


Abstract
Mazindol has been explored as a possible agent in cocaine addiction pharmacotherapy. The tetracyclic compound inhibits both the dopamine transporter and the serotonin transporter, and simple chemical modifications considerably alter target selectivity. Mazindol, therefore, is an attractive scaffold for both understanding the molecular determinants of serotonin/dopamine transporter selectivity and for the development of novel drug abuse treatments. Using molecular modeling and pharmacologic profiling of rationally chosen serotonin and dopamine transporter mutants with respect to a series of mazindol analogs has allowed us to determine the orientation of mazindol within the central binding site. We find that mazindol binds in the central substrate binding site, and that the transporter selectivity can be modulated through mutations of a few residues in the binding pocket. Mazindol is most likely to bind as the R-enantiomer. Tyrosines 95 and 175 in the human serotonin transporter and the corresponding phenylalanines 75 and 155 in the human dopamine transporter are the primary determinants of mazindol selectivity. Manipulating the interaction of substituents on the 7-position with the human serotonin transporter Tyr175 versus dopamine transporter Phe155 is found to be a strong tool in tuning the selectivity of mazindol analogs and may be used in future drug design of cocaine abuse pharmacotherapies.
Introduction
Abuse of psychostimulants is a huge burden to society, resulting in financial, criminal, and health problems (Dutta et al., 2003). Cocaine is one of the most powerful reinforcers known to date, often leading to dependence and abuse. Cocaine is known to inhibit all three human monoamine transporters: the serotonin (hSERT), norepinephrine, and dopamine transporters (hDATs). The most important site of action for cocaine is the hDAT, which has led to the “dopamine hypothesis” (Kuhar et al., 1991) stating that cocaine binds to the transporter and blocks the reuptake of dopamine, leading to an increased concentration of this neurotransmitter in the synaptic cleft. Consequently, enhancement and prolonged stimulation of the dopaminergic system occur (Kuhar et al., 1991), causing the strongly reinforcing effect of cocaine. This hypothesis has been verified by knock-in of a cocaine-insensitive DAT (Chen et al., 2006; Thomsen et al., 2009). Several approaches to treat cocaine addiction have been tried; however, none of these has led to compounds used clinically (Kharkar et al., 2008), and an urgent need for the development of drugs against cocaine abuse remains.
The tetracyclic compound mazindol was first synthesized in 1968 (Fig. 1). Like cocaine, mazindol binds to all human monoamine transporters (Javitch et al., 1984; Angel et al., 1988; Eshleman et al., 1999) and has been shown to be useful as an appetite suppressant and has therefore been used against obesity (Acri, 2008).
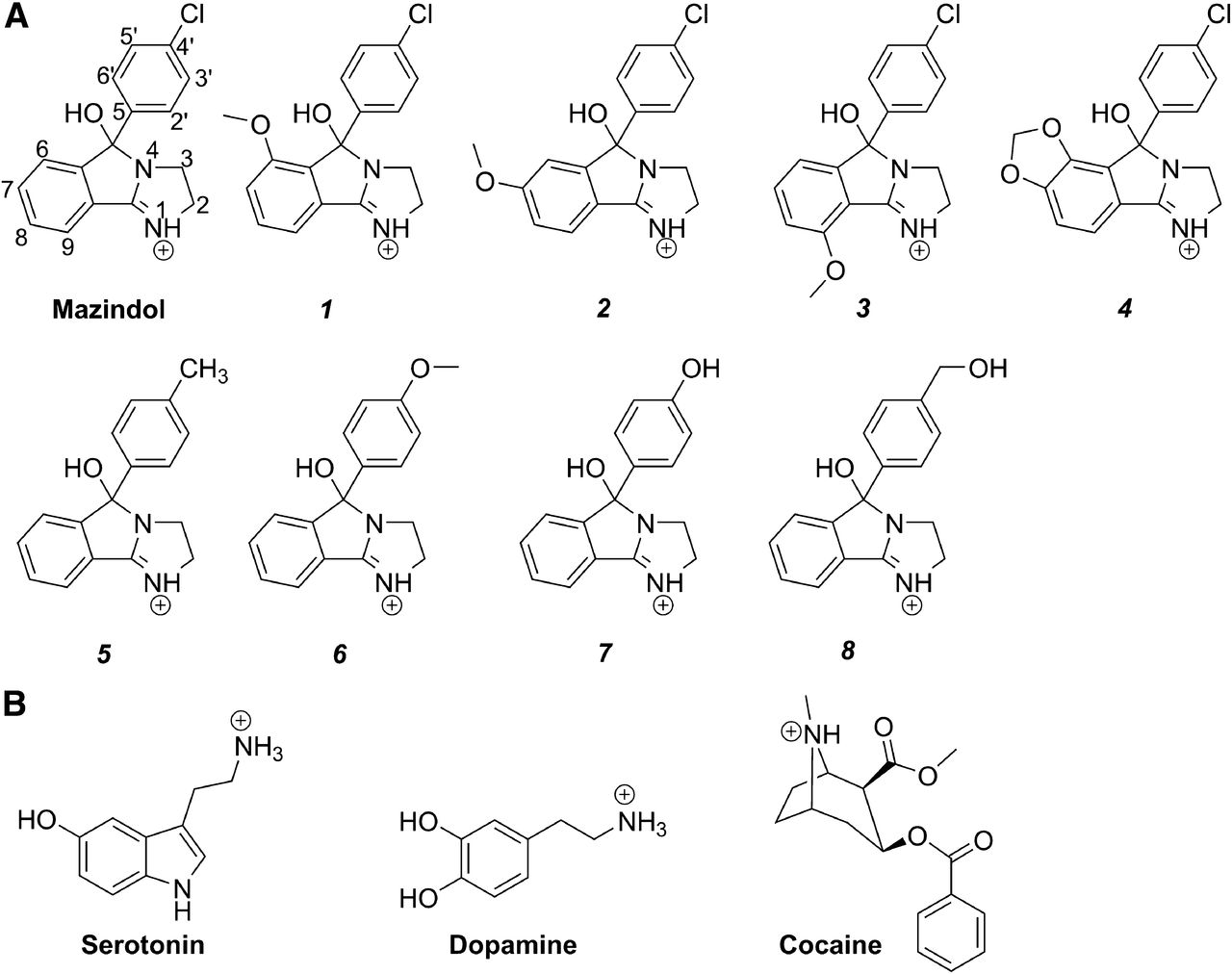
(A) Chemical structures and atom numbering of mazindol and analogs used in this study, and (B) chemical structures of protonated serotonin, dopamine, and cocaine.
Mazindol functions with a favorable rapid rate of onset and binds with a higher affinity to hDAT than cocaine without having significant side effects (Berger et al., 1989; Eshleman et al., 1999; Chen and Reith, 2002), leading to the speculation that mazindol could be used in the treatment of cocaine addiction. Promisingly, mazindol reduces craving and euphoria in cocaine-abusing patients in short-term studies (Berger et al., 1989), although results from longer-term studies are not significantly better than those observed in placebo-treated patients (Stine et al., 1995). Mazindol was shown to cause psychostimulation in monkeys, which might be assigned to a different route of administration, pointing to pharmacokinetics as an important aspect of abuse potential in addition to DAT inhibition (Chait et al., 1987; Dutta et al., 2003).
Mazindol is selective toward hDAT over hSERT and has been used as a starting molecule in the synthesis of different functionalized analogs by Houlihan et al. (2002). Novel analogs were synthesized and tested in this study (Fig. 1). Here we explore the binding of mazindol to hDAT and hSERT homology models to determine the molecular determinants of affinity and selectivity. Inhibitory potencies of mazindol and eight analogs (Fig. 1) for hSERT and hDAT mutants were explored. Of the original mazindol analogs from Houlihan et al. (2002), three have a significant preference for wild-type hDAT over wild-type hSERT; compounds 1, 2, and 3 show a more than 100-fold preference for hDAT, whereas analog 4 only shows 36-fold selectivity. The novel analogs reported here were chosen to aid experimental validation of the mazindol binding mode and they generally show lower potency; however, two analogs show increased selectivity for hSERT. By mutagenesis of a few residues lining the central binding pocket of hSERT, we show that the selectivity of analog 2, and to some extent analog 3, can be reversed from being predominantly hDAT selective to equipotent against hSERT and hDAT. This tendency can be rationalized from computational studies by means of induced fit docking (IFD) (Sherman et al., 2006) and quantum mechanics polarized ligand docking (QPLD) (Cho et al., 2005) calculations of mazindol into homology models of hDAT and hSERT.
The two enantiomers of mazindol and its analogs exist in a dynamic equilibrium between three isomers (the keto and the R and S–ol forms, respectively) with the R or S–ol being the only relevant forms at physiologic pH (Houlihan et al., 2002). These two enantiomers may have markedly different protein binding affinities, which cannot easily be measured experimentally due their presumed easy interconversion. It was necessary to computationally treat the two enantiomeric –ol forms separately, which predicts that the two enantiomers of mazindol are orientated in similar ways in both proteins. The difference in affinity between the transporters can thereby be assigned to the difference in amino acid composition in the binding pocket at primarily two positions and not to differences in ligand binding. This allows us to present the first biochemically validated binding mode of mazindol and analogs inside the central binding site of hSERT and hDAT pointing to R-mazindol as the biologically relevant enantiomer, which may facilitate progress toward the development of antiabuse drugs.
Materials and Methods
The synthesis of analogs 1–4 is described by Houlihan et al. (2002).
Organic Synthesis
Mazindol analogs 5, 6, and 8 were prepared by procedures analogous to those described by Houlihan and Parrino (1982). See the Supplemental Experimental Procedures for details.
Crystallography of Analog 5
Please see Supplemental Fig. 1 for details.
Mutagenesis
Mutations in hSERT or hDAT pcDNA inserted into the pcDNA3.1 vector were introduced by mismatched primer pairs in a polymerase reaction using Phusion (Finnzymes, Vantaa, Finland). DNA was purified from overnight XL10 Gold Escherichia coli cultures grown in LB media supplemented with 200 ng/ml ampicillin using the PureYield Midiprep Kit (Promega, Madison, WI). Mutant DNA was sequenced across the entire transporter open reading frame using BigDye v3.1 chemistry (Applied Biosystems, Foster City, CA) analyzed on an ABI 3100 Sequencer (Applied Biosystems) to verify that the transporter gene contained the desired mutations and that no unwanted mutations had been introduced.
Cell Culture
Human embryonic kidney 293MSR cells (Invitrogen, Carlsbad, CA) were cultured in Dulbecco’s modified Eagle’s medium (BioWhitaker, Mississauga, ON, Canada) supplemented with 10% fetal calf serum (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin (BioWhitaker), and 6 μg/ml Geneticin (Invitrogen) at 95% humidity and 5% p(CO2) at 37°C. Two days prior to the uptake assay, cells were detached by Versene and trypsin/EDTA treatment and mixed with a preformed complex of transporter DNA and LipofectAMINE 2000 (Invitrogen). This transfection mix was dispensed into TC-treated white 96-well plates (Nalgene Nunc International, Penfield, NY) at a cell density of 50–70% confluence and 0.167–0.333 µg DNA per cm2 and incubated for 50–60 hours.
Uptake Inhibition Assay
Adherent transfected cells were washed with phosphate-buffered saline with calcium and magnesium (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, 0.1 mM CaCl2, and 1 mM MgCl2, pH 7.4) and preincubated for 25 minutes with a dilution series of the inhibitor. Uptake was initiated by adding a mixture of 50–100 nM [3H]serotonin or [3H]dopamine (PerkinElmer, Waltham, MA) and the inhibitor at the same concentration as in the preincubation. Radioactive neurotransmitter uptake was allowed to proceed for 10 minutes at 22°C and was terminated by washing with phosphate-buffered saline with calcium and magnesium. Aspirated cells were lysed with Microscint 20 (Packard BioScience Co., Meriden, CT) and the accumulated radioactive neurotransmitter was quantified on a Packard Topcounter.
Data Analysis
Radioactive counts from accumulated neurotransmitters were fitted to a sigmoidal dose-response curve in GraphPad Prism 3 software (GraphPad Software, Inc., La Jolla, CA). The resulting IC50 values were transformed to Ki values using the Cheng–Prusoff equation. Statistical comparison of Ki values was conducted using the t test.
Molecular Modeling
Homology Modeling.
One homology model of hDAT and one homology model of hSERT were used in this study. Both models were constructed using the crystal structure of the Aquifex aolicus leucine transporter (LeuT) as a template (Yamashita et al., 2005). The alignment used between LeuT and hSERT and LeuT and hDAT, respectively, was the thoroughly refined one of the neurotransmitter sodium symporters published by Beuming et al. (2006) and was previously used by us with success (Celik et al., 2008b; Koldsø et al., 2010, 2011, 2013a,b; Sinning et al., 2010; Severinsen et al., 2012).
The hSERT homology model used in the present study is a further refined model based on our previously validated model of hSERT with serotonin bound (Celik et al., 2008b), including the loop optimization described by Koldsø et al. (2010, 2013b). The hDAT homology model was constructed by the use of LeuT and the optimized extracellular loop 2 of hSERT as templates and built in MODELLER9v5 (Fiser et al., 2002; Eswar et al., 2007) as previously described (Koldsø et al., 2013b). During the modeling procedure of hDAT, 20 models of the protein were built. The main validation criteria for these models were as follows: 1) the model quality as measured by the probability density function, molpdf (Eswar et al., 2007); 2) the stereochemical quality illustrated by a Ramachandran plot using PROCHECK (Laskowski et al., 1993); 3) the size of the central cavity binding site of the protein measured the solvent accessible area method with a 1.2 Å probe in Molegro Virtual Docker (version MVD2008.2.4; http://www.molegro.com) (Thomsen and Christensen, 2006); and 4) the χ1 angle of Asp79 in hDAT, which should be ±gauche as was previously suggested (Jørgensen et al., 2007; Celik et al., 2008b; Koldsø et al., 2010, 2013b).
The sodium ions found in the LeuT crystal structure were imported with the same coordinates into the final hDAT and hSERT models, and the chloride ion was manually introduced in both models at the proposed binding site (Forrest et al., 2007; Zomot et al., 2007) followed by a brief minimization.
Ligand Preparation.
Both enantiomers of mazindol were built in Maestro (version 8.5; Schrödinger, LLC, New York, NY). See Supplemental Fig. 2 for MacroModel atom types for mazindol. The pKa values of ionizable groups were predicted by Epik (version 1.6; Schrödinger, LLC) within the Schrödinger software. The nitrogen at position 1, N1 (see Fig. 1 for atom labeling), had a predicted pKa value of 9.12 and should be charged at physiologic pH. Furthermore, because of the possibility of conjugation and charge delocalization between N1 and N4 in the imidazoline ring, N1 and N4 were chosen to be sp2-hybridized and thus planar. To investigate the structure of the two enantiomers of mazindol more precisely, a quantum mechanics (QM) optimization was made using the density functional theory method B3LYP (Lee et al., 1988; Becke, 1993; Stephens et al., 1994) with the 6-31+G(d) basis set (Hehre et al., 1972). The QM optimized structure deviates slightly from planarity around the charged conjugated nitrogen, N1, with a few degrees, which is most likely caused by the intramolecular strain of the tricyclic ring system. The energy minimized structure from the QM optimization was then used as the input for a Monte Carlo conformational search, using the MCMM methodology in MacroModel (version 9.6; Schrödinger, LLC) with the MMFFs force field in an implicit water model. This specific force field was selected because it has been optimized to keep sp2-hybridized nitrogens planar (Halgren, 1996, 1999) with atom types N4 and N2 to describe the two nitrogen atoms of the imidazoline ring (see Supplemental Fig. 2 for details). The conformational search was performed to identify all low energy conformations of mazindol and the global minimum of each enantiomer was used as the input structure for the following IFD simulation.
IFDs with the positive charge localized on N4 were also performed (see the summary in the Supplemental Experimental Procedures). However, these results do not differ from the results from IFD with N1 modeled as positively charged and thus only the results from the latter will be analyzed in detail here.
Protein Preparation.
The protein complexes were prepared for docking calculations by the Schrödinger 2008 Suite Protein Preparation Wizard. Herein, the protonation states and bond orders of the amino acids were initially predicted and afterward optimized. The automatically assigned protonation states were used in most cases; however, two residues in both protein complexes were changed manually. These residues were Asp524 and Glu508 in hSERT and Glu396 and Glu491 in hDAT, which, according to PROPKA 2.0 predictions (Li et al., 2005; Bas et al., 2008), were both suggested to be neutral at physiologic pH. The protonation state of Glu508 in hSERT and Glu491 in hDAT as neutral is further supported by the finding that similar Glu–Glu pairs are conserved in the NNS family as revealed in the LeuT crystal structure (Forrest et al., 2007). Most histidines in hSERT were retained as the δ-tautomer, except His240, which was modeled as charged. In the model of hDAT, His165, His179, His225, His375, and His444 were all modeled as the ε-tautomer. Subsequently, each protein was subjected to a constrained minimization within the OPLS-AA force field (Jorgensen and Tirado-Rives, 1988) and converged to a root mean squared deviation of 0.3 Å. The refined protein structures were then used as the input structures for the following IFD studies.
IFD Simulations.
Since mazindol is significantly larger than leucine, which was bound inside the binding pocket of the template LeuT, it was thus necessary to include protein flexibility in the docking simulations. The Schrödinger Suite 2008 Induced Fit Docking protocol (Sherman et al., 2006) was used throughout this study. In the IFD workflow, some side chain flexibility is introduced for residues with at least one atom within a distance of 5 Å from the ligand. The homology models of hDAT and hSERT do not contain a bound ligand so the binding site was defined by residues previously shown to be important for inhibitor binding, which were Asp98 and Ile172 in hSERT (Henry et al., 2006) and Asp79 and Val152 in hDAT (Lee et al., 2000; Beuming et al., 2008), respectively. The number of poses to save in both the initial and the redocking stages of the IFD protocol was changed from the default value of 20 to 100. Furthermore, the energy window of the Prime energy in the sorting and filtering step was changed from the default value of 30 kcal/mol to 50 kcal/mol to allow for more diversity among the saved docking poses. The scoring function applied in the initial soft docking was standard precision (Friesner et al., 2004), whereas the extra precision scoring function (Friesner et al., 2006) was applied in the redocking stage of the IFD protocol.
Binding Site Optimization.
The binding site of the best docking poses from the IFD calculations, judged from GlideScore and Emodel, were subsequently energy minimized in MacroModel (version 9.6; Schrödinger, LLC) with the conjugated gradient method until convergence using the OPLS-AA (Jorgensen and Tirado-Rives, 1988) force field with no solvent. The ligand was used to define the center and complete residues in a radius of 8 Å are allowed to move freely during minimization. Surrounding the freely moving area, a shell with a radius of 10 Å was constrained with a force constant of 200 kJ/mol⋅Å2 hereby allowing only moderate flexibility of this part of the protein structure, leaving the rest of the protein frozen during this calculation. The refined protein/ligand complexes were used in the following QPLD docking simulations.
QPLD.
The binding modes identified from the IFD calculations of each enantiomer bound to both of the protein models were then further evaluated by the Schrödinger QM-Polarized Ligand Docking protocol. QPLD is a quantum mechanics/molecular mechanics–based docking method (Cho et al., 2005) combining Glide (Friesner et al., 2004) and QSite (Murphy et al., 2000). During the QPLD procedure, the ligands are initially docked into a rigid protein using Glide. The resulting binding modes of the ligands are then used for calculation of partial charges of the ligand by a single-point calculation in QSite. By this method, the effect of the polarization from the protein experienced by the bound ligand is taken into account in the final docking stage in which the partial charges obtained from the QM calculations of the ligand are used. No protein flexibility is possible during the QPLD calculation; however, the ligands are treated as flexible in each of the two docking stages. Three different setups were tested for these calculations exploring the two scoring functions in the initial docking and final redocking stages. In all setups, the center for the grid calculations for the quantum mechanics/molecular mechanics step was defined by the position of the ligand in the minimized best structure from the IFD calculations. The number of poses to be returned in each setup is set to 20. The QM level in the QPLD protocol is set to Accurate, which implies that the density functional theory method B3LYP (Lee et al., 1988; Becke, 1993; Stephens et al., 1994) is applied with the 6-31G(d) basis set.
Results
Molecular Modeling.
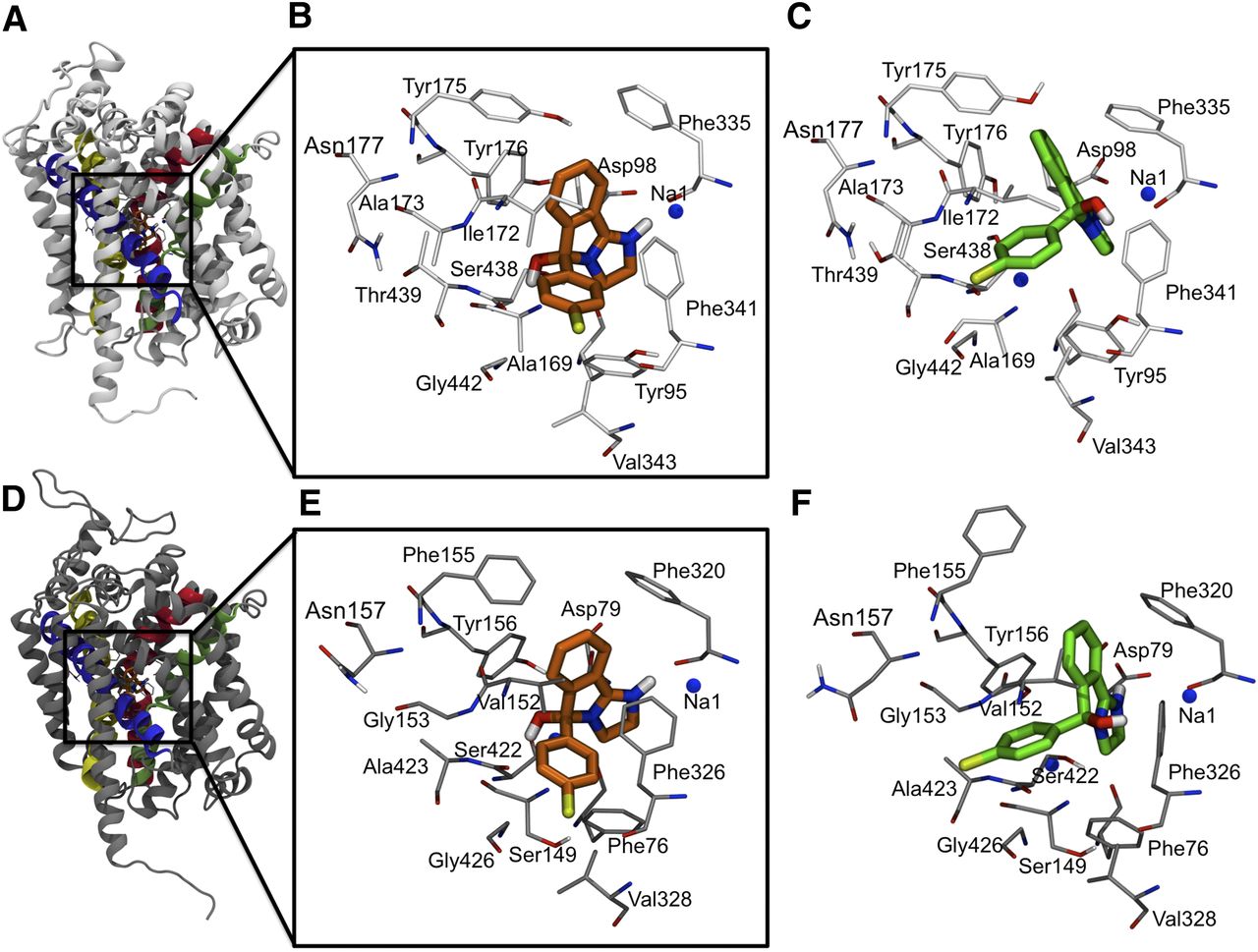
Since mazindol has the positive charge delocalized over two nitrogen atoms, two binding modes can be expected where either N1 or N4 interacts with Asp98 in hSERT and Asp79 in hDAT assuming this ionic interaction to be present also for mazindol, similarly to what was previously found for 5-hydroxytryptamine/serotonin, imipramine, and citalopram (Celik et al., 2008b; Andersen et al., 2009; Koldsø et al., 2010; Sinning et al., 2010) in hSERT and for dopamine and cocaine in hDAT (Koldsø et al., 2013b). It was previously proposed that cocaine binds in a similar fashion to both hDAT and hSERT (Beuming et al., 2008), and it can thus be hypothesized that mazindol also does this. Our experimental data (see below) support these findings, and only binding modes identified in both proteins will thus be analyzed in detail here. One dominating binding mode of R-mazindol in hSERT, as well as hDAT, was identified from the docking calculations in which N1 forms an ionic interaction with Asp98 in hSERT, or the equivalent Asp79, in hDAT. This binding mode is termed R-C1 in the following sections of this article (Fig. 2, B and E). For R-mazindol, no poses with an ionic interaction below 4.5 Å between N4 and Asp79 in hDAT were found. However, two poses in hSERT were different from R-C1 and these two, termed R-C2, showed an ionic interaction with a distance of less than 4 Å between N4 and Asp98 in hSERT. Since the R-C2 binding mode is only present in hSERT, it will not be analyzed further at this time. The R-C1 binding mode of mazindol furthermore shows a hydrogen bond between the positive N1 atom of mazindol and the backbone carbonyl group of Phe335 in hSERT and Phe320 in hDAT. Phe335 is one of the two aromatic residues that form an aromatic lid on top of the binding site as observed in the crystal structures of LeuT and other studies (Celik et al., 2008a,b) and it is conserved among almost all neurotransmitter sodium symporters (Yamashita et al., 2005; Beuming et al., 2006). Furthermore, the hydroxyl group at the spiro-center in R-C1 can form a hydrogen bond with the backbone of Ser438 in hSERT and Ser422 in hDAT. The chloro substituent of mazindol in R-C1 resides in a pocket lined by residues Ile165, Ile168, Ala169, Ile172, Phe341, Val343, and Gly442 in hSERT and Phe76, Ile148, Ser149, Val152, Phe326, Gly327, Val328, Gly425, Gly426, and Ser429 in hDAT, all within 5 Å of the chloro group.

The binding mode of mazindol in hSERT and hDAT. (A) The homology model of hSERT. The transmembrane (TM) parts forming the binding site are highlighted with TM1 in pink, TM3 in purple, TM6 in green, and TM8 in yellow. (B) R-Mazindol bound in hSERT is shown with selected residues lining the binding pocket. R-C1 is shown in orange and the protein in light gray. (C) S-Mazindol bound in hSERT is shown with selected residues around the binding pocket. S-C1 is shown in green and the protein in light gray. (D) The homology model of hDAT. The TM parts forming the binding site are highlighted with TM1 in pink, TM3 in purple, TM6 in green, and TM8 in yellow. (E) R-Mazindol in hDAT is shown with selected residues of the binding pocket. R-C1 is shown in orange and the protein in dark gray. (F) S-Mazindol in hDAT is shown with selected residues of the binding pocket. S-C1 is shown in green and the protein in dark gray.
Comparable with observations for R-mazindol, one similar binding mode was observed for S-mazindol in both hSERT and hDAT with an ionic interaction between N1 and Asp98 and Asp79, respectively. This binding mode is termed S-C1. From the IFD calculations in hDAT, another binding mode (S-C2) was observed with distances between N4 and Asp79 falling below 4.2 Å. This cluster will not be analyzed further because of the lack of its presence in both transporters. From the IFD with the positive charged localized on N4, another binding mode (S-C3), only found in hSERT, was observed. These poses will not be analyzed further because of the experimental data below and the absence of this binding mode in hDAT. The S-C1 binding mode was almost identical between the two different proteins, with only a very small variation in the binding pattern (compare Fig. 2, C and F). In S-C1, the hydroxyl group is orientated toward Phe341 in hSERT and Phe326 in hDAT. The positively charged nitrogen in S-C1 forms charge-reinforced hydrogen bonds with Asp98 in hSERT and Asp79 in hDAT. Furthermore, the chloro substituent in S-C1 is located in the pocket surrounded by Ala169, Ala173, and Gly442 in hSERT and Gly153, Asn157, Ala423, and Met427 in hDAT. The statistics from the different docking setups are listed in Supplemental Table 1 and Table 1. It is evident that the docking scores are similar for both enantiomers in the dominant clusters S-C1 and R-C1 in the two proteins hSERT and hDAT (see column 8 in Table 1). In addition, the ionic interaction distance between the N1+ of the ligand and Asp79 and Asp98 in hDAT and hSERT, respectively, is similar among each enantiomer (see column six in Table 1).
Statistics for the clusters identified from the different setups applied
The mean values [S.D.] are listed for distance, GlideScore, and Emodel. Data for all poses are included in Supplemental Table 1.
In summary, the “core” of mazindol, the fused tricyclic ring system, is predicted to be located in the same orientation inside both proteins for both enantiomers. The major difference between the predicted bindings of the two enantiomers is the orientation of the hydroxyl group and the chlorophenyl group; thus, unequivocal answers may not be expected related to substitutions at the 4′-position if both enantiomers indeed bind to the protein. From the docking scores, it seems most likely that the two enantiomers of mazindol bind with similar affinities.
Experimental Validation of Mazindol Orientation in hSERT and hDAT.
To validate the predicted binding modes, structure-activity measurements were conducted for a battery of mazindol analogs against hSERT and hDAT mutants. The biochemical experiments were performed with racemic mixtures; however, this is appropriate since the two enantiomeric forms are presumed to be readily interchangeable and to bind with similar affinities.
The paired mutation ligand analog complementation (PaMLAC) paradigm was used to validate the binding modes of mazindol in hSERT and hDAT predicted from the docking studies (S-C1, R-C1; Fig. 2). PaMLAC provides a complementary method in which a battery of mazindol analogs and complementary protein mutants allows us to extensively investigate the position of specific ligand-protein interaction points. In this study, we used racemic analogs previously described by Houlihan et al. (2002) with four novel mazindol analogs (4′ substituent analogs 5–8). Compound 5 was furthermore crystallized and the structure was solved by crystallography (see the Supplemental Experimental Procedures). The analogs used to study the binding are substituted either on the “core” of the ligand at positions 6, 7, and 9 (see Fig. 1) or at the para-position (4′-position) of the phenyl group. The mutants used were selected so that the mutated amino acid residue lines the putative binding pocket of the protein. The measured Ki values are listed in Supplemental Tables 2–4 and Tables 2, 3, and 4.
Ki values from 3H-serotonin uptake inhibition experiments in wild-type hSERT and hSERT mutations of residues in the central binding site diverging between hSERT and hDAT
Confidence levels are provided in Supplemental Table 2.
Ki values from 3H-dopamine uptake inhibition experiments in wild-type hDAT and hDAT mutations of residues in the central binding site diverging between hSERT and hDAT
Confidence levels are provided in Supplemental Table 3.
Ki values from serotonin and dopamine uptake inhibition experiments in wild-type hDAT, wild-type hSERT, and mutants
Confidence levels are provided in Supplemental Table 4. Residues Val343, Thr439, Leu443, and possibly Asn177 are suggested by S-C1 to interact with the 4′-substituent on mazindol, whereas A173, and possibly Ile165 are suggested by R-C1 to interact with the 4′-substituent on mazindol. Ala169 is located between the 4′-substituent on mazindol in R-C1 and S-C1.
Orientation of the Tertiary Ammonium Group of Mazindol.
The possible ionic interaction between the charged N1 position of mazindol and Asp98 in hSERT and Asp78 in hDAT is the dominant docking poses of both enantiomers in both proteins. This type of charge-reinforced interaction was previously seen in other compounds that interact with hSERT (Celik et al., 2008b; Koldsø et al., 2010, 2011, 2013a; Sinning et al., 2010), hDAT (Beuming et al., 2008), or both (Severinsen et al., 2012; Koldsø et al., 2013b) and the presence of a protonated ammonium group is a hallmark of high-affinity inhibitors of SERT and DAT.
hSERT Tyr95/hDAT Phe76 Is the Primary Determinant of Mazindol Selectivity for hDAT over hSERT.
Mazindol is 3.4-fold more selective for wild-type hDAT than for wild-type hSERT (Ki,hDAT wt = 140 nM versus Ki,hSERT wt = 480 nM; P < 0.0001). This selectivity can be fully reversed to a selectivity of 0.11 when introducing the corresponding hDAT residue on hSERT position 95 and the corresponding hSERT residue at the equivalent hDAT position 76 (Ki,hDAT Phe76Tyr = 1700 nM versus Ki,hSERT Tyr95Phe = 182 nM; P = 0.0002). This shows that Tyr95 in hSERT and the corresponding Phe76 in hDAT is the primary determinant of mazindol selectivity, as previously shown by Barker et al. (1998). No other single mutant in the binding site of residues diverging between wild-type hDAT and wild-type hSERT (see Tables 2 and 3) shows a similar response; however, secondary and tertiary mutations in the hSERT Tyr95Phe or hDAT Phe76Tyr background, in particular hDAT Val152Ile, hDAT Met427Leu, hSERT Ile172Val, and hSERT Leu443Met, further enhance the response (Tables 2 and 3).
Location of the 6-Position by Use of Analog 1.
The location of the 6-position of mazindol in hSERT has been investigated by measuring inhibitory potencies of analog 1 for wild-type transporters and single mutant constructs in uptake inhibition experiments. The affinity of analog 1 in hDAT is 160-fold better than in hSERT, with Ki values of 14,800 nM in hSERT (Table 2) and 92 nM in hDAT (Table 3). When comparing the inhibitory potency of analog 1 with that for mazindol, a 31-fold decrease is observed in hSERT, with Ki values of 14,800 nM (analog 1) and 480 nM (mazindol). The opposite selectivity is observed in hDAT with a very small 1.5-fold increase in potency upon binding of analog 1 compared with mazindol, with Ki values of 92 nM (analog 1) and 140 nM (mazindol). However, some of the lost affinity can be regained in hSERT when introducing the Tyr175Phe mutation. In this situation, the affinity of analog 1 is increased 7-fold compared with wild-type hSERT (Ki,Tyr175Phe = 2200 nM versus Ki,wt = 14,800 nM; P < 0.0001), whereas the effect of introducing the Tyr175Phe mutation is only a limited 1.6-fold increase for mazindol (Ki,Tyr175Phe = 310 nM versus Ki,wt = 480 nM; P = 0.0225). This indicates that the 6-position must be located in a pocket close to Tyr175, which is also observed in the S-C1 and R-C1 binding models.
7-Position by Use of Analog 2.
The 7-substituted analog 2 inhibits hDAT 113-fold more potently than hSERT, with Ki values of 1940 nM in hSERT and 17.1 nM in hDAT. In addition, the 7-position of S-C1 and R-C1 from IFD and QPLD are located in close proximity to Tyr175 in hSERT and Phe155 in hDAT (see Fig. 2). In wild-type hSERT, the introduction of the 7-methoxy as in analog 2 yields decreased inhibitory potency compared with mazindol corresponding to a 0.25-fold selectivity (Ki,mazindol = 480 nM versus Ki,2 = 1940 nM; P < 0.0001), whereas the complete opposite selectivity is observed for wild-type hDAT with a 8.2 selectivity (Ki,mazindol = 140 nM versus Ki,2 = 17.1 nM; P < 0.0001). These opposite selectivities can be used to orient mazindol within the binding site. Ideally, a single mutation in hSERT that could introduce the hDAT-like selectivity in hSERT and conversely the corresponding single mutation in hDAT that could introduce a hSERT-like selectivity for analog 2 compared with mazindol would be very strong support for a direct interaction between this residue and the 7-position of mazindol. The hSERT Tyr175Phe and corresponding hDAT Phe155Tyr mutations fully satisfy this PaMLAC criterion. For hSERT, the mazindol/analog 2 selectivity shifts from 0.25 for wild-type hSERT to 4.1 for Tyr175Phe (Ki,mazindol = 310 nM versus Ki,2 = 75 nM; P = 0.0045). Similarly, for hDAT, the mazindol/analog 2 selectivity shifts from 8.2 for wild-type hDAT to 0.64 for Tyr175Phe (Ki,mazindol = 61 nM versus Ki,2 = 96 nM; P = 0.0029). These mirroring reversals of selectivity are highly significant [selectivity of 0.25 versus 4.1 (P < 0.0001) and selectivity of 8.1 versus 0.63 (P < 0.0001)] and experimentally support the computational models, in which the 7-position of S-C1 and R-C1 mazindol point toward hDAT Phe155 or hSERT Tyr175 and interact directly with substituents on the 7-position of mazindol.
Location of the 9-Position by Use of Analog 3.
We also used analog 3 to elucidate the effect of the Tyr175Phe mutation in hSERT compared with the reverse mutation Phe155Tyr in hDAT to discern between model R-C1 and S-C1. In S-C1, the 9-position of mazindol is located less than 3.5 Å from the hydroxyl group on Tyr175 and any substituents on the 9-position would be parallel to this hydroxyl and likely to interact. In R-C1, the ring system of mazindol is tilted away from Tyr175 and 9-substituents are not likely to interact with the Tyr175 hydroxyl group.
The inhibitory potency of analog 3 in the wild-type hSERT compared with mazindol is decreased 2.4-fold, with Ki values of 1140 nM (analog 3) and 480 nM (mazindol). On the contrary, the inhibitory potency of analog 3 against wild-type hDAT is 12-fold higher for analog 3 compared with mazindol, with Ki values of 11.5 nM (analog 3) and 140 nM (mazindol). This analog thus clearly has a distinct selectivity for hDAT. When introducing the Tyr175Phe mutation in hSERT, the potency of analog 3 remains unchanged compared with wild-type hSERT, with Ki values of 1240 nM (Tyr175Phe) and 1140 nM (wild-type). Similarly, when introducing the corresponding mutation in hDAT, an approximately 2-fold increase in potency of mazindol and analog 3 is observed compared with wild-type hDAT, with Ki values of 61 nM and 5.6 nM (Phe155Tyr) compared with 140 nm and 11.5 nM (wild-type), but no changes in drug selectivity are observed upon introduction of the mutation (wild-type hDAT selectivity of 12 versus hDAT Phe155Tyr selectivity of 10.8). Similarly, hSERT selectivity for analog 3 compared with mazindol remains constant upon introduction of the hSERT Tyr175Phe mutation, with wild-type hSERT selectivity of 0.42 (480 nM/1140 nM) versus hSERT Tyr175Phe selectivity of 0.25 (310 nM/1240 nM). This indicates that there is no strong interaction between the 9-position of mazindol and Tyr175 in hSERT and that this residue is not conferring the different selectivity pattern between hSERT and hDAT with respect to 9-substituted analogs, which again supports the 9-position as pointing away from the pocket lined by Tyr175 in hSERT and Phe155 in hDAT. These findings do not support the S-C1 model and point to the R-C1 model because they are suggested from the computational studies, in which the 7-position, and not the 9-position, is oriented toward Tyr175 in hSERT and the corresponding position Phe155 in hDAT (Fig. 2).
Substituents at the Phenyl Group.
The R-C1 dockings of R-mazindol in hSERT place the electrophilic 4′-chloro substituent in a subpocket lined by Ile168, Ala169, Val343, and partly Phe341, whereas the S-C1 docking of S-mazindol in hSERT instead suggests a subpocket lined by Ala173, Gly442, Leu443, and partly Ala169. We decided to identify which subpocket harbors the chloro substituent to determine the most likely stereochemistry of mazindol when bound to hSERT.
To determine the correct binding mode, we decided to focus on mutations of Ala169, Asn177, and Val343. Mutations of Gly442 were deemed unsuitable to determine the correct binding mode because mutations of a glycine within a α-helix can have profound effects on the dynamics of the helix, and also because Gly442 is located approximately halfway between the two subpockets. Ala169 is also located between the two subpockets and as such cannot be used to unambiguously validate S-C1 over R-C1 or vice versa, although the models would suggest that the A169 side chain would interact more with the 4′-substituent in R-C1 than S-C1. We find that the introduction of a hydrophilic residue, in the form of the Ala169Ser mutant, does not significantly affect the potency of mazindol or analogs 5, 6, or 8 compared with wild-type hSERT, indicating that the –Cl, −CH3, −OCH3, or −CH2OH is either unable or too large to form a hydrogen bond with the hydroxyl on the serine side chain. However, the introduction of the smaller hydrophilic 4′-hydroxy in analog 7 allows for a new hydrogen bond that increases the potency of analog 7 by a factor of 3.4 (Ki.hSERT wt = 12.3 nM versus Ki,A169S = 3.6 nM; P = 0.0016) and supports the plausibility of both S-C1 and R-C1 and lends support to the notion that R-C1 may be preferred over S-C1.
Asn177 is located deep in the subpocket harboring the 4′-chloro group in S-C1 at a distance of more than 6 Å. It is therefore unlikely that any direct interaction between the side chain and the 4′-substituent is possible but mutation of Asn177 might change the overall hydrophilicity or volume of this subpocket. All mutants with smaller residues (Ala, Ser, Thr) at position 177 exhibit significantly increased affinity for the studied analogs (Table 4) but no pattern of hydrophilic side chains pairing favorably with hydrophilic 4′-substituents or hydrophobic side chains pairing favorably with hydrophobic 4′-substituents are found. For the larger but conservative Asn177Gln mutation, the same trend of generally improved affinity is observed but much less pronounced than for the Ala, Ser, or Thr mutants, indicating that increased volume of the subpocket harboring the 4′-substituent in S-C1 is favorable but that no direct interaction between the side chain of residue 177 and the 4′-substituent can be established as predicted by both models.
Val343 is located at the bottom of the subpocket harboring the 4′-Cl in the R-C1 pose at a distance of less than 3.5 Å from the chlorine, whereas the same distance is in excess of 5.5 Å for S-C1. Mutation of the hydrophobic valine to the hydrophilic serine does not in itself affect mazindol potency significantly. Despite the presence of a potential hydrogen bonding partner in Val343Ser, the selectivity for the four different 4′-substituent analogs 5–8 compared with mazindol remains unchanged, suggesting that the side chain of Val343Ser is unable to interact with the 4′-substituent. This observation disfavors R-C1 as the correct binding mode.
The models predict that the side chain of Ala173 should have considerable impact on the selectivity for the 4′-substituted analogs for the S-C1 pose. We observe that the Ala173Leu mutation has a considerable impact on mazindol potency, improving it 37-fold compared with wild-type hSERT. However, the selectivity for the two hydrophobic analogs (5 and 6) or the two hydrophilic analogs (7 and 8) of Ala173Leu remains unchanged compared with wild-type hSERT. Similarly, the selectivity of Ala173Ser for analogs 5–8 is unchanged, suggesting that the Ala173 side chain does not interact with 4′-substituents, disfavoring S-C1.
Discussion
The IFD and the QPLD calculations of S- and R-mazindol in hSERT and hDAT resulted in one dominating common binding model for each enantiomer bound to the primary binding site in each of the refined homology models with an ionic interaction between N1 of mazindol and Asp98 (hSERT), or the equivalent Asp79 (hDAT). The predicted binding modes of R-mazindol in hSERT and hDAT were similarly oriented. Furthermore, the S-mazindol binding mode was also the same in the two protein homology models except for a small spatial displacement. In addition, the tricyclic scaffold of mazindol molecule was located similarly in both enantiomers and in both protein structures, with all forming an ionic interaction with Asp98 in hSERT and Asp79 in hDAT, respectively.
From extensive structure–activity relationship studies, it was possible to validate the predicted structure for mazindol in hSERT as well as hDAT using the PaMLAC paradigm. The method does not only rely on isolated changes in inhibitory potency of analogs but as a secondary measure also links them to the ability of complementary mutations to reverse these changes. Thus, this approach, when successful, adds an extra layer of evidence that provides strong experimental data supporting a direct interaction between a substructure of the ligand and a particular residue side chain. Conversely, if structural changes to the analog or mutations should induce a different binding mode, this is unlikely to result in PaMLAC-derived conclusions at both levels of the evaluation but instead yield incoherent patterns. For example, we used the PaMLAC methodology to map three key ligand–protein interactions with high certainty to arrive at an experimentally validated orientation of tricyclic antidepressants in hSERT (Sinning et al., 2010). The accuracy of this orientation was verified in a recent crystal structure (Penmatsa et al., 2013) demonstrating the resolution and power of the PaMLAC methodology. By utilizing several analogs, we were able to identify the difference between Tyr175 in hSERT and Phe155 in hDAT as being a key reason for the difference in affinity between these two proteins. Mazindol and the analogs all bind to hDAT with greater affinity than to hSERT.
We show that the 6-position is vicinal to hSERT Tyr175Phe just as predicted by poses R-C1 and S-C1. This means that this phenyl group penetrates the aromatic lid composed of hSERT Tyr176 and Phe335 just as has been observed for other inhibitors (Koldsø et al., 2010; Sinning, et al., 2010) and suggests the same inhibitory mechanism (i.e., inability to close the aromatic lid prevents translocation of the ligand bound to the central substrate site). This proposal is seconded by Gabrielsen et al. (2012), who included R-mazindol in the utilization of a flexible docking protocol for different inhibitors of SERT and found that one of its aromatic groups forced Tyr176 and Phe335 apart. Furthermore, we observe that a substituent on the 9-position is not sensitive to hSERT Tyr175 mutation as predicted from poses R-C1 and S-C1.
However, the single most striking result is that it was possible to reverse the selectivity of analog 2 in hSERT by introduction of the Tyr175Phe mutation. This mutation makes this position in hSERT similar to the corresponding position in hDAT, and a large gain in affinity of analog 2 is observed (26-fold). Similarly, potency decreases when introducing the Phe155Tyr mutation in hDAT, thereby constructing a hSERT-like architecture of the binding site at this position. Consequently, we suggest that the 7-position of mazindol is oriented toward Tyr175 in hSERT and Phe155 in hDAT as observed in S-C1 and R-C1. Mazindol must, accordingly, bind in very similar ways in both hSERT and hDAT and the difference in amino acid composition must be the key difference in determining the selectivity of these types of compounds inside the binding pocket.
With the phenyl part of the tricyclic system extending out through the aromatic lid of the binding site, the chlorophenyl moiety is likely to be buried inside the central substrate site. Both R-C1 and S-C1 predict this to be the case, however, with different location of the chloro group. The inhibition data for Ala169Ser are most consistent with R-C1. However, data for Val343Ser could not corroborate this observation, whereas inhibition data for Ala173 mutants is mostly consistent with R-C1. Therefore, the preponderance of the inhibition data points to R-C1 as the most likely binding mode and could therefore indicate a preference for the R-enantiomer of mazindol. The docking results by Gabrielsen et al. (2012) suggest an alternative orientation of R-mazindol in which the 4′-chlorophenyl protrudes out of the binding site through the aromatic lid in an orientation that is dissimilar to both S-C1 or R-C1 in our model. These differing results could be the consequence of the difficulties of different docking protocols to differentiate between two similar aromatic systems, and minor differences in the protocols might lead to the aromatic system interchanging in the computations. These discrepancies point to the importance of validation by biochemical experiments. In biochemical experiments, we find that a hydroxy group on the 4′-position can interact favorably, likely through a hydrogen bond, with residue 169 when mutated to a serine. This observation points to the 4′-position being buried in the central binding site, consistent with both S-C1 and R-C1.
Also worth mentioning is another disparity in the binding site compositions between hDAT and hSERT, in which Tyr95 in hSERT corresponds to Phe76 in hDAT. The binding data suggest that it is preferable for mazindol analogs to have a phenylalanine at this position since mazindol and all analogs gain affinity when making the Tyr95Phe mutation in hSERT, whereas a decrease in affinity is observed for mazindol and all analogs when introducing the inverted Phe76Tyr mutation in hDAT. This indicates that the property of this aromatic residue is a very important determinant in dictating the selectivity difference between hSERT and hDAT toward mazindol and analogs. In this respect, we were able to replicate the results of Barker et al. (1998), who showed that hSERT Tyr95/hDAT Phe76 is an important determinant of SERT/DAT selectivity.
The same trend also holds true when we examine the affinity of cocaine. There is a large increase in affinity of cocaine toward hSERT when the Tyr95Phe mutant is introduced, and the opposite effect is observed when the Phe76Tyr mutant is incorporated in hDAT. Mazindol has been suggested as an anti-cocaine abuse drug (Berger et al., 1989). Cocaine binds to hSERT with affinity that is very similar to mazindol; however, mazindol binds 11-fold better than cocaine to hDAT (Eshleman et al., 1999; Chen and Reith, 2002; Houlihan et al., 2002). Since hDAT is the main target for cocaine and mazindol apparently binds to the same site as cocaine (Beuming et al., 2008), it might be able to compete with cocaine and displace it from the binding site. Combined with this favorable profile of mazindol, our findings about mazindol orientation may be used to develop novel ligands based on a mazindol core structure that have a desirable pharmacological profile with respect to potential cocaine addiction pharmacotherapies.
Acknowledgments
The authors thank the late Professor William Houlihan for readily sharing analogs 1–4 with them. Jacob Overgaard is also gratefully acknowledged for help with solving crystal structures.
Authorship Contributions
Participated in research design: Severinsen, Koldsø, Wiborg, Jensen, Sinning, Schiøtt.
Conducted experiments: Severinsen, Koldsø, Thorup, Schjøth-Eskesen, Møller, Sinning.
Performed data analysis: Severinsen, Koldsø, Thorup, Schjøth-Eskesen, Møller, Jensen, Sinning, Schiøtt.
Wrote or contributed to the writing of the manuscript: Koldsø, Jensen, Sinning, Schiøtt.
Footnotes
- Received August 2, 2013.
- Accepted November 8, 2013.
K.S., H.K., K.A.V.T., C.S.-E., P.T.M., and S.S. contributed equally to this work.
This work was supported by the Lundbeck Foundation; the Carlsberg Foundation; the Novo Nordisk Foundation; the Danish Natural Science Research Council; and the Danish National Research Foundation [Grant DNRF59]. Computations were possible through allocations of time at the Centre for Scientific Computing (Aarhus, Denmark).
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- hDAT
- human dopamine transporter
- hSERT
- human serotonin transporter
- IFD
- induced fit docking
- LeuT
- Aquifex aolicus leucine transporter
- PaMLAC
- paired mutation ligand analog complementation
- QM
- quantum mechanics
- QPLD
- quantum mechanics polarized docking
- Copyright © 2013 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}