared with CTR (Table 3). In addition, slow inactivation was more complete in the presence of ranolazine (residual availability 38 ± 7% for RAN compared with 55 ± 6% for CTR, P < 0.001). Ranolazine also delayed the recovery of hippocampal NaV channels from slow inactivation induced by a maximally inactivating prepulse (Supplemental Fig. 1E). The time constants of channel recovery were significantly larger in the presence of ranolazine without alterations to the amplitude of either the fast- or slow-recovery components (Table 3). These data suggest that ranolazine may interact with NaV slow-inactivated states similarly to the mechanisms of action proposed for lacosamide (Errington et al., 2008). However, a contribution of fast-inactivated state binding cannot be excluded because these voltage protocols engage both fast- and slow-inactivation mechanisms.
Biophysical parameters for slow inactivation
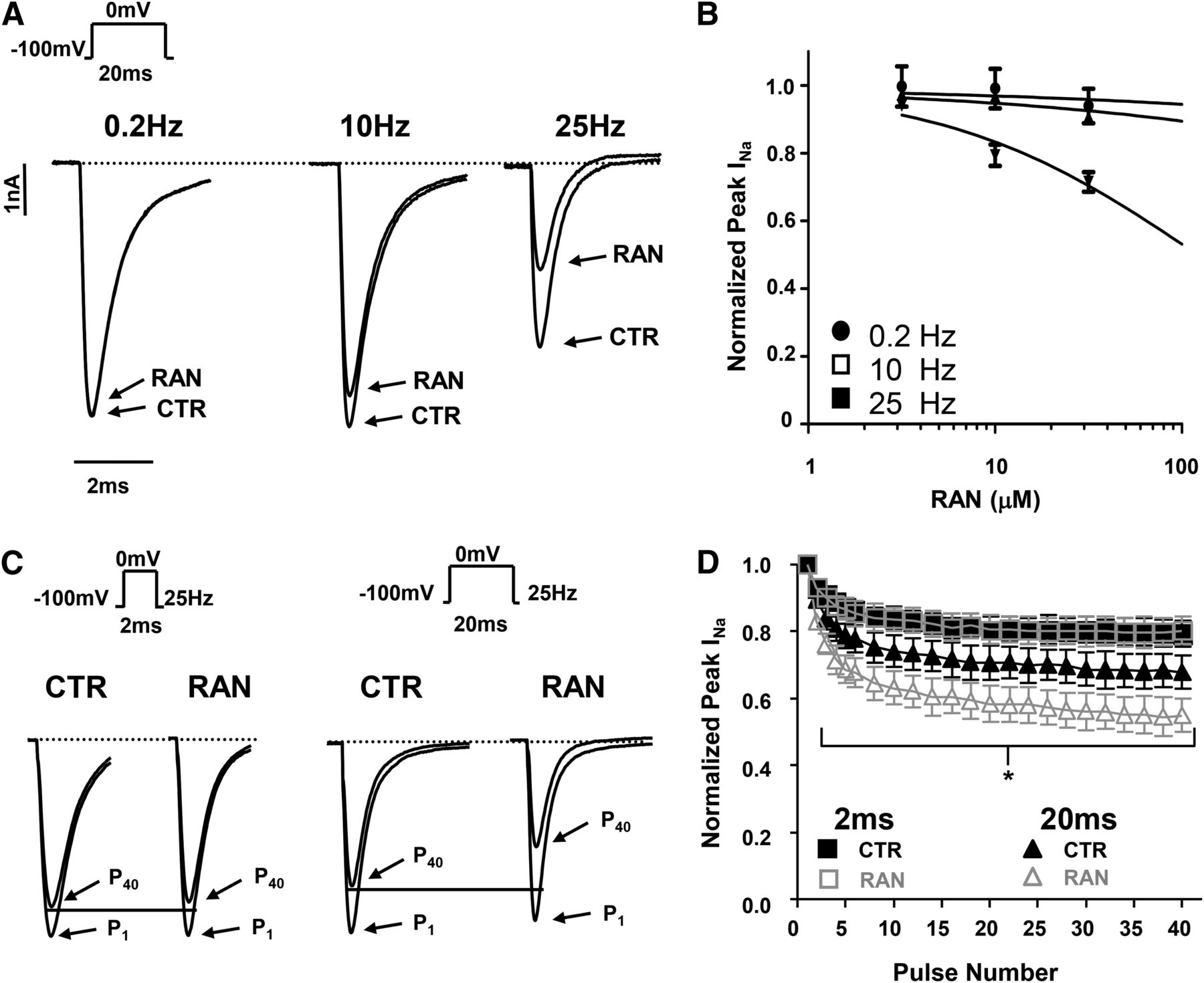
Evaluation of Tonic Block and Use-Dependent Block.
NaV channel inhibitors typically exhibit minimal interaction with closed conformations, which can be assessed using hyperpolarizing holding potentials (tonic block). The inhibition potency increases with rapid, repetitive stimulation (use-dependent block; UDB). Figure 4A shows the minimal level of INa tonic block (0.2 Hz, left) in a representative cell measured in the presence of 10 μM ranolazine compared with the CTR. Increasing the stimulation frequency to 10 Hz or 25 Hz increased the level of inhibition. Figure 4B shows the levels of tonic block and UDB for increasing concentrations of ranolazine. The minimal inhibition observed for tonic block and 10 Hz preclude determination of the IC50. Fitting the concentration-inhibition data for 25 Hz resulted in an estimated IC50 of 114 ± 83 μM (Hill coefficient = 0.6 ± 0.1).

Ranolazine use-dependent block is modulated by frequency and step duration. (A) Representative current traces illustrating tonic block (TB; 0.2 Hz) or use-dependent block 10 Hz or 25 Hz assessed at the end of a 40-pulse train. Hippocampal neuronal INa was measured before (CTR) or after the application of 10 μM RAN. (B) The average tonic block and use-dependent block measured for various ranolazine concentrations. (C) Representative experiment showing the use-dependent inhibition of NaV currents by 10 μM RAN in response to step durations of either 2 milliseconds (left) or 20 milliseconds (right). Note the increased inhibition at pulse 40 (P40) for a step duration of 20 milliseconds compared with 2 milliseconds. (D) The average normalized peak current for each step in a 25-Hz train plotted for either a 2 milliseconds (squares) and 20 milliseconds (triangles) step duration (n = 5). For clarity, after step 5 only even numbered data points are shown. A statistically significant reduction in channel availability was observed for the 20 milliseconds duration step for pulses 2–40. *P < 0.05. Dotted lines in A and C represent zero current.
Previous reports investigating ranolazine block of nonbrain NaV channel isoforms have found evidence of open- or preopen-state inhibition using voltage pulse trains with variable step durations (Rajamani et al., 2008; Wang et al., 2008; Zygmunt et al., 2011). Short voltage steps (2 milliseconds) increase the available sites for preopen and open states by minimizing the presentation of inactivated conformations. Longer steps (20 milliseconds) would allow for additional inactivated states to become available. Equal potency for 2- or 20-millisecond step durations would suggest minimal effects of RAN on fast-inactivated states. Thus, the potency of ranolazine UDB (25 Hz, 40 pulses) in hippocampal neurons using voltage step trains of variable step duration (2 or 20 milliseconds) was determined. Figure 4C plots current records from a representative neuron showing the increased potency of UDB for a step duration of 20 milliseconds (right) compared with 2 milliseconds (left). Figure 4D shows the average UDB at pulse 40 was significantly greater for a step duration of 20 milliseconds (13.0 ± 0.9%) than of 2 milliseconds (1.8 ± 1.4%, P < 0.001). These data suggest that ranolazine does not selectively interact with preopen or open states of the NaV channels expressed in hippocampal neurons, and NaV-inactivated states contribute significantly to the observed UDB.
Ranolazine Suppresses Epileptiform Activity in Hippocampal Cultures.
To investigate the antiepileptic potential of ranolazine, we determined the effect of ranolazine on epileptiform activity evoked by NMDA receptor activation within hippocampal neuronal cultures. Figure 5A shows a representative experiment in which Mg2+ was removed from the bath solution to activate NMDA-dependent hyperexcitability (Rogawski and Loscher, 2004). Ranolazine (10 μM) was able to reduce action potential firing, which recovered following washout. The lower traces in (A) show representative epileptiform activity on an expanded time scale. Figure 5B shows the average reduction in the frequency of the epileptiform bursts by 10 μM ranolazine (0.45 ± 0.07 Hz for RAN compared with 0.78 ± 0.08 Hz for CTR, P < 0.05). These data suggest ranolazine may be capable of suppressing neuronal hyperexcitability during a seizure.
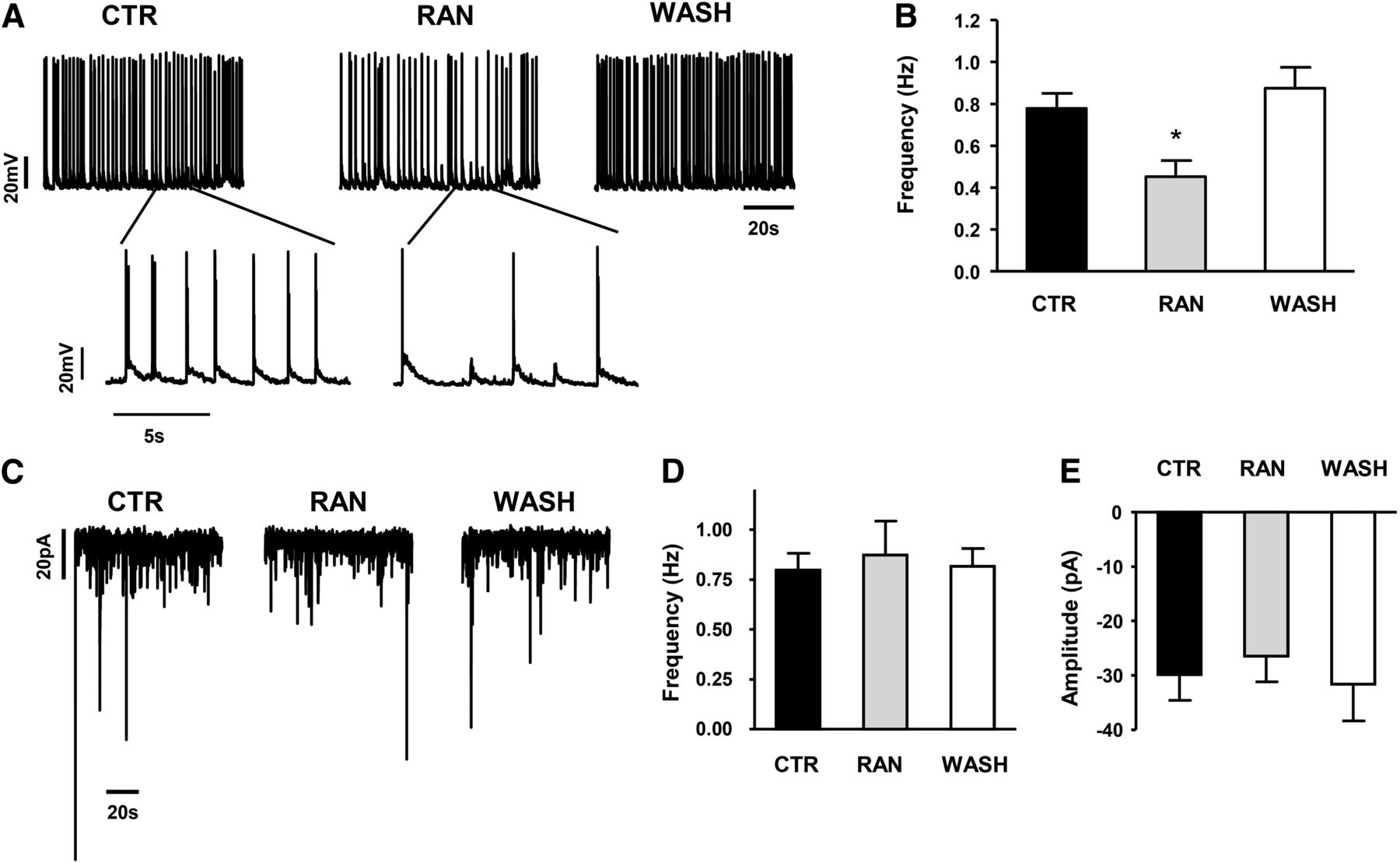
Ranolazine inhibits epileptiform activity by acting extra-synaptically. (A) Spontaneous epileptiform activity was induced in the hippocampal neuronal culture by NMDA receptor activation with low extracellular Mg2+ (CTR). This epileptiform activity was reversibly inhibited by application of 10 μM RAN. Lower panels show representative epileptiform bursts on an expanded time scale. The resting membrane potential in the absence of drug was –60.4 ± 1.7 mV. (B) Average of five experiments illustrating the statistically significant reduction of epileptiform activity (*P < 0.05). (C–E) Miniature synaptic currents were isolated using 0.3 μM TTX to block action potential generation. (C) Representative experiment showing the application of 10 μM RAN did not affect spontaneous miniature synaptic currents. Ranolazine had no effect on either the frequency (D) or amplitude (E) of the events (n = 7) demonstrating minimal interaction with either pre- or postsynaptic targets.
Ranolazine Does Not Alter Miniature Synaptic Activity or Non-Nav Mechanisms.
The reduction in hippocampal epileptiform activity observed for ranolazine may reflect inhibition of NMDA transmission, potentiation of GABA transmission or alterations in voltage-gated potassium (KV) channel activity. The effect of ranolazine to modulate brain NMDA, GABA, and KV channels was directly investigated using hippocampal neurons in culture. These experiments demonstrate that ranolazine exerts minimal/no effect on each of the systems (Supplemental Fig. 2).
We next tested if the reduction of epileptiform activity by ranolazine required NaV inhibition. Tetrodotoxin (TTX; 0.3 μM) was used to block all NaV channels expressed by the hippocampal neurons and spontaneous miniature synaptic currents reflecting action potential independent synaptic transmission were measured at –70 mV. Figure 5C illustrates representative miniature synaptic currents measured before (CTR) and after superfusion with 10 μM RAN, or following washout. Ranolazine had no effect on either the frequency (Fig. 5D) or amplitude (Fig. 5E) of the miniature synaptic currents. This suggests that ranolazine has little direct effect on synaptic vesicle release machinery.
Markov Model of Brain Nav Channels.
To investigate the state dependent interaction of ranolazine with brain NaV channels, we developed a Markov model consisting of three closed states (C4, C3, C2), a preopen state (PO), a fast-inactivated state (FI), a slow-inactivated state (SI), and an open state (O). The open state is the only conducting state and the rate constants connecting the states were optimized to reproduce the generalized behavior of hNaV1.1 and hNaV1.2 channels. Figure 6A shows a diagram describing the Markov model. The model generates rapidly activating and inactivating inward currents in response to 20-millisecond voltage steps to between –80 and +60 mV from a holding potential of –120 mV. The behavior of the model was further validated using standard voltage protocols investigating fast activation, steady-state fast inactivation, recovery from fast inactivation, voltage-dependence of slow inactivation, development of slow inactivation, and recovery from slow inactivation (Supplemental Fig. 3). The blue data points in each figure represent data recorded from heterologously expressed NaV1.1 and represent standard responses used to develop the model (Kahlig et al., 2006).
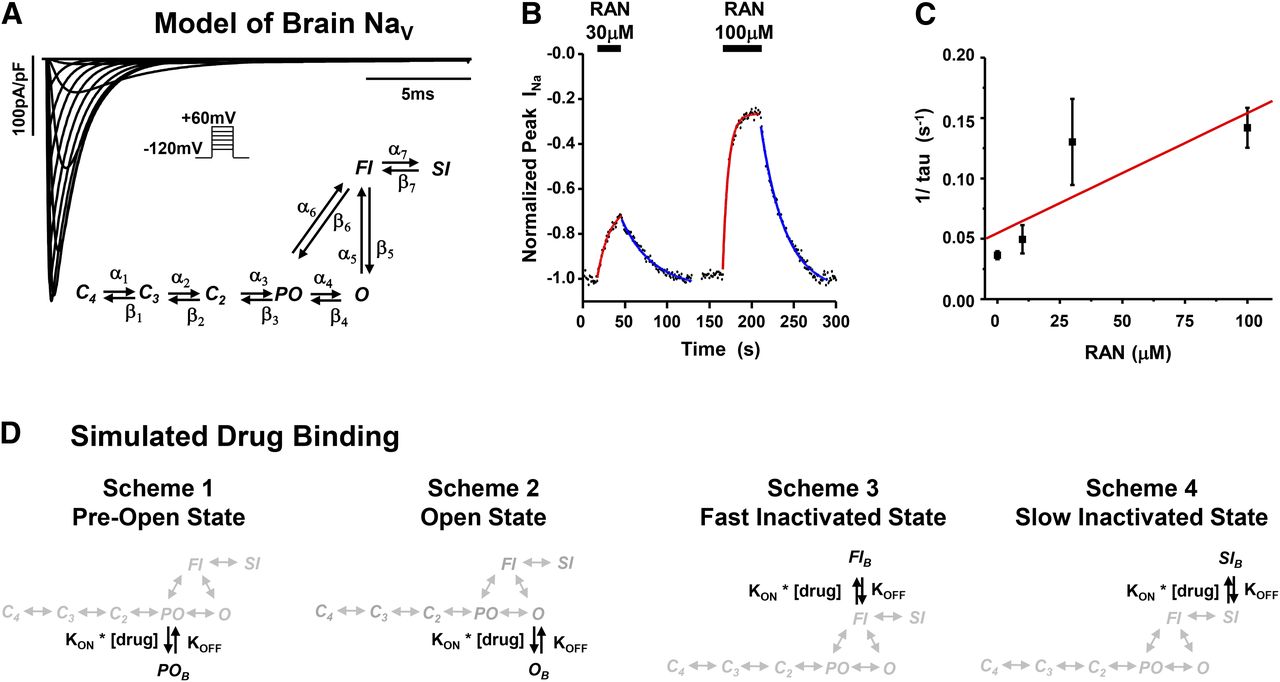
Brain NaV computational model and simulation of ranolazine binding. (A) Inset, diagram of a Markov model for brain NaV channels developed to examine ranolazine binding. Top, the model generates rapidly activating and inactivating inward current similar to native and heterologously expressed NaV channels. (B) Apparent binding kinetics for RAN were measured using hNaV1.2 and step depolarizations (0 mV, 5 milliseconds, 1 Hz) from a holding potential of –70 mV. Fast application of either 30 or 100 μM ranolazine (black bars) resulted in a rapid reduction in the peak current that was reversible upon return to drug-free superfusion. The red and blue lines are single exponential fits of either the binding or disassociation of ranolazine, respectively. KOFF (∼5 × 10−5ms−1) was calculated from the inverse of the average disassociation rate constants (tau). (C) The inverse of the binding rate constants were plotted against the concentration of ranolazine and the apparent KON (∼1 M–1ms−1) was calculated as the slope of the best fit line (n = 4–9 for each point). (D) Four schemes were used to probe the binding site(s) of ranolazine to the Markov model. Binding to the pre-open state (PO, Scheme 1), open state (O, Scheme 2), fast-inactivated state (FI, Scheme 3), or slow-inactivated state (SI, Scheme 4).
Ranolazine Interacts Slowly with Nav Channels.
To quantitate the interaction between ranolazine and brain NaV channels, we directly measured the apparent binding rates for ranolazine to hNaV1.2. Figure 6B show the results of a representative experiment in which the voltage was stepped to 0 mV (5 milliseconds, 0.2 Hz) from a holding potential of –70 mV. The black bars denote rapid application of either 30 μM or 100 μM RAN. The inhibition (red lines) and recovery (blue lines) of peak INa were well fit with a single exponential equation, which estimates the time constant of apparent binding or unbinding, respectively. In separated experiments, the speed of superfusion solution exchange was confirmed using 1 μM TTX, which yielded an inhibition time constant of 0.6 ± 0.1 seconds (n = 6). This was an order of magnitude faster than the average apparent binding rate measured for 100 μM ranolazine (7.9 ± 1.0 seconds, n = 9).
In Fig. 6C, we determined the apparent KON and KOFF of ranolazine binding at –70 mV and the inverse of the average inhibition time constants were plotted against ranolazine concentration. The inverse of the average recovery time constant was plotted as the drug-free condition. A linear regression provided an apparent KON (slope) of 0.99 M–1ms−1 and a KOFF (y-intercept) of 5.5 × 10−5ms−1 for ranolazine binding and unbinding, respectively. The microscopic KON was calculated as 2.9 M–1ms−1. Unless otherwise noted, simulation studies used a KON of 3 M–1ms−1 and KOFF of 5 × 10−5ms−1. With a Markov model it is possible to predict the effect of drug binding to individual states. Figure 6D shows the four schemes tested for ranolazine binding to brain NaV channels: Scheme 1, preopen-state binding; Scheme 2, open-state binding; Scheme 3, fast-inactivated state binding; Scheme 4, slow-inactivated state binding. To simply the computational simulations, the binding schemes do not allow transitions between drug-bound conformations. The binding of ranolazine to the Markov model was validated using each binding scheme and the apparent or microscopic binding kinetics (Supplemental Fig. 4). Restricting binding to PO or O states had minimal effect on peak INa, while binding to either the FI or SI state caused a block of peak INa that exhibited the appropriate magnitude and kinetics (Fig. 6B).
Computational Modeling of Ranolazine Effects on Neuronal Excitability.
We constructed a cellular model based on the somatic compartment of a previously published representation of a hippocampal pyramidal neuron (Uebachs et al., 2010). Figure 7A shows the response of the model to a +160 pA depolarizing current injection from a resting membrane potential of –77.6 mV. In the absence of drug the model generated evoked action potentials during the entire 4-second depolarization. Drug binding was simulated using the microscopic KON and KOFF (3 M–1ms−1 and of 5 × 10−5ms−1, respectively) and a concentration of 10 μM as in Fig. 2. Restricting binding to PO or O states had minimal effect, while binding to either the FI or SI state caused firing cessation late in the depolarization. Figure 7B shows the same experiment performed with KON increased to 10 M–1ms−1 in an attempt to force binding to PO or O states. Significant effects on excitability were only observed for binding to FI or SI states. Figure 7C shows the instantaneous frequency calculated between each action potential for the sweeps in A and B. Drug binding to either the FI or SI state progressively reduced the rate of action potential generation leading to firing cessation (arrows). The number of action potentials evoked during 4 seconds was plotted for each binding scheme (Fig. 7D). Additional simulations were performed with KON values of 1 (apparent KON), 6, or 20 M–1ms−1. Even at the highest KON, binding to either the PO or O state did not affect the number of evoked action potentials. In contrast, binding to either the FI or SI state reduced the number of action potentials depending on the rate of binding, which determines the rate of inhibition accumulation.

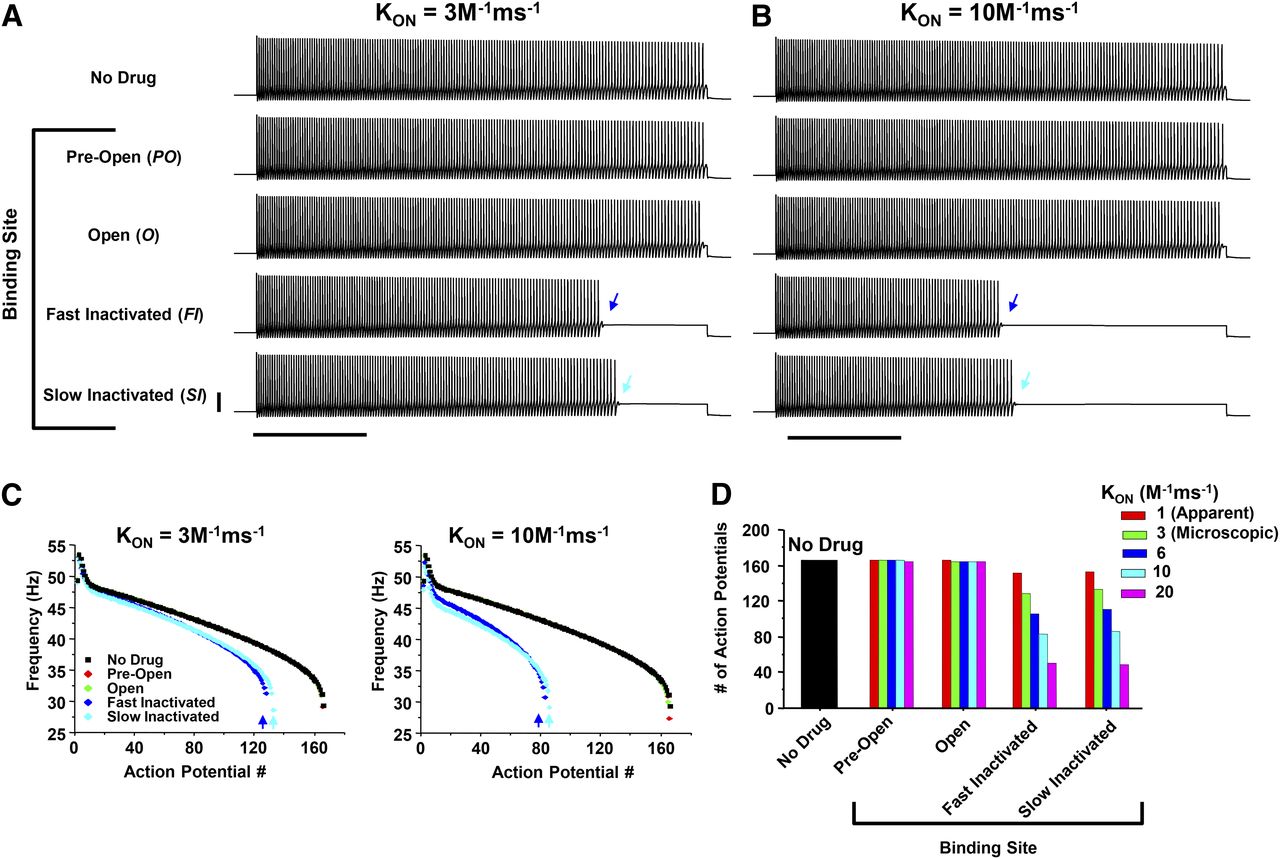
Simulated ranolazine binding to NaV-inactivated states is required to reduce neuronal excitability. Action potential simulations were performed to investigate the mechanism of delayed inhibition of neuronal excitability by ranolazine. (A and B) Depolarizing current pulses (4 seconds) resulted in a train of evoked action potentials in the drug-free condition (No Drug, top trace). Ranolazine binding was investigated using the microscopic binding rate constant (KON) of 3 M–1ms−1 (left) or an elevated KON of 10 M–1ms−1 in an attempt to force binding to PO or O stats (right). Note cessation of firing for binding to either FI or SI states (dark blue and light blue arrows, respectively) with minimal effect of simulated binding to either PO or O states. (C) Instantaneous frequency calculated for each condition in A, left, and B, right, illustrating a slowing in firing rate prior to spike cessation. (D) Number of action potentials evoked during the 4 seconds of depolarizing current injection. Binding targeting the FI or SI state reduced neuronal excitability in a KON dependent manner, while binding to PO or O states had no effect. In A and B, scale bars represent 40 mV and 1 second.
We next simulated the repetitive current injection experiments from Fig. 3 to investigate the binding scheme(s) capable of reproducing the inhibition profile observed with ranolazine. Figure 8 shows the action potentials evoked using a depolarizing current injection train (1 second, +100 pA, 0.667 Hz) for pulses 1 and 9. In the drug-free condition the model generated action potentials during each pulse and the number of action potentials is denoted at the end of the pulse. Drug binding was then simulated at a concentration of 3 μM using the microscopic KON (3 M–1ms−1) and the binding schemes in Fig. 6D. Binding to the FI or SI states reduced the instantaneous frequency of action potential generation at pulse 9 with a concomitant decrease in the number of evoked action potentials (Fig. 8A). The accumulated binding to FI or SI states resulted in firing cessation at the end of pulse 9 (arrow). The simulations were repeated with a drug concentration of 10 μM as in Fig. 3, and only binding to the FI or SI states reduced the excitability of the model (Fig. 8B). At this higher concentration, both binding schemes produced firing cessation during pulse 9 (arrows). Binding to the PO or O states had no effect on the number of evoked action potentials, even at this higher concentration. These data reproduce well the empirical data reported in Fig. 3 and further suggest that inhibition of hippocampal neurons by ranolazine can be reproduced by simulated binding to either fast-inactivated or slow-inactivated states.

Ranolazine induces cessation of stimulated evoked action potential during the repetitive depolarization. Repetitive pulses of depolarizing current (1 second, 0.67 Hz) evoked trains of action potentials in the hippocampal computational model (No Drug, top). The number of evoked action potentials is shown at the end of each trace. Ranolazine binding was investigated using the microscopic KON (3 M–1ms−1) and a drug concentration of either (A) 3 μM or (B) 10 μM. The top panels show the evoked action potentials generated during Pulse 1 (left) or Pulse 9 (right). The bottom panels illustrate the instantaneous frequency calculated during Pulse 1 (left) or Pulse 9 (right). The symbols for PO (red) or O (green) state binding are under the symbols denoting the drug-free condition (black). Binding to either the FI or SI state resulted in a pulse dependent decrease in excitability reflected by a reduction in the number of evoked action potentials and firing cessation (dark blue and light blue arrows, respectively). Simulated drug binding to either the PO or O state had no impact on action potential number or firing frequency. The scale bars represent 40 mV and 100 milliseconds.
Discussion
In this report, hippocampal neurons were used to investigate the effect of ranolazine on central neuron excitability. Ranolazine reduced the excitability of hippocampal neuronal cultures with a slow time course, which was similar to phenytoin and lacosamide. Moreover, ranolazine reduced the epileptiform activity induced by removal of extracellular Mg2+. Both experimental and computer simulations predict that the inhibition of INa could result from fast- and/or slow-inactivation state binding, in contrast to the predicted open-state binding of ranolazine proposed for other nonbrain NaV channel isoforms, including the cardiac NaV1.5 (Wang et al., 2008; Nesterenko et al., 2011).
NaV channels are common targets for antiepileptic drugs due to their role in the initiation and propagation of action potentials in most excitable tissues (George, 2005). Phenytoin and lacosamide are antiepileptic drugs with well characterized actions on NaV channels (Kuo and Bean, 1994; Errington et al., 2008). Kuo and Bean (1994) reported that phenytoin selectively binds to the fast-inactivated state with slow binding rates (KON of ∼10 M–1ms−1 and KOFF ∼6 × 10−5ms−1) and with minimal interactions to other states (Kuo and Bean, 1994). In contrast, lacosamide was shown to selectively bind to slow-inactivated conformations with little or no binding to other conformations, including fast-inactivated states (Errington et al., 2008). Although differing mechanistically, phenytoin and lacosamide each suppressed hippocampal action potentials firing with a similar time course to that of ranolazine (Figs. 1 and 2). These results implicate both fast- and/or slow-inactivated states as potential targets for ranolazine binding. In fact, both inactivation processes (fast and slow) were enhanced in the presence of ranolazine (Supplemental Fig. 1). Ranolazine progressively potentiated entry into inactivation as the inactivating prepulse was extended from 100 to 10,000 milliseconds. This time course correlates well with the slow inhibition profile of ranolazine to hNaV1.2 (Fig. 6). The recovery of hippocampal NaV channels from slow inactivation was delayed in the presence of ranolazine, suggesting the rate-limiting transition is drug dissociation. In the drug-free condition, the voltage protocols employed selectively assess the independent kinetic processes, termed fast inactivation and slow inactivation. However, the sequential presentation of open, fast-inactivated, and slow-inactivated states makes definitive determination of the binding site of ranolazine impossible using only this approach. However, we can conclude that ranolazine exhibits minimal interaction with closed conformations of brain NaV channels as evidenced by a low level of tonic block observed at a holding potential of –100 mV [Fig. 4 and (Kahlig et al., 2010a)].
A common feature of NaV targeting drugs is an increase in potency with repetitive stimulation, which is thought to reflect either: 1) altered presentation of or 2) differential interaction with the binding site(s) during the repetitive activation/inactivating gating cycle (Hille, 1977; Starmer et al., 1987). Ranolazine exhibited UDB (at 25 Hz) of hippocampal NaV channels with an IC50 of 114 ± 83 μM. Repetitive stimulation mimics the rapid neuronal firing associated with seizure and suggests ranolazine may be effective during periods of high neuronal discharge. Previous reports have also inferred open-state binding by using a modified UDB protocol in which the stimulation frequency is fixed and the voltage-step duration is varied (Rajamani et al., 2008; Wang et al., 2008; Zygmunt et al., 2011). With this design, the time of open-state presentation is fixed while the presentation of fast/slow-inactivation states increases with step duration. In our experiments, the block of hippocampal neurons by ranolazine was more potent with a UDB step duration of 20 milliseconds (13.0 ± 0.9%) compared with 2 milliseconds (1.8 ± 1.4%), suggesting an interaction with inactivated states (Fig. 4).
Minimal open-state binding to brain NaV channels contrasts with our previous report, which found no correlation between step duration and inhibition potency for the cardiac NaV1.5 (Zygmunt et al., 2011). Our previous work used a maximum stimulation frequency of ∼6.5 Hz to drive ranolazine binding. It is tempting to speculate that preferential open-state binding to nonbrain NaV isoforms results from differences in the binding site and/or lower firing rates compared with central nervous system neurons. Alternatively, this discrepancy may reflect the chosen experimental conditions as a more rapid stimulation frequency (25 Hz as in this study) may produce pulse duration-dependent UDB. Moreover, our previous computational modeling efforts assumed that ranolazine requires a hydrophobic pathway to the binding site, which is blocked by closure of the inactivation gate (Nesterenko et al., 2011). These mechanistic limitations were coded into the Markov model to the exclusion of inactivation-state binding. This approach reproduces several features of the inhibition of NaV1.5 by ranolazine, including preferential binding to NaV1.5 during atrial versus ventricular cardiac action potential waveforms (Nesterenko et al., 2011). However, subsequent publication of the first crystal structure of a NaV channel revealed prominent and concentric hydrophobic pathways surrounding the channel’s conduction pathway through intramembrane fenestrations (Payandeh et al., 2011). These new structural data support work from Hille in 1977 that found both hydrophilic and hydrophobic pathways for the interaction of NaV channels and local anesthetics (Hille, 1977). With insights from the crystal structure, further investigation will be required to determine if selective open-state block of NaV1.5 is necessary as well as sufficient to describe ranolazine’s actions.
The data presented in this report support inactivated-state binding for ranolazine to brain NaV channels. Targeted binding to inactivated states is a common theme of antiepileptic drugs, and this approach is predicted to maintain normal neuronal responsiveness to incoming stimuli by sparing resting and open conformations (Rogawski and Loscher, 2004). Computational modeling of a hippocampal neuron (Uebachs et al., 2010) was used to explore the NaV binding sites that were sufficient to reproduce the inhibition of evoked action potentials observed by ranolazine. A simplified Markov model of a brain NaV channel was developed that accurately reproduced the behavior of a NaV channel and was based on our previously reported model of NaV1.1 (Kahlig et al., 2006). Simulated binding of ranolazine to either PO or O states had minimal effect on evoked action potentials even at a KON 20-fold greater than the measured apparent KON (1 M–1ms−1 at –70 mV; Fig. 6). This lack of effect could reflect an underestimation of the true KON at physiologic temperatures and potentials. In fact, the binding rate for phenytoin to hippocampal neurons is ∼3 M–1ms–1 at –70 mV but increases to ∼5 M–1ms−1 at –50 mV and ∼14 M–1ms–1 at +40 mV (Kuo and Bean, 1994). However, the microscopic KON calculated for ranolazine is 3 M–1ms−1 and simulated binding of ranolazine to either FI or SI states using this microscopic rate produced a robust reduction in simulated action potentials (Figs. 7 and 8) in parallel to that observed using hippocampal neuronal cultures (Figs. 2 and 3). In addition, the binding rates used in our simulations compare well with those previously measured for carbamazepine (Kuo et al., 1997; KON of 38 M–1ms−1 and KOFF of 6 × 10−4ms−1) and lamotrigine (Kuo and Lu, 1997; KON of 10 M–1ms−1 and KOFF of 8 × 10−5ms−1). Therefore, it is likely that the inhibition exerted by ranolazine simply reflects binding to inactivated states of neuronal NaV channels. The slow inhibition time course of ranolazine may reflect binding to either fast-inactivated or slow-inactivated states because the reduction of action potential generation was equally potent for simulated FI or SI binding.
A common experimental model of seizure is the induction of epileptiform activity by removal of extracellular Mg2+, a maneuver which activates excitatory NMDA ion channels (Rogawski and Loscher, 2004). In this model, ranolazine was able to reduce the epileptiform activity generated in the hippocampal neuronal cultures (Fig. 5). The molecular target of this novel antiepileptic action is likely brain NaV channels because ranolazine had no effect on the miniature synaptic potentials (Fig. 5), GABA, or NMDA neurotransmission or KV channels (Supplemental Fig. 2).
Ranolazine does not exhibit NaV channel isoform selectivity. This feature has been leveraged by previous in vitro studies demonstrating ranolazine can normalize the excessive NaV channel and/or neuronal activity underlying an array of pathological conditions, such as neuropathic pain, paramyotonia congenita, migraine, and epilepsy (Wang et al., 2008; Estacion et al., 2010; Kahlig et al., 2010a; El-Bizri et al., 2011; Hirakawa et al., 2012). Additional studies, including animal models of epilepsy, are necessary to determine if the results presented here will translate into a therapeutic benefit.
Acknowledgments
The authors thank Steven Nguyen for valuable technical assistance.
Authorship Contributions
Participated in research design: Kahlig, Hirakawa, Rajamani.
Conducted experiments: Kahlig, Hirakawa, Liu.
Performed data analysis: Kahlig, Hirakawa.
Wrote or contributed to the writing of the manuscript: Kahlig, Hirakawa, George, Belardinelli, Rajamani.
Footnotes
- Received July 12, 2013.
- Accepted November 7, 2013.
K.M.K. and R.H. contributed equally to this work.
This study was supported by Gilead Sciences.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- CTR
- control drug-free condition
- DPH
- phenytoin
- HEK
- human embryonic kidney
- hNaV1.2
- human brain NaV1.2
- INa
- Na+ current
- KV
- voltage-gated potassium
- LCM
- lacosamide
- NaV
- voltage-gated sodium
- NMDA
- N-methyl d-aspartate
- RAN
- ranolazine
- TTX
- tetrodotoxin
- UDB
- use-dependent block
- V1/2
- half-maximal activation/inactivation
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}