


Abstract
The biologic activity induced by ligand binding to orthosteric or allosteric sites on a G protein–coupled receptor (GPCR) is mediated by stabilization of specific receptor conformations. In the case of the β2 adrenergic receptor, these ligands are generally small-molecule agonists or antagonists. However, a monomeric single-domain antibody (nanobody) from the Camelid family was recently found to allosterically bind and stabilize an active conformation of the β2-adrenergic receptor (β2AR). Here, we set out to study the functional interaction of 18 related nanobodies with the β2AR to investigate their roles as novel tools for studying GPCR biology. Our studies revealed several sequence-related nanobody families with preferences for active (agonist-occupied) or inactive (antagonist-occupied) receptors. Flow cytometry analysis indicates that all nanobodies bind to epitopes displayed on the intracellular receptor surface; therefore, we transiently expressed them intracellularly as “intrabodies” to test their effects on β2AR-dependent signaling. Conformational specificity was preserved after intrabody conversion as demonstrated by the ability for the intracellularly expressed nanobodies to selectively bind agonist- or antagonist-occupied receptors. When expressed as intrabodies, they inhibited G protein activation (cyclic AMP accumulation), G protein–coupled receptor kinase (GRK)–mediated receptor phosphorylation, β-arrestin recruitment, and receptor internalization to varying extents. These functional effects were likely due to either steric blockade of downstream effector (Gs, β-arrestin, GRK) interactions or stabilization of specific receptor conformations which do not support effector coupling. Together, these findings strongly implicate nanobody-derived intrabodies as novel tools to study GPCR biology.
Introduction
The conversion of extracellular cues into specific intracellular responses is often mediated by G protein–coupled receptors (GPCRs) and is critical for regulating nearly all physiological processes. For these reasons, GPCRs have become popular therapeutic targets. Even though the GPCR superfamily consists of nearly 800 genes, the general mode of activation and desensitization remains remarkably conserved (Lagerstrom and Schioth, 2008).
Agonist binding to the orthosteric pocket stabilizes specific receptor conformations leading to the sequential binding of three main protein families: heterotrimeric GTP-binding proteins (G proteins), G protein–coupled receptor kinases (GRKs), and β-arrestins. The agonist-induced interaction between receptor and G protein promotes exchange of GDP for GTP, leading to dissociation of the heterotrimeric G protein subunits (Gαβγ). The free subunits bind to and regulate downstream effectors that commonly generate second messengers such as cAMP (Neves et al., 2002). Receptor desensitization is initiated by GRK-dependent phosphorylation of residues in the receptor carboxyl tail and/or in an intracellular loop, which subsequently leads to the recruitment of the multifunctional adaptor protein β-arrestin (Benovic et al., 1987). Importantly, receptor phosphorylation and β-arrestin binding require specific active receptor conformations. The coupling of β-arrestin promotes desensitization by sterically blocking further G protein binding and facilitating receptor internalization by acting as an adaptor for the endocytic machinery (reviewed in Shenoy and Lefkowitz, 2011). In addition to their role in controlling receptor silencing and trafficking, β-arrestins can also interact with a variety of signaling proteins leading to a unique mechanism of receptor-mediated signal transduction (see Shukla et al., 2011, for a comprehensive review).
The biologic activity of most GPCRs is regulated by ligand-mediated stabilization of specific receptor conformations that activate or inhibit downstream signaling events. In the case of the β2-adrenergic receptor (β2AR), these ligands have been primarily small molecules that bind to the orthosteric binding site. Alternatively, allosteric ligands, which bind sites topographically distinct from the orthosteric binding pocket, could be useful in further stabilizing specific receptor conformational states. With particular relevance to the β2AR, a single-domain Camelid heavy-chain–only antibody (nanobody) was recently described that allosterically binds to and stabilizes a specific agonist-activated β2AR conformation, which was essential for capturing the active state by crystallography (Rasmussen et al., 2011a).
More broadly, the use of antibodies as GPCR allosteric modulators is well founded given their ability to bind a vast array of protein epitopes with high specificity and affinity (reviewed in Gupta et al., 2008). The use of antibodies to fine-tune receptor function in a whole-cell context has mainly been restricted to those that bind extracellular epitopes, since antibody delivery into the cytoplasm is a challenging endeavor. Intracellular expression of correctly folded antibodies is also very difficult due to the reducing cytoplasmic environment. Although intracellular expression of antibodies (intrabodies) can be achieved by conversion into single-chain fragment variants, antibody function and specificity is often lost (Lo et al., 2008).
Heavy-chain antibodies from the Camelid family, which lack the light-chain polypeptide and first constant domain, have become of interest since they can be converted into functional single-domain–containing antibodies (nanobodies) consisting of a single monomeric variable domain (Vincke and Muyldermans, 2012). Their small size (∼12 kDa), enhanced stability, high protein yields in bacterial expression systems, and high propensity for antigen recognition have made nanobodies very useful tools in protein biology. It has been well documented that nanobodies are capable of binding cryptic epitopes such as those found in the catalytic site of enzymes or ligand binding pockets of receptors (reviewed in Muyldermans, 2013). Furthermore, specific nanobodies have been found to exert agonistic or antagonistic effects on their antigens either as orthosteric or allosteric modulators, making them interesting pharmacological tools.
In light of the demonstrated ability of nanobodies to bind distinct conformational states of the β2AR and their single-domain nature, we hypothesized that intracellular expression of β2AR-specific nanobodies could yield novel tools for the study and regulation of receptor function.
Materials and Methods
Reagents, Cell Culture, and Plasmids.
Human embryonic kidney 293 (HEK293) and U2OS cells were purchased from the American Type Culture Collection (Manassas, VA) and grown under the recommended conditions. FLAG-human β2AR and rat β-arrestin-2 were subcloned into the mammalian expression plasmids pBK-CMV or pEGFP, respectively, using standard procedures. To generate intrabodies, nanobody cDNA was amplified using the 5′-GGGGGATCCAGTATGTACCCATACGATGTTCCAGATTACGCTCAGGTGCAGCTGCAGGA GTCTG-3′ and 5′-GGGCTCGAGCTATGAGGAGACGGTGACCTGGG-3′ primers and subcloned into pcDNA3.1 with an N terminus hemagglutinin (HA) or Myc tag. The pcDNA3.1 β2AR-Gsα fusion plasmid was constructed based on the strategy by Bertin et al. (1994). HEK293 cells stably expressing 3 pmol/mg β2AR were maintained as previously described (Shenoy et al., 2006). M2 antibody–conjugated agarose beads, horseradish peroxidase (HRP)–conjugated M2 antibody, tubulin antibody, and all β2AR ligands were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies against total (H20) and p355/6 (sc-22191-R) β2AR were obtained from Santa Cruz Biotechnology (Dallas, TX). HA-tag antibody, peptide-N-glycosidase F, and all secondary immunofluorescent reagents were purchased from Cell Signaling Technology (Danvers, MA), New England Biolabs (Ipswich, MA), and Invitrogen (Carlsbad, CA), respectively.
Nanobody Generation and Purification.
Camelid single-chain antibody fragments (nanobodies) show promise for stabilizing active GPCR conformations and as chaperones for crystallogenesis. β2AR-specific nanobodies were generated as previously described (Steyaert and Kobilka, 2011). In brief, one llama (Lama glama) received six weekly injections of 100 μg of lipid reconstituted β2AR truncated at residue 365 bound to the high-affinity agonist BI-167107 (5-{2-[1,1-dimethyl-2-(o-tolyl)ethylamino]-1-hydroxyethyl}-8-hydroxy-4-oxa-1-aza-1,3-dihydronaphthalen-2-on). The nanobody coding sequences were amplified from total lymphocyte RNA and subcloned into a phage display vector. After two rounds of panning, 96 individual colonies were randomly picked, and the nanobodies were produced as a soluble His-tagged protein in the periplasm of Escherichia coli. The initial solid-phase enzyme-linked immunosorbent assay (ELISA) screen identified 16 nanobodies that recognized native but not heat-denatured β2AR. All nanobodies (Nbs) were produced in E. coli and purified from the periplasmic extract via nickel-agarose. Final nanobody buffer composition consisted of 20 mM HEPES (pH 7.4) and 100 mM NaCl (buffer A).
Nanobody-β2AR ELISA.
Maxisorp (NUNC) 96-well plates (Thermo Scientific, Indianapolis, IN) were seeded with 100 µl of 10 µg/ml purified nanobody overnight at 4°C. Each new reagent addition was preceded by three 5-minute washes with 300 µl of wash buffer [buffer A plus 0.02% DDM (N-dodecyl β-D-maltopyranoside)]. The plate was blocked for 60 minutes at room temperature (RT) in wash buffer with 3% nonfat milk. Solubilized full-length human β2AR was purified as previously described (Kahsai et al., 2011) and preincubated with dimethylsulfoxide (DMSO), 10 µM BI-167107 or ICI-118551 [3-(isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol] for 30 minutes in buffer A, 0.1% DDM, and 0.5% bovine serum albumin (BSA). Immobilized nanobodies were incubated with 100 µl of 0.5 µg/ml purified β2AR for 90 minutes at RT. Captured β2AR was detected using M2-HRP (1:5000) diluted in buffer A, 0.02% DDM, and 0.5% BSA. Antibody was incubated for 1 hour at RT, plates were subsequently treated with 100 µl Ultra-TMB (Pierce, Rockford, IL), and absorbance was measured at 450 nm.
Intrabody and β2AR Expression.
HEK293 cells were transfected with Fugene (Roche, Indianapolis, IN) as described by the manufacturer. In brief, subconfluent HEK293 cells were transfected with a 3:1 ratio of pcDNA-HA-intrabody and pBK-FLAG-β2AR, respectively, and scraped into lysis buffer [20 mM HEPES, 100 mM NaCl, 0.5% maltose-neopentyl glycol (MNG)] 48 hours post-transfection. For β2AR deglycosylation, 250 units of peptide-N-glycosidase F (New England Biolabs) were incubated with MNG-extracted lysate at room temperature for 1 hour. Intrabody and β2AR expression were assessed by running equivalent total protein on SDS-PAGE and subsequently immunoblotting with HA-HRP (Cell Signaling Technology) or M2-HRP (Sigma-Aldrich) antibodies, respectively. Equal loading was assessed using an anti-tubulin antibody (Sigma-Aldrich). To determine functional receptor expression, HEK293 cells were transfected as described earlier, and 48 hours post-transfection, cells were removed from the culture dishes using a 0.5% EDTA solution (Sigma-Aldrich). One well of a 6-well dish was equally divided into four binding reactions and incubated with 30 nM [3H]CGP-12177A (4-[3-[(1,1-dimethylethyl)amino]2-hy\x{200b}droxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one hydrochloride) plus either assay buffer (Dulbecco’s modified Eagle’s medium plus 20 mM HEPES) or 10 µM propranolol (nonspecific).
Coimmunoprecipitation.
HEK293 cells stably expressing FLAG-β2AR were transfected with pcDNA-HA-intrabodies and treated with vehicle (DMSO) or 10 µM isoproterenol (ISO) for 15 minutes before scraping cells in MNG-containing lysis buffer (as described earlier). Cells were lysed while rotating at 4°C for 1 hour, and insoluble material was separated by centrifugation at 14,000g for 15 minutes. FLAG-β2AR was immunoprecipitated via FLAG-M2 beads (Sigma-Aldrich), and intrabodies were detected using a HA-tag–specific antibody.
Flow Cytometry.
Sf9 cells were infected with a baculovirus encoding an N-terminal FLAG-tagged β2AR construct or a control virus. Three days postinfection, cells were resuspended in assay media [ESF921 media (Expression Systems, Davis, CA) containing 2.5 mM CaCl2 and 0.5% BSA) at a density of 8 × 106 cells/ml and kept at 4°C for the remainder of the experiment. Cells were preincubated with DMSO, 1 μM ICI-118551, or 1 μM BI-167107. Purified nanobodies were diluted in assay media to a working concentration of 100 μg/ml, and 20 μl of cells and 20 μl of nanobody were combined. Following a 1-hour incubation, cells were harvested, washed three times with 100 μl of assay media, and resuspended in 20 μl of Dylight488-labeled anti–6× His tag antibody (1:500) (Abcam, Cambridge, MA). In parallel, cells that had not been incubated with nanobody were incubated with Dylight488-labeled anti-FLAG M1 antibody (10 μg/ml). Following a 1-hour incubation, cells were washed three times with 100 μl of assay media and resuspended in 100 μl of media containing SYTOX AADvanced Dead Cell Stain (Life Technologies, Carlsbad, CA). Flow cytometry data were collected on a FACSCantoII flow cytometer (BD Biosciences, Carlsbad, CA) and analyzed using CyFlogic software (CyFlo Ltd., Kirkkonummi, Finland).
Competition Radioligand Binding Assays.
Competition radioligand binding assays were performed on both freshly isolated whole cells and frozen membranes. Briefly, HEK293 cells expressing the β2AR were harvested using a 0.05% EDTA solution (Sigma-Aldrich) and collected via centrifugation at 500g for 1 minute. The cells were then washed and resuspended in cold whole-cell binding buffer (minimum Eagle’s medium and 20 mM HEPES, pH7.5) prior to the assay. Purified membrane stocks were prepared from β2AR -expressing Sf9 or HEK293 cells via differential centrifugation. The cells were first washed with cold phosphate-buffered saline and then dounce homogenized (100 strokes on ice) in cold homogenization buffer [75 mM Tris-HCl, pH 7.4, 2 mM EDTA, and protease inhibitor cocktail (Roche)]. The cell debris was pelleted at 500g for 10 minutes at 4°C, and the microsomal membrane fraction was subsequently recovered from the supernatant via centrifugation at 35,000g for 1 hour at 4°C. The P2 membrane pellet was resuspended in homogenization buffer plus 12.5 mM MgCl2, aliquoted, and stored at −80°C until further use. Pilot studies ensured that less than 10% of the radioligand input was bound in all assays.
Whole-cell binding assays contained 1 nM [3H]CGP-12177A and either isoproterenol (0.01 nM to 100 µM) or 10 µM propranolol (nonspecific binding) diluted in cold whole-cell binding buffer. Equilibrium was reached by incubating the 96-well plates at 4°C for 4 hours. Purified membrane binding assays contained 60 pM [125I]iodocyanopindolol, 10 nM [3H]dihydroalprenolol, or 4 nM [3H]methoxyfenoterol and either isoproterenol (0.01 nM to 100 μM) or 10 µM propranolol (nonspecific binding) diluted in assay buffer (50 mM Tris-HCL, pH 7.4, 2 mM EDTA, 12.5 mM MgCl2, 0.05% BSA). Equilibrium was reached by incubating the 96-well plates at room temperature for 90 minutes. When required, nanobodies were included at a final concentration of 1 µM. Binding assays were terminated by rapid filtration onto GF/B glass-fiber filters and washing with 8 ml of cold binding buffer using a harvester (Brandel, Gaitherburg, MD). Bound [125I] was quantified using a Packard Cobra Quantum gamma counter (Packard, San Diego, CA), and bound [3H] was quantified on a Tri-Carb 2800 liquid scintillation counter (PerkinElmer, Waltham, MA) following extraction with Lefko Fluor scintillation cocktail (RPI, Mount Prospect, IL).
[3H]Methoxyfenoterol Binding Assay.
We developed the [3H]methoxyfenoterol binding assay to determine the relative amount of β2AR in active conformations in the absence and presence of nanobodies. We based this approach on the allosteric principle that binding of agonist and transducer to distinct sites on a GPCR reciprocally enhances one another’s affinity for the receptor. Thus, agonists possess both low affinity for the uncoupled receptor and high affinity for the transducer-coupled receptor (DeLean and Lefkowitz, 1980; Gurevich et al., 1997). More generally, this principle applies to other modulators of GPCR function, including nanobodies. For example, the binding of Nb80 to the β2AR increased agonist affinity by 95-fold and was comparable to the 100-fold shift observed with Gαs (Rasmussen et al., 2011a). Based on the previous report that [3H]methoxyfenoterol binds the active β2AR with ∼100-fold greater affinity than the inactive β2AR (Toll et al., 2012), we posited that using [3H]methoxyfenoterol at a concentration roughly equal to its high affinity Kd (i.e., 4 nM) would selectively label the active pool of β2AR. Proof-of-concept experiments using a fusion between the β2AR and the Gαs subunit (Bertin et al., 1994) confirmed that 4 nM [3H]methoxyfenoterol was sensitive to increases in active β2AR (Supplemental Fig. 1). [3H]Methoxyfenoterol binding assays were performed on HEK293 and Sf9 cell membranes in the absence and presence of nanobody exactly as described earlier.
cAMP Assays.
A GloSensor cAMP biosensor (Promega, Madison, WI) which contains a modified form of firefly luciferase was used to indirectly measure G protein activation (Fan et al., 2008). In brief, enzyme complementation as a result of cAMP binding to the GloSensor biosensor results in luminescence following incubation with a luciferase substrate. HEK293 cells were plated at 35,000 in 96-well dishes, and 24 hours thereafter, each well was transiently transfected with 10 ng of GloSensor biosensor and 40 ng of pcDNA-HA-intrabody using the Fugene6 transfection reagent (Roche). Cells were incubated with GloSensor reagent for 90 minutes at 28°C 48 hours post-transfection and then treated with a dose response of ISO for 10 minutes. Luminescence was measured using a NOVOstar microplate reader (BMG Labtech, Ortenberg, Germany).
β-Arrestin Recruitment.
The recruitment of β-arrestin to the β2AR was assessed using the Tango Assay as previously described (Barnea et al., 2008). To amplify β-arrestin recruitment, β2AR with C-terminal tail replaced with that of the vasopressin-2-receptor (β2V2) (Rajagopal et al., 2011). HEK293-T cells stably expressing a tetracycline transactivator (tTA) protein driven luciferase reporter and β-arrestin-2 fused to the tobacco etch virus (TEV) protease were plated at 35,000 in 96-well dishes and transiently transfected with 40 ng of pcDNA-HA-intrabody and 10 ng of β2V2 followed by a TEV protease site and tTA transcription factor. Upon agonist stimulation, recruitment of β-arrestin–TEV leads to cleavage of the tTA fused to β2V2, resulting in nuclear translocation and transcription of the luciferase reporter. Cells were treated with a dose response of ISO 36 hours post-transfection, and the Bright-Glo luciferase substrate (Promega) was added 16 hours thereafter.
Results
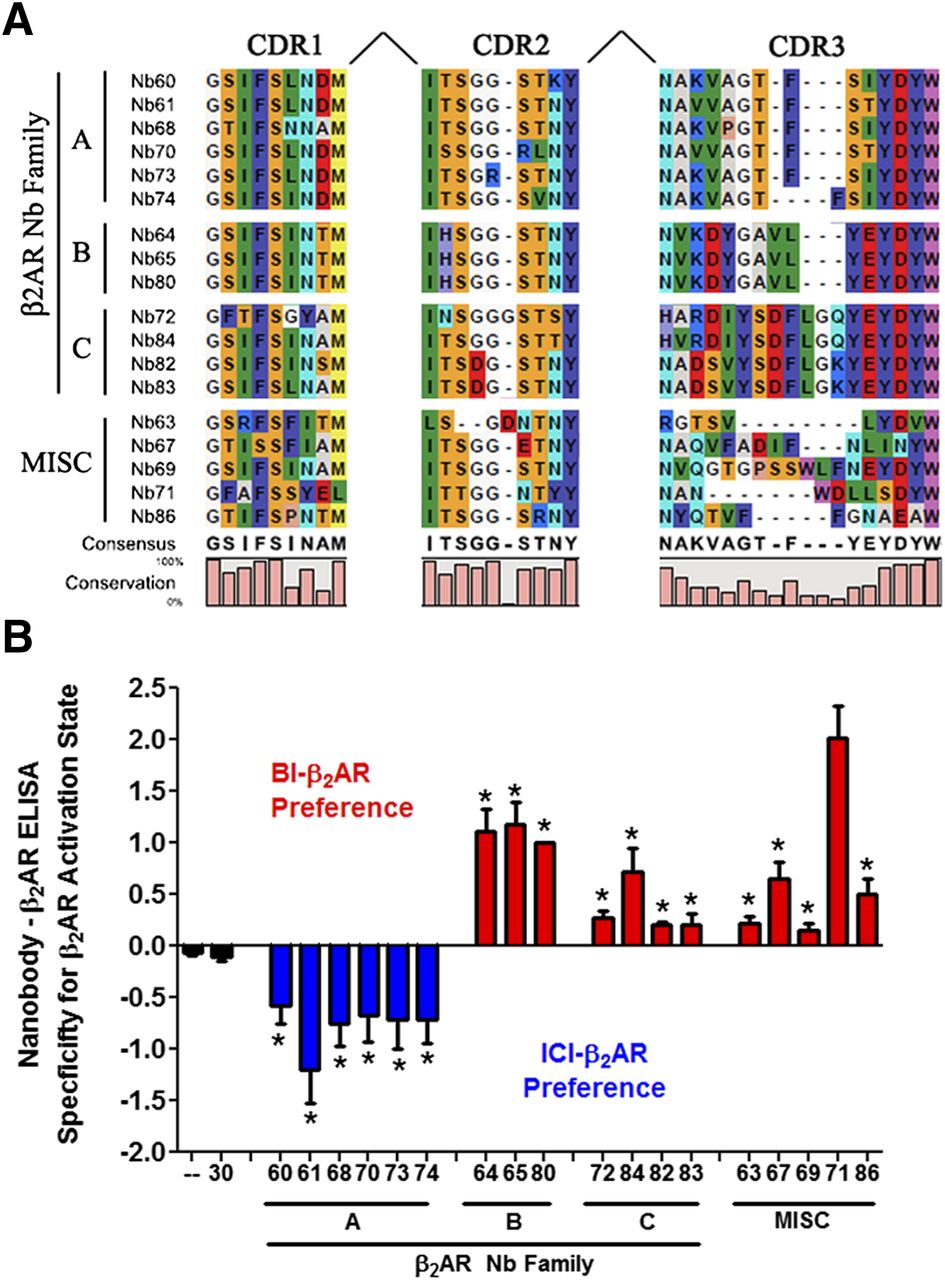
To test whether intracellular expression of Nbs could be used as a novel tool to modulate receptor function, we set out to characterize a panel of nanobodies previously found to specifically bind the β2AR. In brief, these nanobodies were obtained by immunizing one llama (L. glama) with β2AR reconstituted in lipid vesicles bound to the high affinity agonist BI-167107 (Rasmussen et al., 2011a). Sequence alignment placed the 18 unique nanobody clones into four distinct families [A, B, C, and miscellaneous (MISC)] based on complementarity determining region 3 (CDR3) conservation (Fig. 1A). Nanobodies from all four families were subsequently purified from E. coli and tested in an ELISA assay to characterize their propensity to bind agonist- or antagonist-occupied β2AR. Consistent with the demonstrated ability for Nb80 to bind activated β2AR (Rasmussen et al., 2011a), nanobodies in family B showed a clear preference for agonist (BI-167107)-occupied β2AR when compared with the inverse agonist (ICI-118551)-occupied β2AR (Fig. 1B). Similar results were obtained for Nb families C and MISC, although their preferences for the activated receptor were more variable (Fig. 1B). Importantly, the control nanobody (Nb30) that was obtained in a separate immunization using a different antigen did not recognize the β2AR regardless of ligand occupancy.

Identification of conformationally selective β2AR nanobodies. (A) Classification of β2AR nanobodies based on CDR3 conservation. Nanobodies with a divergent CDR3 were categorized as MISC. (B) ELISA assay describing nanobody selectivity for active agonist-bound (BI-167107) or inactive antagonist-bound (ICI-118551) β2AR. Using immobilized nanobody, the relative capture of BI-167107–β2AR was subtracted from that derived with ICI-118551–β2AR; values greater than 0 (blue bars) denote nanobodies that preferentially bound BI-167107–β2AR, whereas negative values represent preference for ICI-118551–β2AR. The absence of nanobody (—) and Nb30 were negative controls. T-tests were performed to determine the significance between each nanobody and (—) (*P < 0.05).
To confirm that these nanobodies are truly stabilizing an active receptor state and not just a unique conformation specific to BI-167107, we measured Nb80 binding to receptor by ELISA in the presence of various β2AR antagonists and agonists. Nb80 robustly bound to agonist (BI-167107, ISO, and formoterol)-occupied β2AR, but this binding was significantly reduced or eliminated in the presence of a partial agonist salbutamol or antagonists (ICI-118551, Carazolol), respectively (Supplemental Fig. 1). Although family A Nbs could bind to the active BI-167107–occupied β2AR, they clearly favored the inactive ICI-118551–occupied β2AR (Fig. 1B). These data indicate that all nanobodies screened have a preference for binding specific active (families B, C, and MISC) or inactive (family A) β2AR conformations. Moreover, the observation that occupancy of the β2AR by BI-167107 or ICI-118551 increased Nb binding suggests they bind to an allosteric site distinct from the orthosteric binding pocket. We next assessed the ability of Nbs to stabilize active or inactive β2AR conformations by measuring their respective abilities to increase or decrease binding of a radiolabeled β2AR agonist, [3H]methoxyfenoterol (Toll et al., 2012). It has long been established that allosteric transducers such as G proteins and β-arrestins promote high affinity agonist binding by stabilizing active receptor conformations (Williams and Lefkowitz, 1977; DeLean and Lefkowitz, 1980). Based on this premise, we selectively labeled the active pool of β2AR using a low concentration of [3H]methoxyfenoterol (i.e., 4 nM; see Materials and Methods for details). As shown in Supplemental Fig. 2, [3H]methoxyfenoterol binding was enhanced upon stabilization of a β2AR high affinity state induced by G protein binding. When tested in this assay, the majority of nanobodies in families B, C, and MISC significantly increased [3H]methoxyfenoterol binding relative to the control Nb30 (Fig. 2). This is consistent with their preferences for binding active β2AR conformations (Fig. 1B). By contrast, family A nanobodies moderately, but significantly, decreased [3H]methoxyfenoterol binding, which was in good agreement with their preference for the inactive receptor (Fig. 1B). Taken together, these data suggest that family A nanobodies stabilize inactive β2AR conformations, whereas families B, C, and MISC stabilize active β2AR conformations.

Nanobodies allosterically stabilize active or inactive β2AR conformations. Sf9 insect cell membranes expressing FLAG-β2AR were incubated with 4 nM [3H]methoxyfenoterol and 1 µM Nb in the absence (total binding) or presence of 10 µM propranolol (nonspecific). Specific binding was determined by subtracting nonspecific from total binding and normalized relative to Nb80. The absence of nanobody (—) and Nb30 were negative controls. t tests were performed to determine the significance between each nanobody and (—) (*P < 0.05).
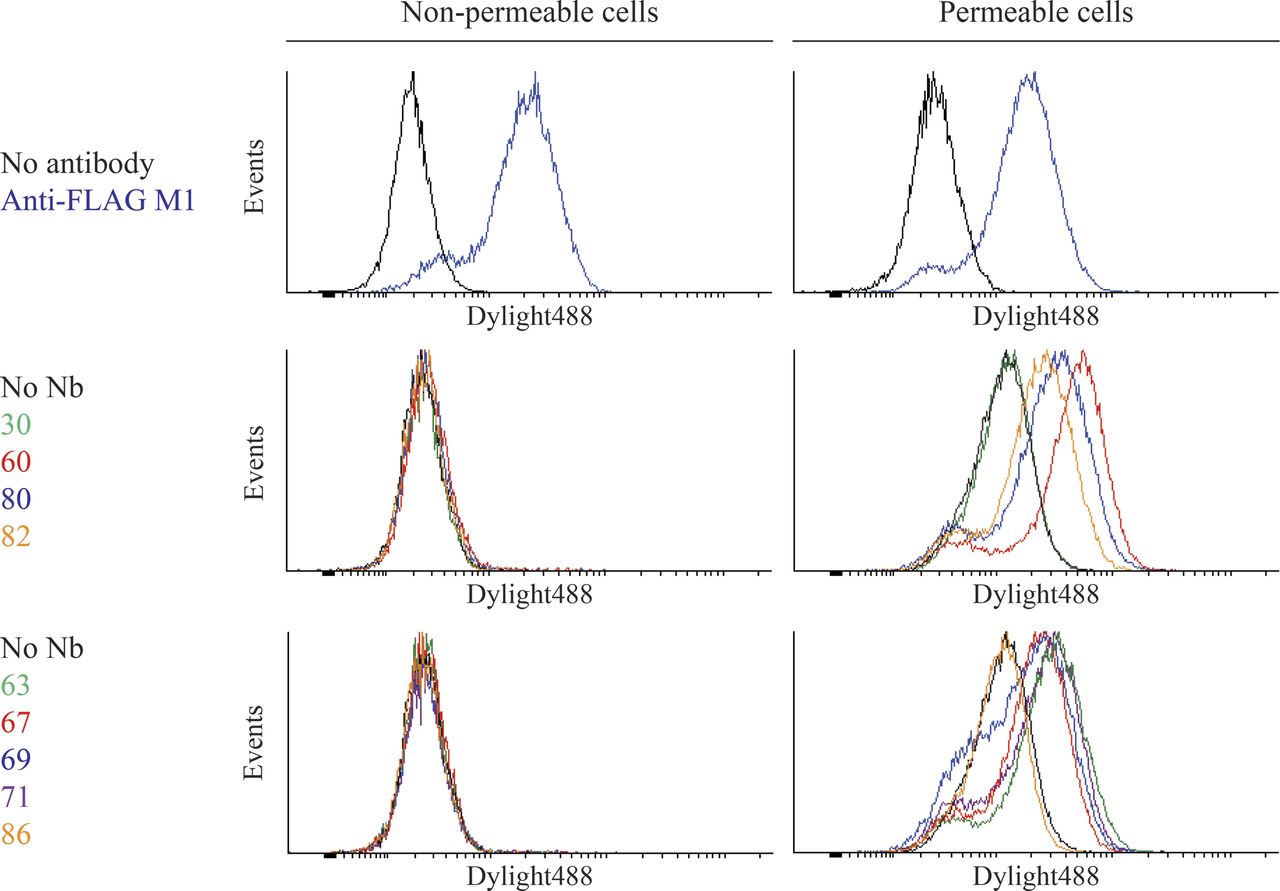
We next used flow cytometry and Sf9 insect cells expressing FLAG-β2AR to determine whether the nanobodies bound intracellular or extracellular β2AR epitopes. As a consequence of baculovirus infection, we observed permeable and nonpermeable cells expressing FLAG-β2AR at similar levels as shown by FLAG-antibody staining (Fig. 3, top panels). However, all nanobodies from the MISC group and representative nanobodies (see Supplemental Fig. 2 for all nanobodies) from families A (Nb60), B (Nb80), and C (Nb82) specifically labeled only permeabilized cells, indicating that they bind intracellular β2AR epitopes (Fig. 3, middle and bottom panels). Importantly, nanobodies did not label permeable or nonpermeable cells infected with a control virus (Supplemental Fig. 3).
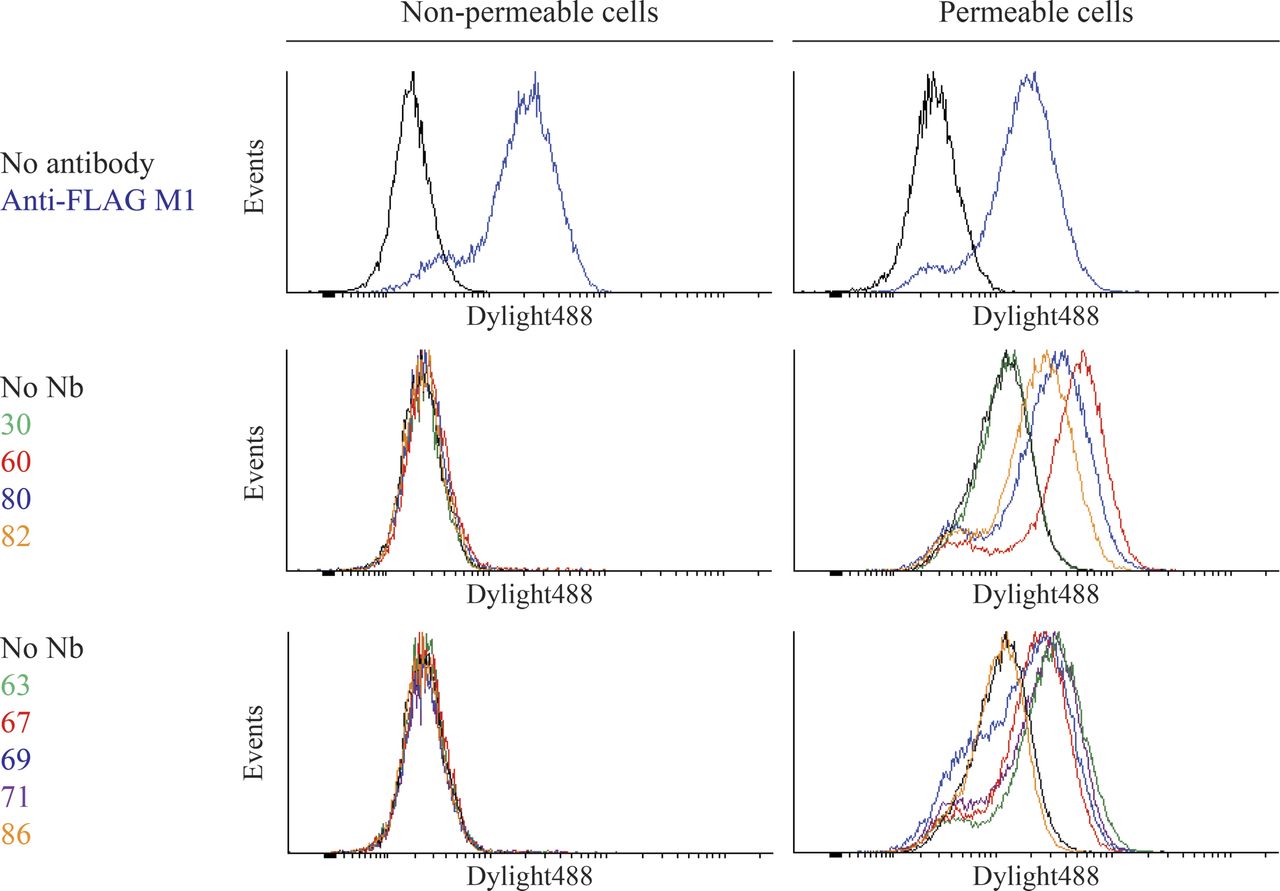
Nanobodies bind specifically to intracellular epitopes of the β2AR. Sf9 insect cells were infected with a baculovirus encoding FLAG-β2AR resulting in cell populations that were partially permeabilized due to viral infection. β2AR-expressing cells were preincubated with 1 µM ICI-118551 (family A nanobodies) or 1 µM BI-167107 (families B, C, and MISC). Binding of purified His-tagged nanobodies to cells was detected with a DyLight488-labeled anti–6× His antibody. FLAG-M1 antibody was labeled with DyLight488. Singlet cells were gated into intact and permeable populations based on staining with SYTOX AADvanced Dead Cell Stain.
Given that all nanobodies bound intracellular epitopes, we next assessed their ability to modulate receptor-dependent signaling by transiently expressing them as intrabodies in HEK293 cells. Due to its well established selectivity for agonist-activated β2AR (Rasmussen et al., 2011a), nanobody 80 was initially chosen to test whether intracellular expression of nanobodies as intrabodies compromised their selectivity for binding specific receptor conformations. Intrabody 30 (Ib30) and Ib80, when expressed in HEK293 cells, had negligible effects on the total and surface expression of FLAG-β2AR as assessed by immunoblotting (Fig. 4A) and radioligand binding using the noncell-permeable antagonist radioligand [3H]CGP-12177A (Fig. 4B). Importantly, Ib80, but not Ib30, selectively coimmunoprecipitated with FLAG-β2AR following stimulation with the agonist ISO (Fig. 4C). Taken together, these data demonstrate that nanobodies can be expressed as intrabodies, and that their remarkable selectivity for specific receptor conformations is conserved.

Intrabody 80 expression and specificity for activated β2AR. (A) Immunoblotting (IB) of HEK293 cells transiently transfected with FLAG-β2AR and pcDNA, HA-Ib30, or Ib80. (B) HEK293 cells were transfected as described earlier, and receptor expression was assessed via whole-cell binding using [3H]CGP-12177A. Specific binding normalized to pcDNA. (C) HEK293 cells stably expressing FLAG-β2AR were transfected with HA-Ib30 or Ib80, stimulated with the agonist ISO for 15 minutes, and then solubilized in lysis buffer. FLAG-β2AR was immunoprecipitated (IP) using FLAG beads, the eluate was subjected to SDS-PAGE, and intrabody was detected using an HA antibody.
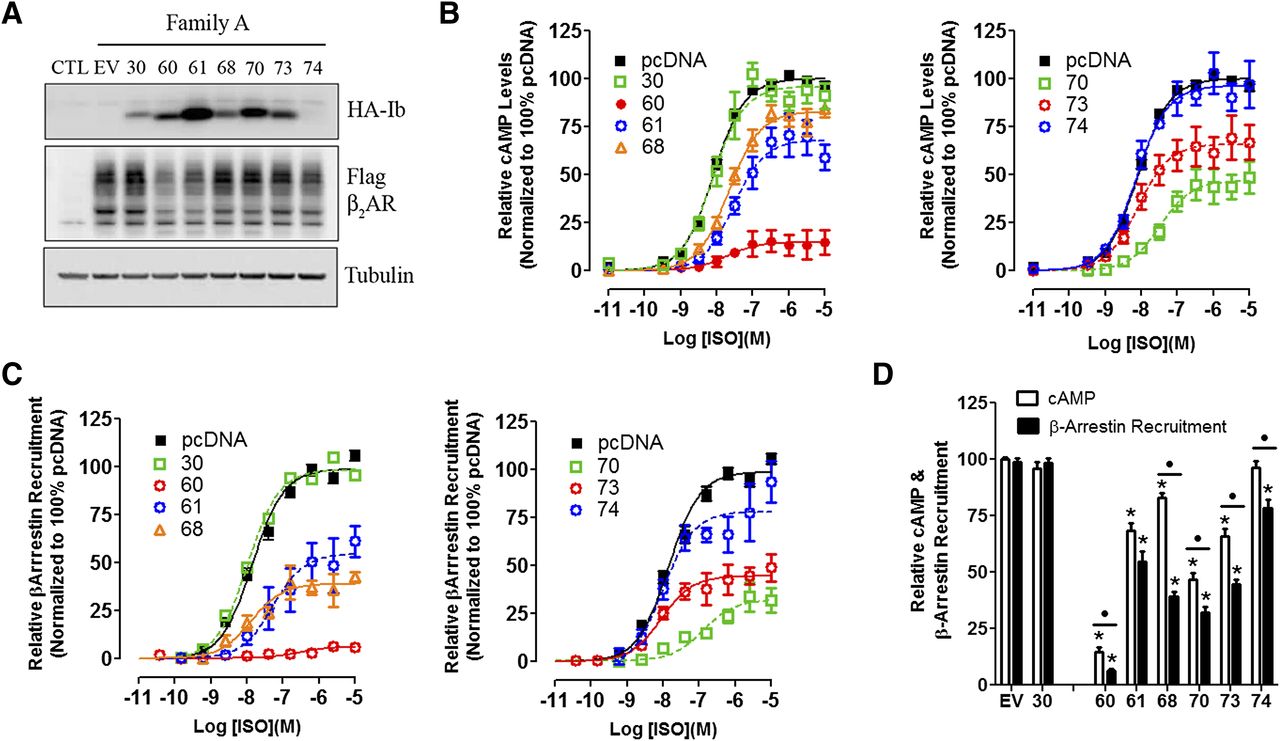
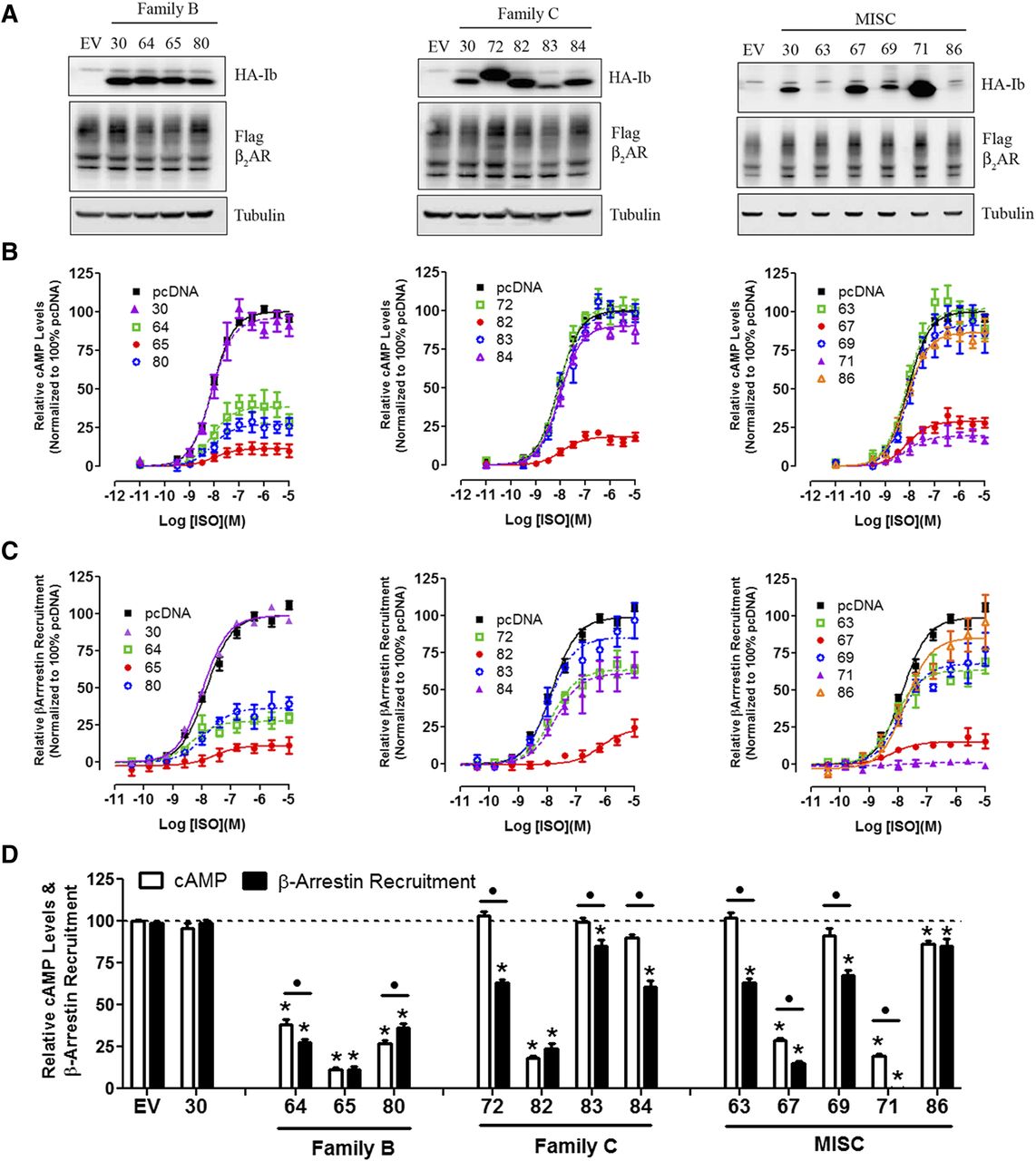
Next, all nanobodies that stabilized an active β2AR conformation (families B, C, and MISC; Fig. 1) were tested as intrabodies for their potential to modulate β2AR-dependent signaling. Intrabody expression varied extensively, but these differences had little effect on β2AR expression (Fig. 5, A–C, left panel). G protein activation and β-arrestin recruitment were measured in HEK293 cells using luminescence-based strategies (see Materials and Methods for details). Using these assays, we found that approximately half of the intrabodies had significant yet variable inhibitory effects on G protein activation and β-arrestin recruitment (Fig. 5, A–C, middle and right panels). The reductions in the maximal effect (Emax) of ISO in both G protein activation (cAMP) and β-arrestin recruitment induced by intrabody expression were calculated and summarized in Fig. 5D.

Regulation of β2AR functions by “active” state stabilizing intrabodies. Receptor expression (A), G protein–mediated cAMP levels (GloSensor) (B), and β-arrestin recruitment (Tango Assay) (C) was measured for intrabody family B, C, and MISC. (A) Immunoblot analysis of HEK293 cells transiently transfected with FLAG-β2AR and the indicated HA-Intrabody (Ib). Tubulin was used to ensure equal total protein loading. (B) HEK293 cells were transfected with the GloSensor cAMP biosensor (Promega) and pcDNA empty vector or individual HA-intrabodies, stimulated with a dose response of isoproterenol (ISO), and luminescence measured 10 minutes thereafter. Data were analyzed using GraphPad Prism program with sigmoidal dose response curve fit and normalized to 100% pcDNA empty vector. (C) HEK293T cells stably expressing β-arrestin-2 fused to the Tobacco Etch Virus (TEV) protease and a tTA transcription factor driven luciferase reporter were transiently transfected with β2AR fused to the tTA transcription factor, but separated by a TEV cleavage site and the indicated HA-intrabody. Cells were stimulated with a dose response of isoproterenol, and luminescence was measured 16 hours thereafter. Data were normalized as described in C. (D) The maximal response (Emax) for cAMP and β-arrestin following isoproterenol treatment in presence of the indicated intrabody as determined by nonlinear regression analysis. Data were normalized to 100% pcDNA. T-tests were used to compare each intrabody to pcDNA control (*P < 0.05), underlined • denotes significance between Bmax of Glosensor and Tango assays for each individual intrabody
When tested in the same assays, the majority of intrabodies that stabilized an “inactive” β2AR conformation (family A; Fig. 1) significantly inhibited G protein activation and β-arrestin recruitment, although to varying levels (Fig. 6, B–D). Unlike family B, C, and MISC intrabodies, expression of several family A intrabodies (i.e., Ib60 and Ib61) reduced β2AR expression when compared with the Nb30 and empty vector controls (Fig. 6D). This suggested that the large inhibitory effects of these family A nanobodies on receptor signaling may therefore be due to stabilization of specific receptor conformations or a reduction in receptor expression, or a combination of both factors.
Intrabodies that stabilize “inactive” β2AR conformation(s) inhibit G protein activation and β-arrestin recruitment. The effects of intrabody family A on expression of β2AR (A), G protein activation (B), and β-arrestin recruitment (C) were analyzed as described in Fig. 4. (D) The maximal response (Emax) for cAMP and β-arrestin following isoproterenol treatment in the presence of indicated intrabody as determined by nonlinear regression analysis. Data were normalized to 100% pcDNA. t tests were used to compare each intrabody to pcDNA control (*P < 0.05); underlined • denotes significance between Bmax of GloSensor and Tango assays for each individual intrabody.
Interestingly, we observed that several members of each nanobody family were more effective at inhibiting β-arrestin recruitment than cAMP generation (Figs. 5D and 6D). To rule out the possibility that the β-arrestin recruitment assay was more sensitive to inhibition than the cAMP assay, we measured the level of assay amplification in both assays. As shown in Supplemental Fig. 4, partial agonists elicited similar submaximal responses for both β-arrestin recruitment and cAMP accumulation, suggesting that both assays were equally coupled. Thus, variations in assay amplification could not explain the differential inhibitory effects of intrabodies such as Ib71.
Given its high expression and marked inhibitory effects on both G protein activation and β-arrestin recruitment assays (Fig. 5), we focused on Ib71 to further investigate the effects of intrabody expression on β2AR-dependent signaling. Consistent with our earlier findings, Ib71had no effect on receptor expression (Fig. 7A), selectively coimmunoprecipitated with FLAG-β2AR after ISO treatment (Fig. 7B), and stabilized a β2AR high affinity state as assessed by whole-cell competition binding experiments (Fig. 7C). Since β-arrestin recruitment is known to be highly dependent on GRK phosphorylation of the β2AR C-terminal tail, we set out to determine if GRK-mediated receptor phosphorylation is altered in the presence of Ib71. As shown in Fig. 7D, dose-dependent phosphorylation of Serine355 and Serine356 (Ser355/356) (canonical GRK5/6 phosphorylation sites) on the β2AR was strongly inhibited by Ib71 in comparison with Ib30 or the empty vector control. To further validate the inhibition of β-arrestin recruitment by Ib71, we conducted β2AR–β-arrestin cross-linking and green fluorescent protein–β-arrestin-2 recruitment experiments in live cells. Cross-linking studies demonstrated that Ib71 markedly decreased the amount of β-arrestin recruited to the β2AR after isoproterenol treatment when compared with the empty vector or Ib30 controls (Fig. 7E). Furthermore, Ib71, but not Ib30, blocked the recruitment of green fluorescent protein–β-arrestin-2 to immunoreactive puncta containing the β2AR following ISO stimulation as assessed by confocal microscopy (Fig. 7F). Taken together, these data demonstrate that Ib71 is a powerful inhibitor of β-arrestin recruitment to the β2AR, presumably exerting this effect by blocking receptor phosphorylation.

Attenuation of β-arrestin recruitment, GRK-mediated receptor phosphorylation, and β2AR internalization by intrabody 71. (A) HEK293 cells were transfected with FLAG-β2AR and pcDNA empty vector (EV) or HA-Ib71, and cell surface expression was measured via [3H]CGP-12177A radioligand binding. Data were normalized relative to pcDNA. (B) HA-Ib71 was transfected in HEK293 cells stably expressing FLAG-β2AR, and coimmunoprecipitation assays were performed after treatment with DMSO (−) or ISO (+). (C) HEK293 cells expressing FLAG-β2AR and pcDNA or HA-Ib71 were subject to whole-cell binding experiments in the presence of [3H]CGP-12177A and a dose response of isoproterenol. (D) HEK293 cells transiently expressing FLAG-β2AR and pcDNA, Ib30, or Ib71 were stimulated with a dose response of isoproterenol and analyzed for GRK-dependent phosphorylation of serine 355/6. (E) Whole-cell cross-linking of β-arrestin-1/2 with FLAG-β2AR in HEK293 cells in the presence of pcDNA, Ib30, or Ib71 after stimulation with 10 µM isoproterenol. (F) U2OS cells were transiently transfected with FLAG-β2AR, green fluorescent protein–β-arrestin-2, and Myc-tagged Ib30 or Ib71, and β-arrestin recruitment and receptor internalization were visualized using immunostaining and confocal microscopy. DAPI, 4′,6-diamidino-2-phenylindole; IB, immunoblots; IP, immunoprecipitation.
Discussion
It has been repeatedly demonstrated that targeting the biologic activity of GPCRs has tremendous therapeutic potential given their indispensable roles in regulating a vast array of physiological and pathological processes. Although most studies have focused on using orthosteric ligands, the use of allosteric modulators to further fine-tune receptor function has become of recent interest (Conn et al., 2009; Wang et al., 2009; Wang and Lewis, 2013). Herein, we explored the ability of a panel of closely related single-domain Camelid heavy-chain antibodies (nanobodies) to allosterically target and stabilize distinct agonist-bound (“active”) and antagonist-bound (“inactive”) β2AR conformations. Our studies uncovered four families of nanobodies with differing abilities to stabilize active or inactive β2AR conformations. Subsequent conversion of these nanobodies to intrabodies revealed a variety of effects on cAMP accumulation and β-arrestin recruitment, supporting their utility as novel tools to study GPCR biology.
One of the major goals of this study was to explore the role of nanobodies in modulating receptor function by expressing them as intrabodies in mammalian cells. Unlike the case for traditional antibodies and antibody fragments which require proper folding of multiple domains, we hypothesized that the unique single-domain nature of nanobodies would allow for their functional expression in the reducing cytoplasmic environment. We found that the active state nanobody-derived intrabodies are selectively recruited to the β2AR upon agonist treatment, resulting in inhibition of both G protein activation and β-arrestin recruitment. Unlike modulators that bind linear epitopes, the remarkable selectivity of these intrabodies for specific β2AR conformations may reduce the probability of having off-target effects within the cell. Furthermore, in addition to directly modulating receptor function, intrabodies were recently used to monitor the activation state of the β2AR and its cognate G protein in live cells (Irannejad et al., 2013). Taken together, using intrabodies as tools to study GPCR biology will likely extend well beyond this study.
There are likely many different mechanisms by which these intrabodies can inhibit β2AR signaling. Since several studies have recently highlighted the ability of an agonist-bound GPCR to reside in an array of conformations (Ghanouni et al., 2001; Yao et al., 2006; Kahsai et al., 2011), it is plausible that this heterogeneity would lead to nanobodies which stabilize a variety of distinct conformations. Therefore, stabilization of specific conformations that are not conducive to effector coupling would result in inhibition of receptor signaling. This is likely the mode of inhibition seen with intrabodies which stabilize an inactive receptor state (family A). However, we cannot rule out the possibility that other intrabody families may stabilize specific conformational states that fail to couple to downstream effectors. Second, intrabody recruitment to the receptor following agonist stimulation could sterically block the binding of G protein and/or β-arrestin. This is most certainly the case with Ib80 since recent crystallographic findings demonstrate that the binding epitope and β2AR conformation stabilized by both Nb80 and the heterotrimeric G protein complex are nearly identical (Rasmussen et al., 2011a,b). Although the receptor binding interface between β-arrestin and nanobodies other than Nb80 is not yet known, steric occlusion could explain the inhibitory effects of intrabodies from families B, C, and MISC.
Nanobodies in families B, C, and MISC were shown to stabilize an active receptor conformation (Figs. 1B and 2), which could result in chronic receptor activation in the absence of agonist, leading to desensitization and/or receptor internalization. Given that we did not observe any decreased levels of receptor expression upon intrabody expression (Figs. 4, A and B, 5A, and 7A), together with the lack of binding of Ib80 or Ib71 to the β2AR in the absence of agonist (Figs. 4C and 7B), we find this possibility unlikely. Additionally, since the β2AR can also couple to Gi (Daaka et al., 1997), we cannot rule out the possibility that decreases in cAMP levels seen here are mediated by modulation of this signaling pathway. Additional studies will be required to delineate the potential mechanisms by which these nanobodies regulate cAMP production and β-arrestin recruitment.
The discovery that β-arrestin signaling can occur independently of G protein activation has led to consideration of the possibility that specific receptor signaling arms can be pharmacologically isolated, a concept which is referred to as “functional selectivity” or “biased agonism” (reviewed in Reiter et al., 2012). Perhaps the best example of this phenomenon is observed with the angiotensin type 1 receptor where two biased agonists, SII (Sar1, Ile4, Ile8-angiotensin II) and most recently TRV027, selectively stimulate β-arrestin–dependent signaling in the absence of Gq protein activation (Wei et al., 2003; Violin et al., 2010). Our findings suggest that numerous intrabodies have a disproportionate effect on inhibition of G protein activation or β-arrestin recruitment, implying that they may be partially biased (Figs. 5 and 6). However, since these two signaling arms were measured using different assay platforms, the quantification of such intrabody bias will need to be further investigated given the confounding variables of receptor, intrabody, and effector expression. Such analysis will likely require the development of novel assays to quantitatively measure G protein activation and β-arrestin recruitment in the same cells. Nonetheless, despite the fact that our β-arrestin recruitment assay used overexpression of both β2AR and β-arrestin (compared with endogenous receptor and G protein in cAMP assays), many of the intrabodies (i.e., Nb63, 71, and 72) were still capable of inhibiting β-arrestin recruitment to a greater extent than G protein activation. Given the current findings, we cannot determine whether this biased inhibition is due to enhanced steric blockade of β-arrestin or stabilization of specific receptor conformations less conducive to β-arrestin coupling in comparison with G protein. Additionally, several studies have now demonstrated precoupling of receptor and G protein which could sterically hinder nanobody binding, resulting in the observed differential inhibition of β-arrestin recruitment and G protein activation (Nobles et al., 2005; Qin et al., 2011). Furthermore, intrabody-mediated inhibition of β-arrestin recruitment may also be attenuated due to diminished levels of GRK-dependent receptor phosphorylation. Further work will be required to delineate the detailed molecular mechanisms governing this potential bias.
Targeting the biologic functions of many receptors and signaling pathways in a cell type–specific manner in vivo remains a challenging endeavor. More specifically, generating cell type–specific knockouts can be quite difficult, and localized or global antagonist treatments can have a variety of off-target effects which make data interpretation problematic, although considerable advances have recently been made using receptor activated solely by a synthetic ligand and designer receptor exclusively activated by designer drugs technologies (Conklin et al., 2008). In this context, intrabody expression driven by cell type–specific promoters may provide a novel strategy to examine the contribution of particular receptors and signaling pathways to regulating numerous physiological and pathological processes. Additionally, the discovery of intrabodies that bias signaling may also be useful in dissecting the influence of specific signaling arms to downstream receptor-mediated events in vivo. Given the recent discovery of a nanobody that stabilizes the active state of the M2 muscarinic acetylcholine receptor (Kruse et al., 2013), the use of nanobodies to modulate receptor function will likely extend well beyond this study. Although we have focused on the use of allosteric intrabodies to manipulate GPCR function, these reagents could also conceivably be used to modulate other intracellular targets. Indeed, nanobodies have been described that allosterically activate or inhibit the catalytic function of specific enzymes (Saerens et al., 2004; Barlow et al., 2009; Oyen et al., 2011). Furthermore, a nanobody that inhibits Clostridium botulinum neurotoxin proteases maintained its antagonist properties when expressed as an intrabody in neuronal cells (Tremblay et al., 2010). The recent development of small-molecule or peptide reagents that facilitate efficient delivery of biologically active proteins across cell membranes could be used to deliver intrabodies, thereby extending their use to acutely regulate protein function in live cells (Milletti, 2012). Although all nanobodies discussed herein bind to intracellular receptor epitopes, the identification of nanobodies that modulate receptor function by binding to extracellular epitopes as orthosteric or allosteric ligands may provide very useful therapeutic agents.
In conclusion, our data demonstrate that nanobody-derived intrabodies function as novel regulators of β2AR-dependent G protein activation and β-arrestin recruitment. These innovative reagents will undoubtedly be powerful tools for furthering our understanding of GPCR biology.
Acknowledgments
The authors thank J. Wisler, J. Violin, and A. Shukla for helpful discussions, and G. Kweon, X. Zhu, X. Jiang, and D. Capel for technical assistance. The authors thank Dr. Irving Wainer (Laboratory of Clinical Investigation, National Institute on Aging Intramural Research Program, Baltimore, MD) for assistance with the 3H-methoxyfenoterol studies and Q. Lennon and D. Addison for excellent secretarial assistance.
Authorship Contributions
Participated in research design: Staus, Wingler, Strachan, Ahn, Kobilka, Lefkowitz.
Conducted experiments: Staus, Wingler, Strachan.
Contributed new reagents or analytic tools: Rasmussen, Pardon, Kobilka, Steyaert.
Performed data analysis: Staus, Wingler, Strachan.
Wrote or contributed to the writing of the manuscript: Staus, Wingler, Lefkowitz.
Footnotes
- Received September 9, 2013.
- Accepted December 5, 2013.
L.M.W. and R.T.S. contributed equally to this work.
This work was supported, in part, by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL16037 and HL70631] (to R.J.L.). R.J.L. is an Investigator with the Howard Hughes Medical Institute.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- β2AR
- β2-adrenergic receptor
- BI-167107
- 5-{2-[1,1-dimethyl-2-(o-tolyl)ethylamino]-1-hydroxyethyl}-8-hydroxy-4-oxa-1-aza-1,3-dihydronaphthalen-2-on
- BSA
- bovine serum albumin
- CGP-12177A
- 4-[3-[(1,1-dimethylethyl)amino]2-hy\x{200b}droxypropoxy]-1,3-dihydro-2H-benzimidazol-2-one hydrochloride
- DDM
- N-dodecyl β-D-maltopyranoside
- DMSO
- dimethylsulfoxide
- ELISA
- enzyme-linked immunosorbent assay
- Emax
- maximal effect
- GPCR
- G protein–coupled receptor
- G protein
- GTP binding protein
- GRK
- G protein–coupled receptor kinase
- HA
- hemagglutinin
- HEK293
- human embryonic kidney 293
- HRP
- horseradish peroxidase
- Ib
- intrabody
- ICI-118551
- 3-(isopropylamino)-1-[(7-methyl-4-indanyl)oxy]butan-2-ol
- ISO
- isoproterenol
- MISC
- miscellaneous
- MNG
- maltose-neopentyl glycol
- Nb
- nanobody
- RT
- room temperature
- TEV
- tobacco etch virus
- tTA
- tetracycline transactivator protein
- β2V2
- β2AR with C-terminal tail replaced with that of the vasopressin-2-receptor
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}