


Abstract
Cytochrome P450 (CYP)2C9 and CYP2C19 are important human enzymes that metabolize therapeutic drugs, environmental chemicals, and physiologically important endogenous compounds. Initial studies using primary human hepatocytes showed induction of both the CYP2C9 and CYP2C19 genes by tert-butylhydroquinone (tBHQ). As a pro-oxidant, tBHQ regulates the expression of cytoprotective genes by activation of redox-sensing transcription factors, such as the nuclear factor E2-related factor 2 (Nrf2) and members of the activator protein 1 (AP-1) family of proteins. The promoter region of CYP2C9 contains two putative AP-1 sites (TGAGTCA) at positions −2201 and −1930, which are also highly conserved in CYP2C19. The CYP2C9 promoter is activated by ectopic expression of cFos and JunD, whereas Nrf2 had no effect. Using specific kinase inhibitors for mitogen-activated protein kinase, we showed that extracellular signal-regulated kinase and Jun N-terminal kinase are essential for tBHQ-induced expression of CYP2C9. Electrophoretic mobility shift assays demonstrate that cFos distinctly interacts with the distal AP-1 site and JunD with the proximal site. Because cFos regulates target genes as heterodimers with Jun proteins, we hypothesized that DNA looping might be required to bring the distal and proximal AP-1 sites together to activate the CYP2C9 promoter. Chromosome conformation capture analyses confirmed the formation of a DNA loop in the CYP2C9 promoter, possibly allowing interaction between cFos at the distal site and JunD at the proximal site to activate CYP2C9 transcription in response to electrophiles. These results indicate that oxidative stress generated by exposure to electrophilic xenobiotics and metabolites induces the expression of CYP2C9 and CYP2C19 in human hepatocytes.
Introduction
There are four cytochrome P450 (CYP)2C genes in humans, as follows: CYP2C9, CYP2C8, CYP2C19, and CYP2C18 (Goldstein and de Morais, 1994). CYP2C9 and CYP2C19 enzymes metabolize ∼20% of clinically prescribed drugs and a number of environmental chemicals and are found primarily in human liver (Zanger and Schwab, 2013). CYP2C19 metabolizes the anticonvulsant drug S-mephenytoin, proton pump antiulcer drugs such as omeprazole, the β-adrenergic receptor blocker propranolol, the anxiolytic diazepam, and clopidigrel, used to prevent heart attacks and strokes (Zanger and Schwab, 2013). CYP2C9 is the most abundant of the CYP2C enzymes in human liver, and its substrates include the anticoagulant warfarin, the hypoglycemic drugs tolbutamide and glipizide, the anticonvulsant phenytoin, the anticancer drug cyclophosphamide, the angiotensin II blocker losartan, the diuretic torsemide, and numerous nonsteroidal anti-inflammatory drugs, such as diclofenac, flurbiprofen, ibuprofen, piroxicam, and mefenamic acid (Goldstein, 2001; Lee et al., 2002). CYP2C9 also metabolizes the endogenous substrate arachidonic acid to the biologically active and vasoactive epoxyeicosatrienoic acids (Fisslthaler et al., 1999).
The CYP2C genes are induced by drugs and xenobiotics, such as rifampicin, St. John’s wort, phenytoin, phenobarbital, and dexamethasone (Gerbal-Chaloin et al., 2001; Gerbal-Chaloin et al., 2002; Chen et al., 2004), leading to enhanced metabolism of many therapeutic drugs and other CYP2C substrates (Rettie and Jones, 2005). This can alter drug efficacy, resulting in tolerance and drug-drug interactions. There is considerable interindividual variability in CYP2C9/CYP2C19 expression and in metabolism of their substrates partly due to genetic polymorphisms and partly due to prior exposure of humans to drugs capable of inducing these enzymes. Studies in humans have reported enhanced clearance of drugs, such as tolbutamide, phenytoin, glyburide, and glipizide after treatment with rifampicin (Zilly et al., 1975; Kay et al., 1985; Niemi et al., 2001). The induction of CYP2C9 by xenobiotics is mediated by three major xenobiotic-sensing nuclear receptors, as follows: the constitutive androstane receptor, the glucocorticoid receptor, and the pregnane X receptor (PXR). The activation of these nuclear receptors by xenobiotics induces CYP2C9 transcription by binding to cis-regulatory elements on the promoter of CYP2C9 gene. Regulatory elements identified in the promoter of CYP2C9 include proximal HNF4α binding sites located at −152, −185, and −211; a glucocorticoid-responsive element at −1675; and proximal and distal constitutive androstane receptor/PXR response elements at positions −1839 and −2899, respectively (Ibeanu and Goldstein, 1995; Ferguson et al., 2002; Gerbal-Chaloin et al., 2002; Chen et al., 2005).
Many drugs and foreign compounds are metabolized in the liver to electrophilic and reactive metabolites (Chang et al., 1997; Erve, 2006; Guengerich, 2006; Takakusa et al., 2008). Moreover, xenobiotics capable of undergoing redox cycling induce oxidative stress by generation of semiquinone radicals and reactive oxygen species. The effects of electrophilic metabolites and oxidative stress on the regulation of the human CYP2C genes have not yet been investigated. The alteration of cellular redox state by reactive intermediates activates transcription factors such as activator protein 1 (AP-1) and nuclear factor E2-related factor 2 (Nrf2). AP-1 factors are basic leucine zipper proteins that regulate the transcription of target genes in response to environmental stimuli as either homodimers of the Jun family (cJun, JunD, and JunB) proteins or heterodimers of the Jun and Fos family proteins (cFos, FosB, Fra1, and Fra2) (Eferl and Wagner, 2003). The dimeric AP-1 proteins bind to a cis-acting 12-O-tetradecanoylphorbol 13-acetate–responsive element on the promoter of target genes. AP-1 proteins also form transcriptionally active heterodimers with Nrf2, which binds to the antioxidant or electrophilic response element (Hayes et al., 2010). The transcriptional activity of AP-1 and Nrf2 is also regulated by phosphorylation via the mitogen-activated protein kinase (MAPK) signaling pathways (Mendelson et al., 1996; Ventura et al., 2003; Sherratt et al., 2004).
We identified two putative AP-1 sites (TGAGTCA) in CYP2C9 promoter at positions −2201 and −1930, which are highly conserved in the promoter of the CYP2C19 gene. In this study, we show that the CYP2C9 and CYP2C19 genes are induced by the electrophilic xenobiotic tert-butylhydroquinone (tBHQ) in cultured human hepatocytes, and we examined the mechanism of this induction using the CYP2C9 promoter. We demonstrate that the pathway for CYP2C9 gene induction by electrophiles involves an AP-1 heterodimer of cFos and JunD. We show for the first time using chromosome conformation capture (3C) assays the formation of a DNA loop on the CYP2C9 promoter possibly allowing formation of a complex between cFos bound to the distal site and JunD to the proximal AP-1 site. Thus, drugs and environmental chemicals capable of inducing oxidative stress enhance the expression of CYP2C9 by this mechanism.
Materials and Methods
Chemicals and Reagents.
t-BHQ, SP600125 [Jun N-terminal kinase (JNK inhibitor)], and PD98059 [mitogen-activated protein kinase kinase (MEK) inhibitor] were purchased from Sigma-Aldrich (St. Louis, MO). Anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (clone 6C5, MAB374) was purchased from Millipore (Temecula, CA). Rabbit polyclonal antibodies against c-Jun (sc-44), JunD (329; sc-74), JunB (N-17; sc-46), c-Fos (sc-52), Nrf2 (C-20; sc-722), Activating transcription factor 2 (ATF2) (C-19; sc-187), and ATF4 (C-20; sc-200) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-JunD (Ser100) antibody (9164), stress-activated protein kinase/JNK antibody (9252), phospho–stress-activated protein kinase/JNK (Thr183/Tyr185) antibody (9251), p42 MAPK [extracellular signal-regulated kinase (ERK2)] antibody (9108), and phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) antibody (9101) were obtained from Cell Signaling Technology (Danvers, MA). The expression plasmids for c-Fos (pRSV-cfos) and c-Jun (pRSV-cjun) were provided by C. B. Pickett and T. Nguyen (Schering-Plough Research Institute, Kenilworth, NJ). The expression plasmids for JunD (pcDNA3.1-JunD) and JunB (pcDNA3.1-JunB) were gifts of S. K. Agarwal (Genetics and Endocrinology Section, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health). The Nrf2 expression plasmid (pCI-Nrf2neo) was provided by K. S. Ramos (University of Louisville, Louisville, KY).
Isolation and Culture of Human Hepatocytes.
One set of human livers was obtained from kidney transplant donors at the Transplantation and Surgical Clinic, Semmelweis University Budapest (Budapest, Hungary). Permission from the Hungarian Regional Committee of Science and Research Ethics was obtained to use human tissues. Clinical histories of the donors are shown in Table 1. Liver cells were isolated by the method of Bayliss and Skett (1996), as previously described (Belic et al., 2009). Hepatocytes having viability better than 90% as determined by trypan blue exclusion were used in the experiments. The cells were plated at a density of 1.7 × 105 cells/cm2 in collagen-precoated plastic dishes in the previously described medium (Ferrini et al., 1998). After overnight attachment, the medium was replaced with serum-free medium. Twenty-four hours after serum deprivation, cells were exposed to tBHQ (50 μM) or rifampicin (5 μM) for 24 and 48 hours, and specific mRNAs for CYP2C9, CYP2C19, and CYP2A6 were measured, as described below. The control cultures received the same volume of the vehicle [dimethylsulfoxide (DMSO)] as the treated cultures.
Clinical histories of human donors
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction.
Total RNA was isolated from human primary hepatocytes and HepG2 cells using TRIzol reagent (Invitrogen, Carlsbad, CA) and RNeasy mini kit (Qiagen, Valencia, CA), respectively, according to the manufacturer’s instructions, and reverse transcribed to cDNA using the SuperScript III First Strand Synthesis System for reverse-transcription polymerase chain reaction (PCR) kit (Invitrogen) with oligo(dT) primers. Quantitative real-time polymerase chain reaction was performed using the ABI Prism 7900 Sequence Detector System (Applied Biosystems, Foster City, CA) with the following primers and probe sets purchased from Applied Biosystems: CYP2C9 (Hs00426397_m1), CYP2C19 (Hs00426380_m1), and GAPDH (Hs03929097_g1). Each cDNA (100 ng) was mixed with 1× TaqMan Universal PCR master mix (Applied Biosystems). All quantitative real-time PCR experiments were performed in triplicate with cDNA samples from independent samples, as described previously (Makia et al., 2012). mRNA levels were normalized with GAPDH as the endogenous control.
Preparation of Microsomes.
Additional cultured human primary hepatocytes in six-well plates were obtained from Triangle Research Laboratories (Research Triangle Park, NC) for immunoblotting experiments with CYP2C proteins before and after treatment with tBHQ or DMSO controls. Microsomes were prepared from cultured human hepatocytes, as follows. Cells were suspended in ice-cold buffer (0.1 M potassium phosphate, pH 7.4, containing 0.25 M sucrose and 1 mM EDTA) and homogenized using a Potter-Elvehjem homogenizer. The 10,000g supernatant was subjected to ultracentrifugation for 2 hours at 112,000g using a TLA-55 rotor (Beckman Coulter, Palo Alto, CA). The microsomal pellet was resuspended in 50 mM potassium phosphate buffer (pH 7.4) containing 20% glycerol and 1 mM EDTA, and stored at −80°C for later use.
Western Blotting.
Total cell and nuclear extracts were prepared from HepG2 cells, as described previously (Makia et al., 2012). The extracts were separated on 4–20% SDS-PAGE gels and transferred onto nitrocellulose membranes. Membranes were probed with antibodies against GAPDH (1:5000) as the endogenous control or 1:1000 dilutions of all other primary antibodies. Horseradish peroxidase–conjugated goat anti-rabbit or anti-mouse secondary (1:10,000) antibodies were used, and the proteins were visualized using SuperSignal West Pico or Dura Western blotting detection system (Thermo Scientific, Rockford, IL). Microsomal proteins were separated using a Protean II xi Cell (Bio-Rad), and Western blot was performed with the following rabbit antibodies: 1590 (raised to recombinant purified CYP2C9 expressed in Escherichia coli, which recognizes CYP2C9 > CYP2C19 > >CYP2C8), 1592 (raised to recombinant purified CYP2C9 expressed in E. coli, which recognizes CYP2C9 > >CYP2C19 and CYP2C8), and 1937 (specific anti-CYP2C8 peptide antibody that recognizes only CYP2C8) (Zhang et al., 2012). The following CYP2C standards were also used: human liver microsomes (Gentest, Woburn, MA) and yeast CYP2C9, CYP2C8, and CYP2C19.
Small Interfering RNA Transfection.
Silencer Select Predesigned small interfering RNA (siRNA) for JunD (s7664), Jun (s7658), and negative control 1 (negative control) were purchased from Ambion (Austin, TX). HepG2 cells cultured in six-well plates were transfected with 50 pmol/well siRNA duplexes using Lipofectamine 2000 (Life Technologies, Carlsbad, CA) for 48 hours. Total cell extracts were prepared for Western blot analysis.
Transcription Factor Binding Site Analysis.
The Genomatrix MatInspector software was used to analyze the human CYP2C9 (−3077/+1) and CYP2C19 (−4722/+1) promoters for putative Nrf2 (TGACNNNGCA) and AP-1 (TGAC/GTCA) binding sites.
Site-Directed Mutagenesis.
A QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) was used to mutate the putative AP-1 binding sites located at positions −2201 and −1930 of the human CYP2C9 promoter, according to the manufacturer’s instructions. The primers for mutagenesis of the putative AP-1 sites were as follows: −2201 (forward, 5′-TGATACTTTGTCTCACTTGGTTGATAATTGCTCATTTCT-3′ and reverse, 5′-AGAAATGAGCAATTATCAACCAAGTGAGACAAAGTATCA-3′); −1930 (forward, 5′-TGTTAGAGTTTAGAGTTTCATTGGTTGGGGACCAAGTTATTGC-3′ and reverse, 5′-GCAATAACTTGGTCCCCAACCAATGAAACTCTAAAC TCTAACA-3′). The mutated sites are underlined. The mutation of the putative AP-1 sites was confirmed by sequencing.
Transfection of HepG2 Cells and Luciferase Reporter Assays.
The human hepatocellular carcinoma cell line, HepG2 (HB8065; American Type Culture Collection, Rockville, MD), was maintained in modified Eagle’s media supplemented with 10% fetal bovine serum (HyClone, Logan UT), 1 mM sodium pyruvate, 2 mM glutamine, and penicillin-streptomycin (Life Technologies). Cells were seeded in 24-well plates and transfected with 200 ng/well CYP2C9 luciferase construct and 20 ng Renilla luciferase plasmid using Lipofectamine 2000. The cells were cotransfected with pRSV-cFos (50 ng), pRSV-cJun (50 ng), pcDNA3.1-JunD (100 ng), and pCI-Nrf2neo (100 ng) plasmids. The cells were resuspended in passive lysis buffer (Promega, Madison, WI), and luciferase activity was assayed with a Dual-Glo luciferase reporter assay system. The data were expressed relative to Renilla luciferase activity to normalize for transfection efficiency. Transfection experiments were performed in triplicate and repeated at least twice for confirmation.
Electrophoretic Mobility Shift Assays.
Nuclear extracts were prepared from HepG2 cells, as described previously (Makia et al., 2012). The sequences of the complementary oligonucleotides were as follows: AP-1 control (forward, 5′-CGCTTGATGAGTCAGCCGGAA-3′ and reverse, 5′-TTCCGGCTGACTCATCAAGCG-3′); distal CYP2C9 AP-1 site (forward, 5′-GTGATACTTTGTCTCACTGAGTCAATAATTGCTCATTTCT-3′ and reverse, 5′-AGAAATGAGCAATTATTGACTCAGTGAGACAAAGTATCAC-3′); and proximal CYP2C9 AP-1 site (forward, 5′-GAGTTTAGAGTTTCATGAGTCAGGGACCAAGTTATTGCT-3′ and reverse, 5′-AGCAATAACTTGGTCCCTGACTCATGAAACTCTAAACTC-3′). The complementary oligonucleotides (1 pmol/μl) were annealed by incubating at 95°C for 5 minutes with annealing buffer (10 mM Tris-HCl, 1 mM EDTA, and 50 mM NaCl, pH 8.0). Double-stranded oligonucleotides were labeled with [γ-32P]ATP using a T4 polynucleotide kinase kit following the supplier’s instructions (Promega). Unincorporated nucleotides were removed by chromatography on microspin G-25 columns (GE Healthcare, Piscataway, NJ). Protein-DNA complexes were formed by incubating 5 μg nuclear proteins and 106 cpm of 32P-labeled oligonucleotide probe for 30 minutes at room temperature in a total volume of 20 μl with binding buffer [10 mM Tris-HCl, 1 mM EDTA, 1 mM MgCl2, 10% glycerol, 1 mM DTT, and 50 ng/μl poly(dI-dC)]. Binding specificity was assessed by addition of 100-fold excess of unlabeled double-stranded oligonucleotides. For supershift analyses, antibodies (4 μg) were added to the binding reactions after the initial 20-minute incubation, and incubation was continued for 2 hours at 4°C. Loading buffer was added to the reactions, and 10 μl binding reactions were resolved by electrophoresis on 5% polyacrylamide gels using 0.5× Tris/borate/EDTA buffer (50 mM Tris, pH 8.3, 50 mM sodium borate, and 1 mM EDTA) at 200 V for 2 hours. The gel was transferred to Whatman 3MM filter paper, dried, and exposed to film overnight at −80°C.
In Vitro Transcription/Translation of cFos and cJun Proteins and Electrophoretic Mobility Shift Assays with the Pure Translated Proteins.
cFos and cJun proteins were synthesized in vitro from 1 μg pRSV-cFos, pCMV-cFos, or pCR2.1-cJun plasmids using a TNT Quick Coupled Transcription/Translation System (Promega) in the presence of unlabeled or labeled 35S-methionine, following the manufacturer’s instructions. Additional controls containing the TNT Quick master mix with no template DNA were also performed. The labeled proteins were separated on a 4–20% Tris-glycine polyacrylamide gel. The gel was dried on a sheet of Whatman 3MM filter paper, and the protein bands were visualized by autoradiography. In vitro translation of unlabeled cFos and cJun proteins was verified by Western blotting using antibodies against cFos and cJun. Electrophoretic mobility shift assay (EMSAs) were performed by incubating 5 μl in vitro translated unlabeled cFos or cJun proteins with labeled double-stranded oligonucleotides containing AP-1 control, CYP2C9 distal and proximal AP-1 sites. EMSAs with the in vitro transcribed proteins were performed essentially as described above.
Chromatin Immunoprecipitation Assay.
Chromatin immunoprecipitation (ChIP) assays were performed, as described previously (Makia et al., 2012), using the MAGnify Chromatin-Immunoprecipitation System (Life Technologies), according to the manufacturer’s protocol, with minor modifications. Briefly, HepG2 cells in 10-cm dishes treated with either DMSO (control) or tBHQ (50 μM) were fixed in 1% formaldehyde at room temperature for 10 minutes to cross-link the nuclear proteins to DNA, and the reaction was stopped by incubation with 125 mM glycine for 10 minutes. Cells were resuspended in lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 0.5% Nonidet P-40) containing 1× complete protease inhibitor (Roche Diagnostics, Indianapolis, IN) and incubated on ice for 30 minutes. The nuclei were harvested and solubilized in buffer containing 50 mM Tris-Cl (pH 8.0), 1% SDS, 10 mM EDTA, and 0.5 mM phenylmethanesulfonyl fluoride with 1× complete protease inhibitor. The homogenate was sonicated on ice at 40% setting (Branson Sonicator, North Olmsted, OH) to shear the chromosomal DNA into fragments of ∼200–500 bp in size. Immunoprecipitation of the sonicated DNA fragments was performed overnight at 4°C with 4 μg antibodies against IgG (negative control), JunD, c-Fos, cJun, or phospho-JunD conjugated to Dynabeads protein A/G. The cross-linked protein-DNA complexes were uncross-linked in the presence of proteinase K, and the purified DNA was analyzed by PCR using PCR SuperMix High Fidelity (Life Technologies) with primers spanning the putative distal (position −2201) and proximal (position −1930) AP-1 sites in the promoter of CYP2C9 gene. The sequences of the primers were as follows: distal AP-1 site (forward, 5′-GAATGGCATGAACCCAGGAGCTGGA-3′ and reverse, 5′-AAGAAATGAGCAATTATTGACTCAG-3′); proximal AP-1 site (forward, 5′-ACAGCAACCGAGCTTATTTTACCCA-3′ and reverse, 5′-ACAGCCCCAGTTGACTAGATTGAGA-3′). PCR products were resolved on a 1.5% agarose gel and visualized by ethidium bromide staining.
In Vitro and In Vivo 3C Assays.
In vitro 3C assays were performed essentially as previously described with minor modifications (Babu et al., 2008; Inoue and Negishi, 2009; Li et al., 2010). Nuclear extracts (100 µg) prepared from HepG2 cells transfected with empty vector or cFos expression vector were incubated with 50 ng linearized CYP2C9 plasmid (containing −3077 to +1 of the CYP2C9 promoter) at room temperature for 45 minutes. The protein-DNA complexes were cross-linked by fixing with 1% formaldehyde for 10 minutes. The cross-linking reaction was quenched with 0.125 M glycine for 10 minutes. After overnight ethanol precipitation at −20°C, complexes were digested with 10 U BsrGI (New England Biolabs, Ipswich, MA) at 37°C for 2 hours. BsrGI enzyme activity was inactivated by incubation at 80°C for 20 minutes. Ligation was performed with 20 ng DNA using 800 cohesive end units of T4 DNA ligase (New England Biolabs) at 16°C for 4 hours. SDS (1%) was added to the ligation reaction, 0.3 M NaCl to reverse cross-link the protein-DNA complexes, and the reaction was incubated overnight at 65°C. Proteinase K (200 μg/ml) was added to the reaction mixture, which was incubated at 55°C for 1 hour. DNA was purified using a PCR purification kit (Qiagen) and amplified with PCR SuperMix High Fidelity kit for the looped and control DNA products. The sequences of the primers spanning the ligated and control regions were as follows: looped product (97 bp: forward, 5′-TACTAGACTGAATTACGAAAT-3′ and reverse, 5′-AACATTGACGCATCATCATCA-3′); control product (194 bp: forward, 5′-ACAGCAACCGAGCTTATTTTACCCA-3′ and reverse, 5′-ACAGCCCCAGTTGACTAGATTGAGA-3′).
In vivo 3C was performed, as previously described (Hagege et al., 2007). Briefly, human hepatocytes were treated with 1% formaldehyde for 10 minutes to cross-link protein-DNA, and the cross-linking reaction was stopped by incubation with 0.125 M glycine for 10 minutes. Cells were harvested and resuspended in lysis buffer (10 mM Tris-HCl, 10 mM NaCl, 0.2% Nonidet P-40) containing 1× complete protease inhibitor (Roche Diagnostics). The nuclei were washed with 1× New England Biolabs buffer 2 and incubated in 1× New England Biolabs buffer 2 containing 0.3% SDS at 37°C for 1 hour. SDS in the reaction was sequestered by incubation with 1.8% Triton X-100 at 37°C for 1 hour. Chromatin (∼20 μg) was digested with 200 U BsrG1 by incubation at 37°C for 2 hours, and enzyme activity was inactivated by incubation with 1.6% SDS at 65°C for 20 minutes. The digested chromatin (3 μg) was incubated at 37°C for 1 hour with 1× T4 ligase buffer and 1% Triton X-100. Two thousand units of T4 DNA ligase were added, and the reaction was incubated at 16°C for 4 hours. The protein-DNA complexes were reverse cross-linked by overnight incubation with 100 μg/ml proteinase K at 65°C. RNA was digested by addition of 25 μg/ml RNase A, and the reaction was incubated at 37°C for 30 minutes. The DNA was purified by phenol-chloroform extraction using phenol:chloroform:isoamyl alcohol (25:24:1) and ethanol precipitated. The purified DNA was amplified for the looped and control DNA products by conventional PCR. The in vivo 3C primer sequences for amplification of the looped product were 144 bp: forward, 5′-ACCTCTCAATCTAGTCAACTG-3′ and reverse, 5′-CAACCAAAGCTCATAGTTTAT-3′, and for control product, 194 bp: forward, 5′-ACAGCAACCGAGCTTATTTTACCCA-3′ and reverse, 5′-ACAGCCCCAGTTGACTAGATTGAGA-3′.
Statistical Analysis.
Data were analyzed with Student’s t tests or one-way analysis of variance (ANOVA), followed by Tukey’s test using Sigma Stat software (Sigma Stat 3.5; SPSS, San Jose, CA). Differences were considered to be statistically significant at P value 0.05.
Results
tBHQ Induces CYP2C9 and CYP2C19 in Primary Human Hepatocytes.
We examined the effect of treatment with 50 μM electrophilic tBHQ on CYP2C9, CYP2C19, and CYP2A6 mRNA expression in primary human hepatocytes after 24 and 48 hours (Fig. 1A, 1B). Maximum induction of CYP2A6, CYP2C9, and CYP2C19 mRNA by 50 μM tBHQ (∼4.0-, 3.0-, and 2.5-fold, respectively) occurred at 48 hours (Fig. 1B). As expected, CYP2C9, CYP2C19, and CYP2A6 mRNAs were also induced by the PXR agonist, rifampicin (Fig. 1A, 1B). CYP2A6 has been reported to be induced via both PXR and electrophilic activation (Abu-Bakar et al., 2013). Treatment of primary hepatocytes with tBHQ also induced expression of CYP2C9 and CYP2C19 proteins (∼2.5-fold and ∼fivefold, respectively) after 48 hours but did not induce CYP2C8 protein (Fig. 1C, 1D). The slightly higher induction of CYP2C19 may partially reflect its low constitutive levels.
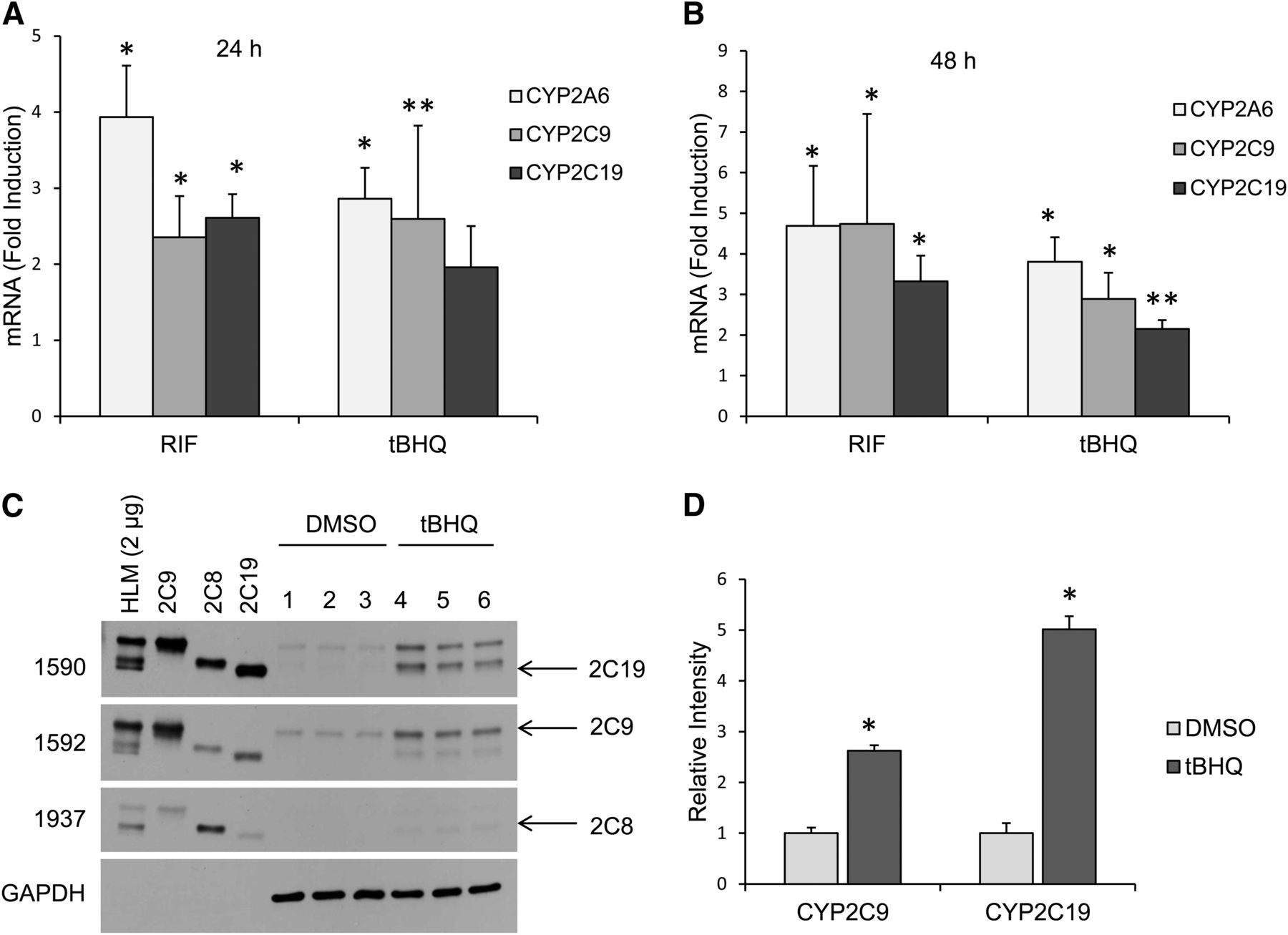
Human CYP2C mRNA and protein levels were induced by tBHQ in primary human hepatocytes. Induction of CYP2C9, CYP2C19, and CYP2A6 mRNA in primary human hepatocytes following treatment with 50 μM tBHQ for 24 hours (A) and 48 hours (B). Results are presented relative to uninduced cells. A PXR inducer, rifampicin (5 μM), was used as a positive control. *P < 0.05, significantly different compared with DMSO control (Student’s t test); **P = 0.06 compared with DMSO control (Student’s t test). (C) Western blot analysis showed CYP2C9 and CYP2C19 protein levels were induced in human primary hepatocytes after treatment with 50 μM tBHQ for 48 hours, whereas CYP2C8 levels were unchanged. (D) Densitometry analysis of Western blot with Image J software. *P < 0.05, significantly different compared with DMSO control (Student’s t test). The following recombinant yeast CYP2C protein controls were used: 2C9 (CYP2C9), 2C8 (CYP2C8), and 2C19 (CYP2C19). HLM, human liver microsomes (Gentest).
Functional Characterization of the Putative AP-1 Binding Sites in CYP2C9 Promoter Constructs in HepG2 Cells.
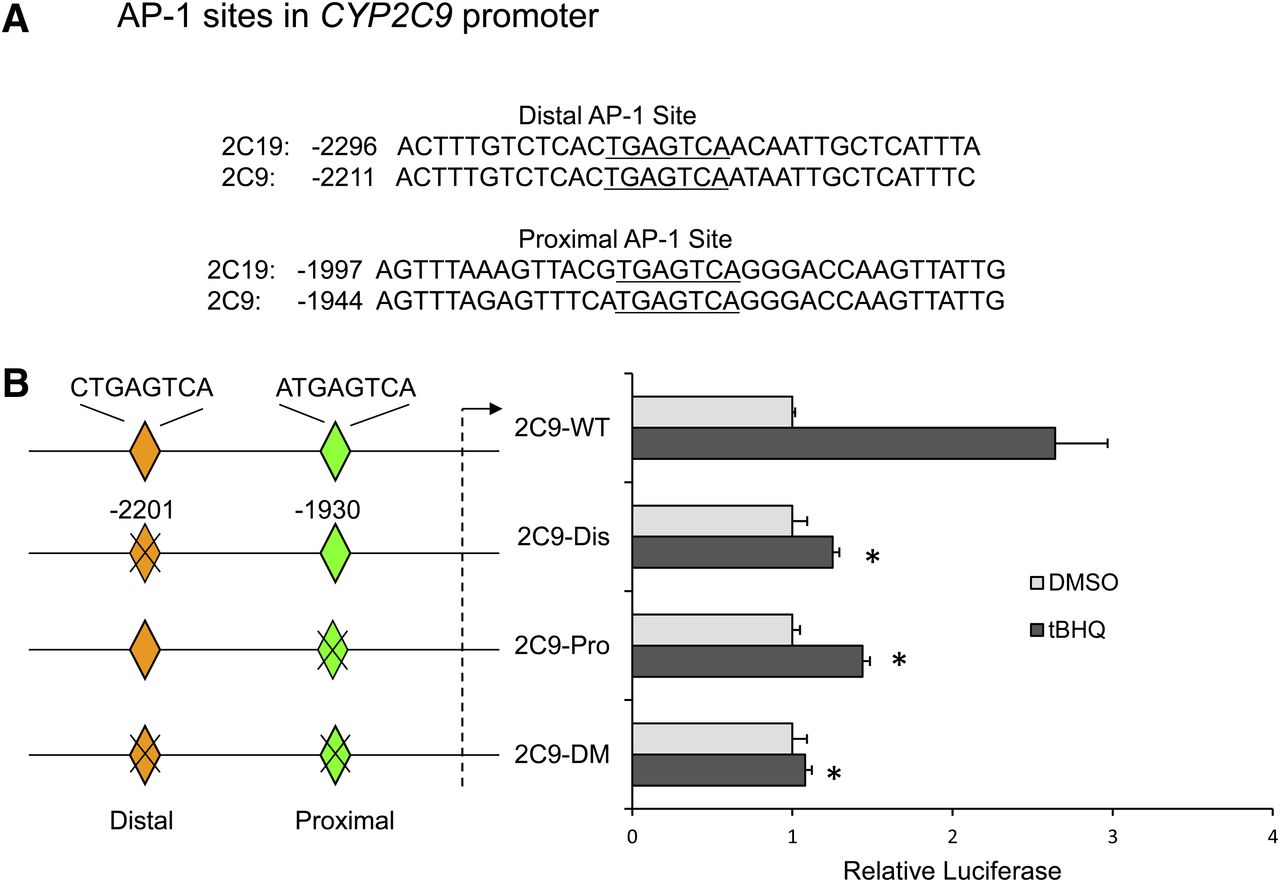
Computer analysis of CYP2C9 gene promoter revealed the presence of two putative AP-1 sites (TGAGTCA) at positions −2201 and −1930. These AP-1 sites are highly conserved between the CYP2C9 and CYP2C19 promoters but were not found in the CYP2C8 promoter (Fig. 2A). Site-directed mutagenesis was used to investigate the relative importance of the two putative AP-1 sites in the transactivation of the CYP2C9 promoter by tBHQ. Mutation of either AP-1 site significantly decreased CYP2C9 luciferase activity in HepG2 cells treated with tBHQ compared with the wild-type plasmid. Double mutation of both the distal and proximal AP-1 sites abolished CYP2C9 reporter activation by tBHQ (Fig. 2B). These results demonstrate that both AP-1 sites are required for maximal activation of the CYP2C9 luciferase promoter construct by tBHQ.

Effect of mutagenesis of the putative proximal and distal AP-1 elements on induction of CYP2C9 promoter luciferase activity after treatment of HepG2 cells with tBHQ. (A) Computer analysis of the CYP2C9 promoter showed two putative AP-1 sites (TGAGTCA) at positions −2201 and −1930, which are highly conserved in CYP2C19. (B) HepG2 cells were transiently transfected with p2C9-3.0Luc (2C9-WT), 2C9-Dis (mutation of distal AP-1 site), 2C9-Pro (mutation of proximal AP-1 site), or 2C9-DM (mutation of both distal and proximal AP-1 site). Twenty-four hours after transfection, cells were treated with either DMSO (0.1%) or tBHQ (50 μM) for 24 hours. Cells were harvested, and firefly and Renilla luciferase activities were measured. Results were expressed as fold induction compared with DMSO-treated cells. *P < 0.05, significantly different compared with tBHQ-treated 2C9-WT (wild-type)-transfected cells (one-way ANOVA, followed by Tukey’s test).
Effect of AP-1 Transcription Factors on the Expression of CYP2C9 in HepG2 Cells.
We next examined the effect of ectopic expression of AP-1 proteins and Nrf2 on CYP2C9 luciferase reporter activity in HepG2 cells (Fig. 3A). Ectopic overexpression of Nrf2 had no significant effect on CYP2C9 luciferase activity. We observed ∼30-, ∼6.7-, and ∼sevenfold increases in CYP2C9 luciferase activity after transfection with cFos, cJun, and JunD, respectively. Ectopic expression of cFos together with cJun resulted in significantly lower CYP2C9 luciferase activity compared with cFos alone. Similar levels of CYP2C9 luciferase activity were observed when cells were transfected with cFos and JunD (∼30-fold) compared with cFos alone. Because Fos proteins are not known to form active transcriptional complexes as a homodimer, but form active heterodimers with Jun proteins, we examined the levels of Jun and ATF proteins in nuclear extracts of HepG2 cells before and after ectopic expression of cFos (Fig. 3B). Western blot analysis of nuclear extracts showed high basal levels of JunD, cJun, ATF2, and ATF4 in contrast to low levels of cFos and JunB. Due to high basal level of JunD and cJun in the nuclear extract of HepG2 cells, we hypothesize that CYP2C9 might be induced by formation of an AP-1 heterodimer of cFos and JunD. The absence of synergism after ectopic expression of cFos and JunD could be due to the high levels of constitutive expression of JunD. Therefore, we examined the effect of silencing either JunD or cJun on cFos-induced transactivation of CYP2C9 luciferase activity in HepG2 cells. Western blot analysis confirmed silencing of JunD (86% knockdown) and cJun (70% knockdown) proteins by JunD- and cJun-specific siRNA, respectively, compared with control siRNA (Fig. 3C, 3D). As predicted, knockdown of JunD significantly decreased cFos-mediated transactivation of CYP2C9 luciferase activity (Fig. 3E), suggesting that both cFos and JunD contribute to the induction of CYP2C9 gene. The residual increase in luciferase activation presumably reflects the small amounts of JunD or another transcription factor present after silencing JunD (Fig. 3D). In contrast, silencing of cJun had no significant effect on cFos activation of CYP2C9 luciferase reporter activity (Fig. 3E).

CYP2C9 luciferase activity is significantly transactivated by ectopic expression of c-Fos. (A) Cotransfection of cJun with cFos, but not JunD, decreased the cFos-mediated increase in CYP2C9 luciferase activity. HepG2 cells were cotransfected with p2C9-3.0Luc (spanning −3077 to +1 of the CYP2C9 promoter) and various expression plasmids, as indicated. Cells were harvested after 48 hours, and luciferase activity was normalized to Renilla luciferase activity. Results were expressed as fold induction from at least three independent experiments compared with that of vector control. *P < 0.05, significantly different compared with vector control; †P < 0.05, significantly different compared with cFos-transfected cells (one-way ANOVA, followed by Tukey’s test). (B) Western blot analysis of AP-1 proteins (40 μg) in nuclear extracts of vector control and c-Fos plasmid-transfected HepG2 cells using antibodies against c-Fos, c-Jun, JunB, JunD, ATF2, and ATF4. (C) Western blot analysis of JunD and cJun in total HepG2 cell extracts after transfection with control siRNA (siControl), JunD-specific siRNA (siJunD), or cJun-specific siRNA (sicJun). (D) Densitometry analysis of the Western blot of JunD and cJun knockdown, as shown in (C), using Image J software. (E) Knockdown of JunD, but not cJun, significantly decreased cFos-mediated transactivation of CYP2C9 luciferase activity. HepG2 cells were transfected with p2C9-3.0Luc reporter plasmid and expression plasmid for cFos together with the indicated siRNAs for 48 hours. Luciferase activity was normalized to Renilla luciferase, and results were expressed as fold induction from at least three independent experiments compared with that of control siRNA-transfected cells. *P < 0.05, significantly different compared with vector-transfected cells; †P < 0.05, significantly different compared with cFos/siControl-transfected cells (one-way ANOVA, followed by Tukey’s test).
Induction and Phosphorylation of AP-1 Proteins in HepG2 Cells Treated with tBHQ.
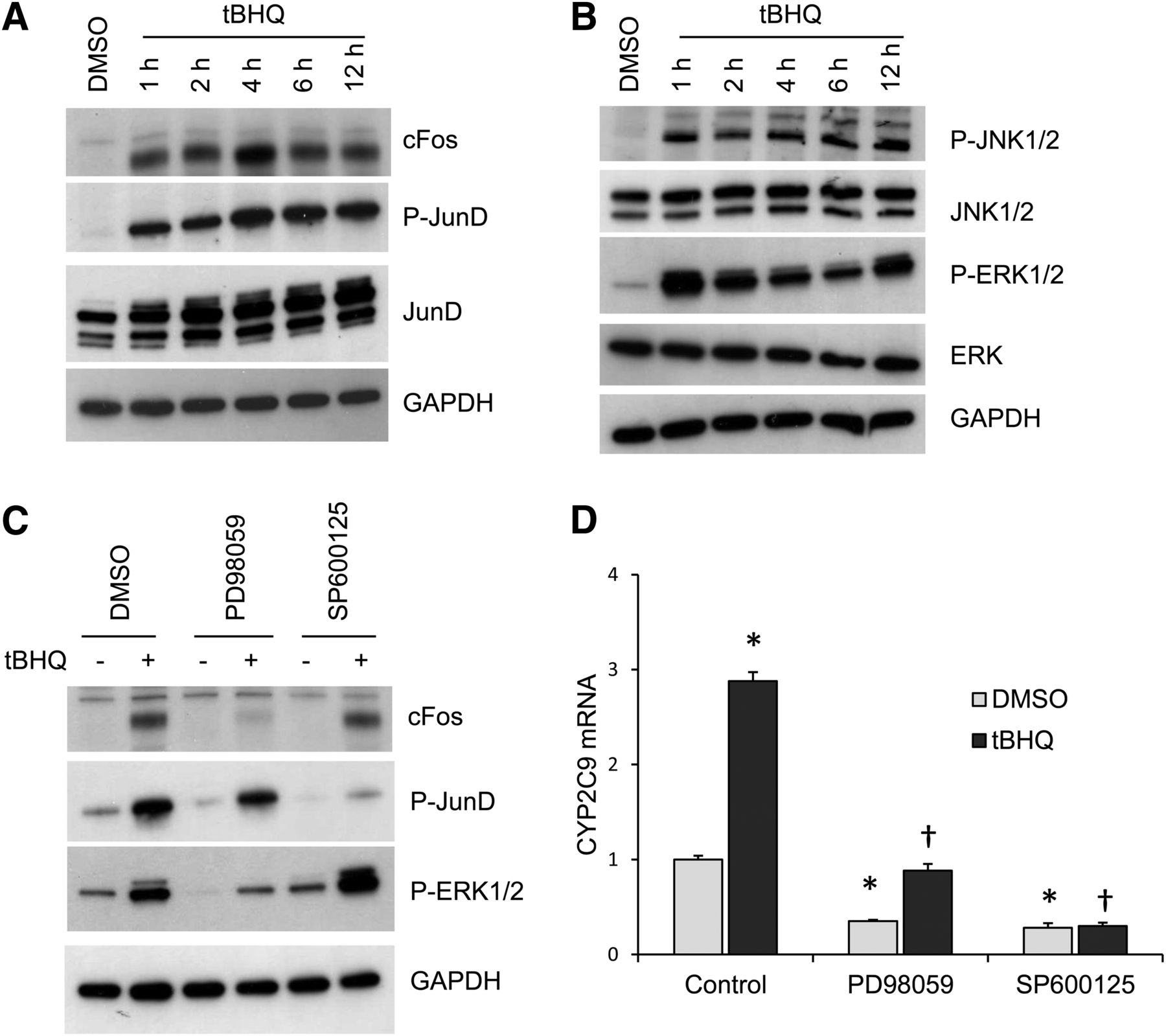
The activation by AP-1 protein usually involves increases in AP-1 mRNA and protein levels as well as increases in phosphorylation of AP-1 proteins and their DNA-binding activities. We examined the effects of 50 μM tBHQ in HepG2 cells on cFos protein after 1- to 12-hour treatment. cFos was dramatically increased as early as 1 hour after tBHQ treatment and maximally after 4 hours (Fig. 4A). We also observed a dramatic increase in phosphorylation of JunD 1 hour after tBHQ treatment with the maximal increase occurring at 4 hours. Alterations in the cellular redox state are known to activate ERK and JNK, which then phosphorylate and activate AP-1 proteins (Eferl and Wagner, 2003). Dramatic increases in phosphorylation of JNK and ERK were present 1 hour after tBHQ treatment (Fig. 4B). Treatment with PD98059 (a specific MEK inhibitor) inhibited tBHQ-induced ERK phosphorylation and cFos expression, whereas SP600125 (a JNK inhibitor) inhibited JunD phosphorylation (Fig. 4C). Moreover, the inhibition of ERK and JNK phosphorylation by PD98059 and SP600125 significantly decreased the tBHQ-mediated induction of CYP2C9 mRNA (Fig. 4D), suggesting that tBHQ-induced expression of CYP2C9 mRNA involves activation of ERK and JNK, which induce expression of cFos expression and phosphorylation of JunD, respectively.

ERK and JNK are essential for tBHQ-mediated induction of CYP2C9 mRNA by increased expression of cFos and phosphorylation of JunD. (A) Treatment of HepG2 cells with tBHQ (50 μM) increased expression of cFos and phosphorylation of JunD. HepG2 cells were treated with tBHQ for various time intervals (0–12 hours). Total cell lysates were analyzed by Western blot analysis with antibodies against cFos, phospho-JunD, and JunD with GAPDH as a loading control. (B) The activation of ERK and JNK by tBHQ. Membranes were probed with antibodies against phospho-p54/46 JNK, p54/46 JNK, phospho-p44/42 ERK, and p42 ERK. (C) Increased cFos expression and JunD phosphorylation were inhibited by PD98059 (MEK inhibitor) and SP600125 (JNK inhibitor), respectively. HepG2 cells were pretreated for 2 hours with 20 μM PD98059 or 50 μM SP600125 and then treated with 50 μM tBHQ for 6 hours. Total cell extracts were analyzed by Western blot with antibodies against phospho-ERK, phospho-JunD, JunD, and GAPDH. (D) Cells were pretreated with 20 μM PD98059 and 50 μM SP600125 for 2 hours, and then treated with 50 μM tBHQ for 24 hours and CYP2C9 mRNA analyzed by quantitative real-time PCR. *P < 0.05, significantly different compared with DMSO-treated cells; †P < 0.05, significantly different compared with tBHQ-treated cells (one-way ANOVA, followed by Tukey’s test).
EMSA Analysis of Putative AP-1 Binding Sites.
EMSA was used to examine whether the AP-1 proteins bind to the CYP2C9 distal and proximal AP-1 sites. 32P-labeled oligonucleotides containing the consensus AP-1 element (Promega, Madison, WI), CYP2C9 distal, and proximal AP-1 sites were incubated with nuclear extracts from both untreated and tBHQ-treated HepG2 cells (Fig. 5A–C). We observed formation of a DNA-protein complex with the labeled AP-1 consensus oligonucleotides, which was effectively inhibited by addition of excess unlabeled oligonucleotide (cold), indicating the binding was specific (Fig. 5A). Supershift experiments showed that the protein complexes formed with the distal and proximal CYP2C9 AP-1 oligonucleotides were shifted by antibodies against cFos and JunD, respectively, in tBHQ-treated nuclear extracts but not detectable in control nuclear extracts. Although cJun did not supershift CYP2C9 proximal or distal AP-1 site in EMSAs (Fig. 5B, 5C), the cJun antibody produced a consistent depletion of the DNA-protein complex at the proximal site in complexes with tBHQ nuclear extracts (Fig. 5B, 5C), suggesting the possibility of minimal binding at these sites. Supershift experiments were performed by incubating radiolabeled oligonucleotides containing CYP2C9 distal, proximal AP-1 sites, and AP-1 consensus element with tBHQ-treated HepG2 cell nuclear extracts and with broad-spectrum AP-1 antibodies or Nrf2 proteins (Fig. 5D–F). Again the complexes were supershifted by antibodies against cFos (for the distal site) and JunD (for proximal site), indicating that cFos interacts with the distal and JunD with the proximal site after incubation with tBHQ-treated nuclei (Fig. 5D, 5E). The DNA-protein complexes with the labeled AP-1 consensus oligonucleotide were competed with excess unlabeled oligonucleotide (Fig. 5F, lane 3), and complexes were supershifted by antibodies against cFos, JunD, and, importantly, cJun (lanes 4–6), indicating the presence of these proteins in the AP-1 complex. These results indicate that, in contrast to the AP-1 consensus control, cFos distinctly binds to the distal site and JunD to the proximal site of CYP2C9. Because the DNA-protein complexes at the proximal site were decreased by antibody to cJun, we cannot rule out cJun binding. However, these EMSAs contrast with the AP-1 consensus sequence, where a distinct supershift is seen with the same antibody for cJun.
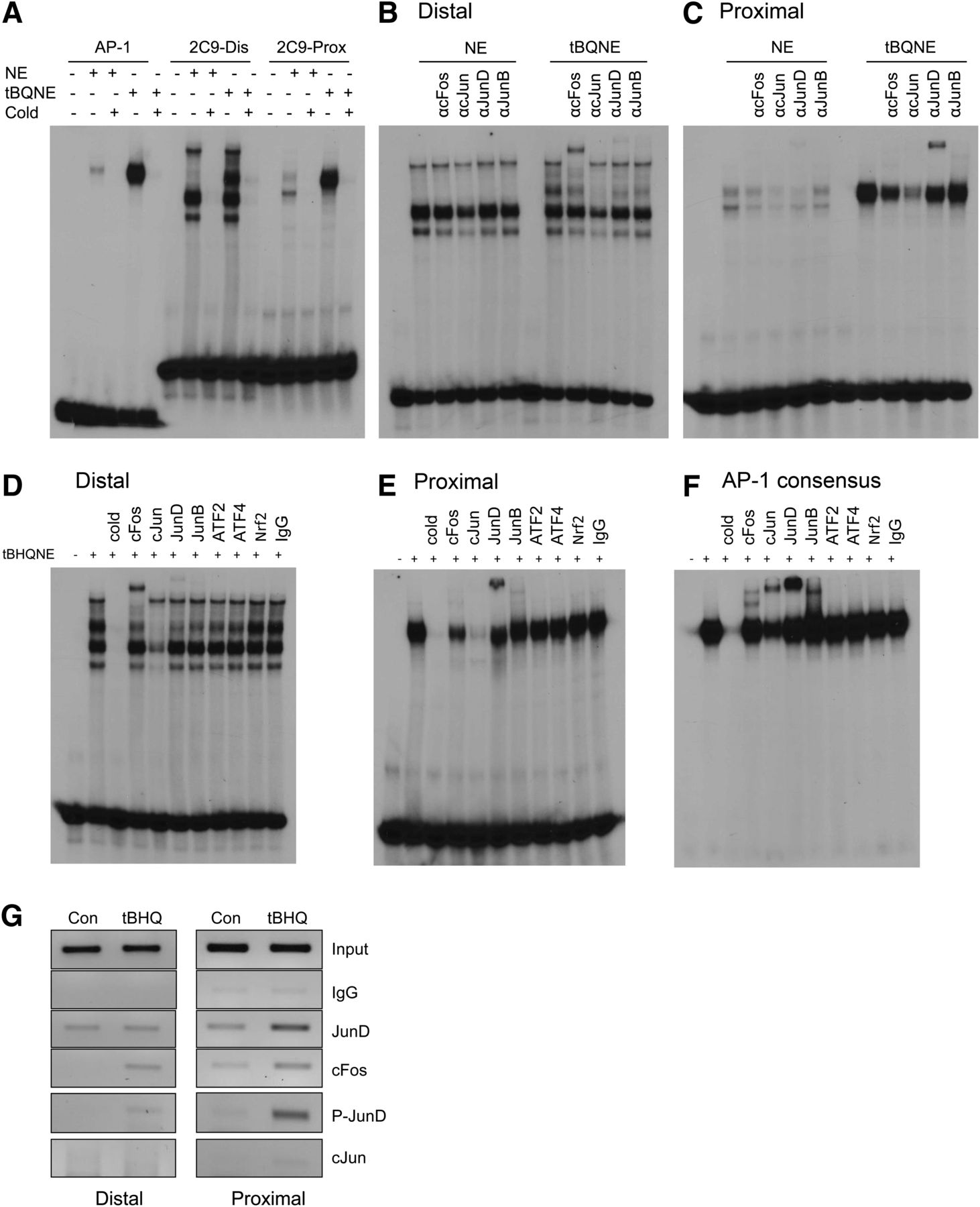
EMSA analysis indicates formation of a nuclear protein complex with the distal (−2201) and proximal (−1930) AP-1 sites of the CYP2C9 promoter, and ChIP analysis confirms the binding of cFos and JunD in vivo. (A) 32P-labeled double-stranded oligonucleotides containing CYP2C9 distal AP-1 site, CYP2C9 proximal AP-1 site, and AP-1 consensus sequence (as a positive control) were incubated for 30 minutes with 5 μg nuclear extracts from control (NE) or tBHQ-treated (tBQNE) cells. In competition experiments, a 100-fold excess of unlabeled double-stranded oligonucleotide inhibited formation of the complex. (B) 32P-labeled double-stranded oligonucleotides containing CYP2C9 distal AP-1 site and (C) CYP2C9 proximal AP-1 site were incubated for 30 minutes with 5 μg nuclear extracts isolated from DMSO (0.1%)- or tBHQ (50 μM)-treated HepG2 cells. Supershift experiments were then performed by incubating the binding reactions with 4 μg antibodies to cFos, cJun, JunD, or JunB for 2 hours at 4°C. (D–F) 32P-labeled oligonucleotides containing the CYP2C9 distal AP-1 site (D), CYP2C9 proximal AP-1 site (E), or AP-1 consensus sequence (F) (as a positive control) were incubated for 30 minutes with 5 μg nuclear extracts from tBHQ-treated HepG2 cells, and supershift experiments were performed by incubating the binding reactions with 4 μg antibodies to a wider spectrum of AP-1 antibodies (cFos, cJun, JunD, JunB, ATF2, ATF4) or Nrf2 antibodies for 2 hours at 4°C. (G) ChIP analysis showed binding of cFos and JunD in vivo to the CYP2C9 distal and proximal AP-1 sites. Chromatin was prepared from the HepG2 cells treated with DMSO control or tBHQ for 24 hours; sheared; immunoprecipitated with IgG (negative control), cFos, cJun, JunD, or phospho-JunD (P-JunD) antibodies conjugated to Dynabeads protein A/G; and analyzed by PCR with primers spanning the distal and proximal AP-1 sites.
EMSAs Using In Vitro Translated cFos or cJun with the Proximal and Distal CYP2C9 AP-1 Sites and the AP-1 Consensus Site.
cFos and cJun synthesized using a TNT Quick Coupled Transcription/Translation System in the presence of 35S-methionine or unlabeled methionine were verified by autoradiography and Western blot using antibodies to cJun and cFos (Supplemental Fig. 1). To determine whether either cFos or cJun binds to the AP-1 sites of CYP2C9, EMSAs were performed using both sites as well as the AP-1 consensus control. Pure translated cFos bound to the distal (−2201) AP-1 site of CYP2C9 either as a monomer or homodimer in the absence of other AP-1 proteins (Supplemental Fig. 2A). In contrast, we found no evidence for binding of in vitro translated cJun to the proximal −1930 CYP2C9 AP-1 site (or the distal site), although it did bind to the AP-1 consensus control (Supplemental Fig. 2B).
ChIP Analysis of Transcription Factor Binding to the Putative TREs in HepG2 Cells before and after Treatment with tBHQ.
ChIP experiments were also used to assess in vivo recruitment of cFos and JunD to the distal and proximal AP-1 sites before and after treatment of HepG2 cells with tBHQ. As shown in Fig. 5G, we observed enhanced recruitment of JunD and phospho-JunD to the proximal AP-1 site after treatment of the HepG2 cells with tBHQ compared with more moderate recruitment of cFos. There was minimal recruitment of JunD and cFos without treatment of the cells with tBHQ and no recruitment of phospho-JunD. The proximal and distal AP-1 sites appeared to bind JunD moderately even in the absence of tBHQ. However, JunD is highly expressed in these cells. Moreover, it should be noted that the distal and proximal sites are only ∼270 bases apart, and the average length of the sonicated fragments is 200–500 bp. Thus, we cannot completely separate the sites. cFos bound to both the distal and proximal sites only after treatment of the cells with tBHQ. Binding to both sites is expected by an in vivo method such as ChIP if the distal and proximal AP-1 sites are brought together by DNA looping as proposed. This is probable because, in ChIP experiments, formaldehyde is used to cross-link the protein to DNA prior to sonication. The binding to both sites may also reflect the incomplete separation of the sites. Binding of cJun was not detected at either site before or after tBHQ treatment.
3C Analysis of DNA Looping.
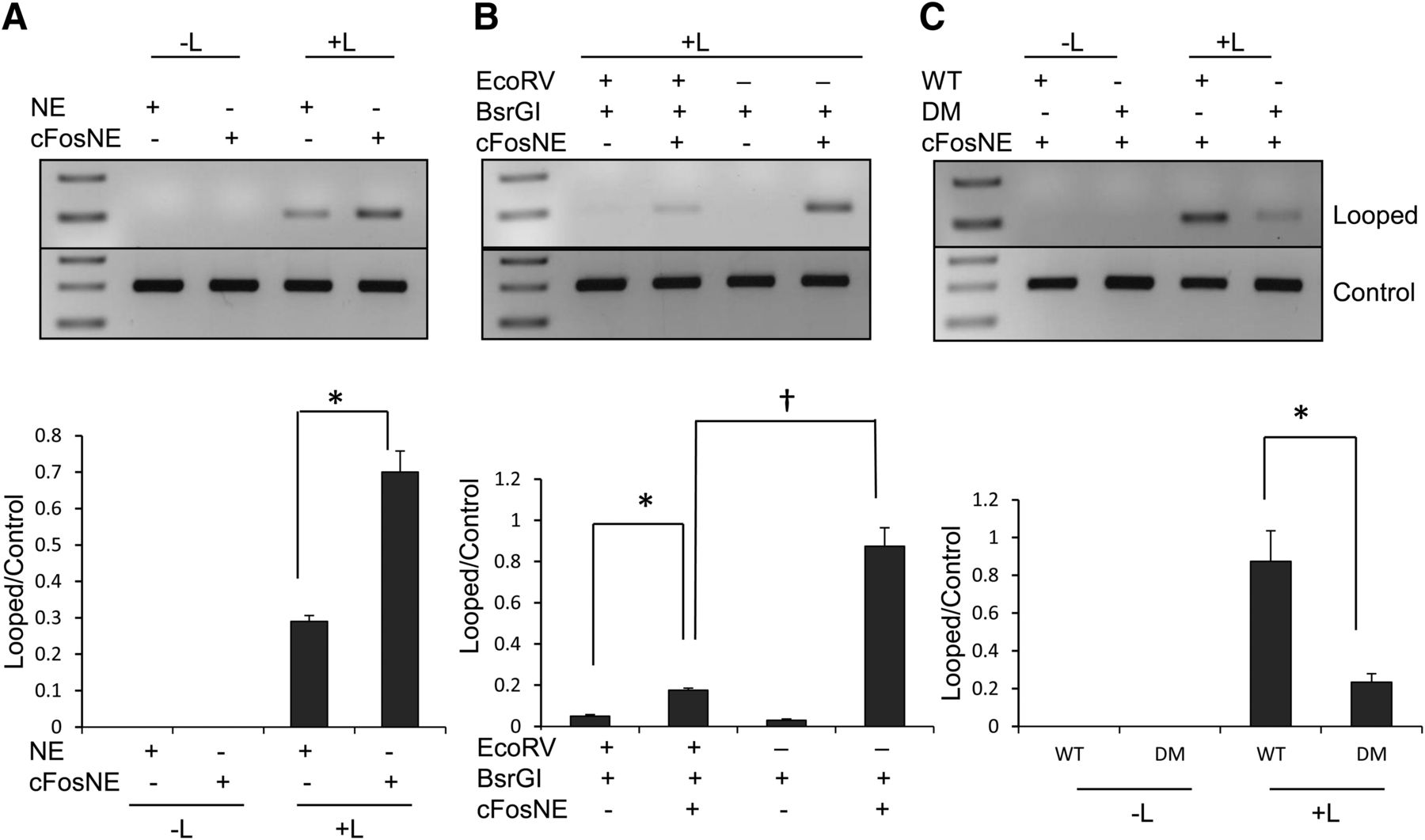
Because CYP2C9 transcription may be induced by a cFos and JunD heterodimer, we propose that DNA looping could bring the distal and proximal AP-1 sites into close proximity, allowing formation of an AP-1 complex containing cFos and JunD. To test this hypothesis, we assessed the interaction between these two AP-1 sites by 3C assays in HepG2 cells after ectopic expression of cFos. The promoter region of CYP2C9 contains BsrG1 restriction sites that closely flank the distal and proximal AP-1 sites (Fig. 6A). Fig. 6B shows a graphical representation of the 3C method explained in Materials and Methods similar to the strategy described by Dekker (2006). The purified DNA was amplified by conventional PCR with primers spanning the ligated region to produce the looped product and control primers for linear DNA product. We showed increased formation of looped product after ectopic cFos expression compared with empty vector control (Fig. 7A). The digestion with EcoRV was used to differentiate looping from end-to-end ligation. Because there is the possibility of end-to-end ligation instead of DNA looping, the linearized plasmid was incubated with cFos-transfected nuclear extracts and digested with either BsrGI or both BsrGI and EcoRV. The increased formation of looped product after ectopic cFos expression was eliminated with EcoRV digestion (Fig. 7B), indicating the formation of predominantly looped product instead of end-to-end ligation product. The formation of the looped product was significantly reduced by mutation of both AP-1 sites compared with wild-type sequence (Fig. 7C).

Schematic diagram of the 3C assay. (A) The CYP2C9 promoter showing the distal and proximal AP-1 sites with restriction enzymes, BsrGI and EcoRV, for the 3C assay. (B) Outline of the 3C method. For in vitro 3C assays, linearized plasmid (−3077 to +1) containing both AP-1 sites were incubated with nuclear extracts. For in vivo 3C, primary human hepatocytes were directly treated with formaldehyde to cross-link protein to DNA. The restriction enzyme, BsrGI, digested the DNA close to the AP-1 sites. The digested DNA was then ligated with or without T4 DNA ligase. The protein-DNA complexes were reversed cross-linked, and the control and ligated DNA were purified using either a PCR purification kit (in vitro 3C) or phenol chloroform (in vivo 3C). The purified DNA was amplified by conventional PCR with primers spanning the ligated region (primers A and B) for looped products and control primers (primers C and D) for linear DNA products. The digestion with EcoRV was used to differentiate looping from end-to-end ligation.

In vitro 3C assays demonstrating interaction of the CYP2C9 distal and proximal AP-1 binding sites by DNA looping. (A) The linearized CYP2C9 promoter (−3077 to +1) was incubated with nuclear extract (NE) from HepG2 cells before or after transfection with cFos (cFosNE). In vitro 3C assays were performed with BsrGI, and the purified DNA was amplified with primers spanning the ligated region compared with products amplified with internal control primers. Below is a densitometry analysis with Image J software. *P < 0.05, significantly different compared with nuclear extract from HepG2 cells (Student’s t test). −L, absence of ligase; +L, ligase. (B) Confirmation of DNA looping by EcoRV restriction analysis. The CYP2C9 promoter (−3077 to +1) was incubated with nuclear extract from cFos-transfected HepG2 (cFosNE). The cross-linked DNA was digested with BsrGI alone or with BsrGI and EcoRV restriction enzymes. The reversed cross-linked and purified DNA was amplified with primers spanning the ligated region. *P < 0.05, significantly different compared with BsrG1/EcoRV digestion in the absence of nuclear extracts; †P < 0.05, significantly different compared with BsrG1 digestion in the presence of cFos-transfected nuclear extracts (one-way ANOVA, followed by Tukey’s test). (C) Wild-type CYP2C9 promoter (WT) or the promoter containing mutated distal and proximal AP-1 sites (DM) was incubated with nuclear extract from cFos-transfected cells (cFosNE). In vitro 3C assays were performed, as described in Materials and Methods. *P < 0.05, significantly different from ligated CYP2C9 WT promoter (Student’s t test).
In vivo 3C experiments were also performed in primary human hepatocytes transfected with control siRNA or JunD-specific siRNA and treated with DMSO or tBHQ. We observed ∼fourfold increase in the formation of looped product by tBHQ in control siRNA-transfected cells by PCR (Fig. 8A). The increased formation of looped products by tBHQ in control hepatocytes was significantly reduced by knockdown of JunD. Consistent with the in vitro 3C, increased looped product formation by tBHQ in cultured hepatocytes was absent when DNA was digested with both BsrGI and EcoRV compared with BsrGI alone (Fig. 8B), indicating the formation of predominantly looped product instead of end-to-end ligation product. The induction of CYP2C9 mRNA levels by tBHQ in primary human hepatocytes was significantly decreased by knockdown of JunD (Fig. 8C). Consistent with experiments in HepG2 cells, the specific inhibitors of ERK (PD98059) and JNK (SP600125) significantly decreased the tBHQ-mediated induction of CYP2C9 mRNA (Fig. 8D).
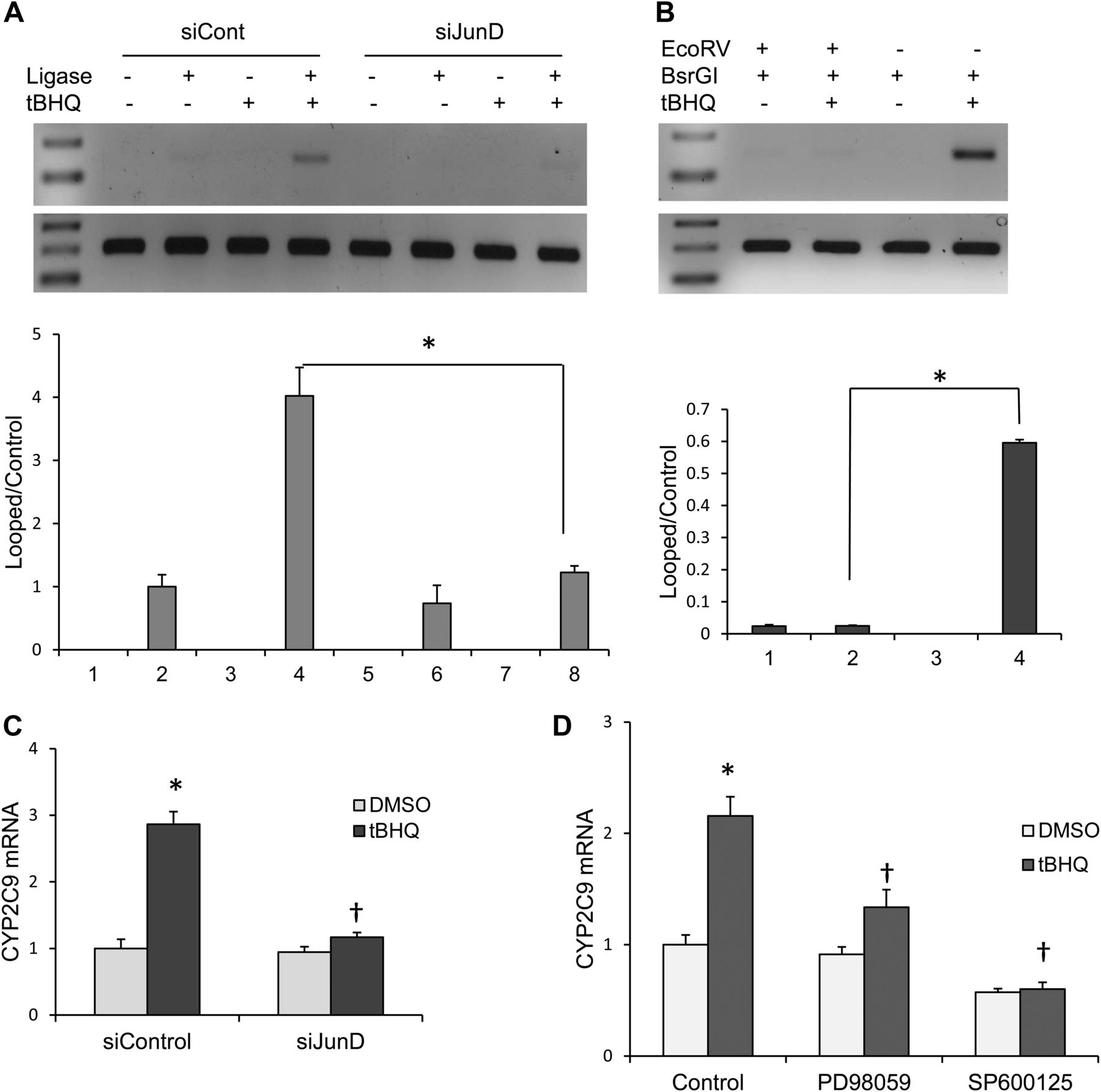
In vivo 3C assay to assess interaction of the distal and proximal AP-1 sites by DNA looping in primary human hepatocytes. (A) Hepatocytes were transfected with either control siRNA or JunD-specific siRNA. Forty-eight hours after transfection, cells were treated with DMSO (control) or tBHQ for 24 hours. Cells were treated with formaldehyde to cross-link protein-DNA. In vivo 3C assays were performed with BsrGI restriction enzyme, and the purified DNA was amplified with primers spanning the ligated region for looped PCR product and internal control primers for control products. Below is a densitometry analysis of the in vivo 3C with Image J software. *P < 0.05, significantly different compared with ligated tBHQ/siControl-transfected cells (one-way ANOVA, followed by Tukey’s test). (B) Confirmation of DNA looping by EcoRV restriction analysis. Primary human hepatocytes were treated with DMSO control or tBHQ for 24 hours. The formaldehyde cross-linked DNA was digested with BsrGI alone or with BsrGI and EcoRV. The reversed cross-linked and purified DNA was amplified with primers spanning the ligated region. *P < 0.05, significantly different compared with BsrG1-digested tBHQ-treated primary human hepatocytes (one-way ANOVA, followed by Tukey’s test). (C) Primary human hepatocytes transfected with either control siRNA or JunD-specific siRNA were treated with DMSO (control) or tBHQ for 24 hours, and (D) primary human hepatocytes were pretreated with 20 μM PD98059 and 50 μM SP600125 for 2 hours, and then treated with 50 μM tBHQ for 24 hours, and CYP2C9 mRNA levels were analyzed by quantitative real-time polymerase chain reaction. *P < 0.05, significantly different compared with DMSO-treated cells; †P < 0.05, significantly different compared with tBHQ-treated cells (one-way ANOVA, followed by Tukey’s test).
Discussion
In this study, we report a new mechanism for CYP2C9 and CYP2C19 induction, but not CYP2C8, by electrophiles and oxidative stress. Many drugs and environmental compounds, such as butylated hydroxyanisole, are metabolized in the liver to electrophilic or reactive metabolites with enhanced production of reactive oxygen species and induction of oxidative stress (Chang et al., 1997; Erve, 2006; Guengerich, 2006; Takakusa et al., 2008). The role of electrophiles and oxidative stress in the regulation of the CYP2C genes has received very little attention. Reactive intermediates are important signaling molecules that induce the expression of cytoprotective genes by directly activating redox-sensitive transcription factors or indirectly activating kinase signaling pathways (Mendelson et al., 1996; Yu et al., 1997; Takakusa et al., 2008). Our laboratories previously reported the regulation of the electrophile detoxification gene, Aldh1a1, by electrophiles such as acrolein and tBHQ in mouse liver and liver-derived cell lines (Makia et al., 2012).
Due to the presence of putative AP-1 sites (TGAGTCA) at positions −2201 and −1930 in the promoter of CYP2C9 and the knowledge that many inducers of CYP2C are capable of forming reactive intermediates, we hypothesized that drugs and xenobiotics capable of inducing oxidative stress may enhance CYP2C9 expression by activation of AP-1 and/or possibly Nrf2 proteins. We show that both CYP2C9 and CYP2C19 genes were induced by tBHQ in cultured human hepatocytes. tBHQ is a cytochrome P450-dependent metabolite of BHA that undergoes autoxidation to tert-butylquinone, generating reactive oxygen species by redox cycling. The pro-oxidant activity of tBHQ was supported by experiments that demonstrated that HepG2 cells treated with tBHQ generated hydroxyl radicals and resulted in decreased levels of intracellular glutathione (Pinkus et al., 1996). Moreover, the induction of GST-Ya expression and AP-1 activity by tBHQ was inhibited by exogenous catalase and the antioxidant, N-acetyl cysteine (Pinkus et al., 1996).
AP-1 transcription factors regulate target gene expression as either homodimers of Jun family proteins or heterodimers of the Jun and Fos family proteins (Eferl and Wagner, 2003). Previously, Fos proteins had not been shown to form active transcriptional complexes as homodimers. However, in this study and another study by one of our laboratories, we find that translated cFos can bind to the distal site as either a homodimer or a monomer (Xu et al., 2014). We showed that tBHQ-induced expression of CYP2C9 is mediated possibly by interaction between cFos and JunD on different AP-1 sites. Additive effects of ectopic cFos and JunD were not observed in induction of CYP2C9 mRNA or CYP2C9-luciferase activity probably due to the high levels of constitutive expression of JunD in HepG2 cells. However, silencing JunD strongly suggested that CYP2C9-luciferase activity is transactivated by a cFos and JunD heterodimer. Until recently, JunD was thought to form a transcriptionally inactive complex with cFos, but an active heterodimer with FosB (Byun et al., 2006). However, Dong and coworkers provided evidence that hematopoietic genes were induced by a transcriptionally active heterodimer of cFos and JunD (Lee et al., 2012). Ectopic expression of Nrf2 had no significant effect on CYP2C9-promoter luciferase activity alone or in combination with cJun. Electrophilic or extracellular stresses are known to activate MAPK consisting of p38, ERK, and JNK (Mendelson et al., 1996; Yu et al., 1997; Cui et al., 2010). AP-1 proteins are major targets of activated MAPK. Activated MAPK pathways regulate AP-1 transcriptional activity by altering gene expression, phosphorylation, and DNA-binding activity. Previous studies have demonstrated that the activation of JunD is mediated by enhanced phosphorylation of Ser100 at the N terminus by JNK and ERK (Gallo et al., 2002; Yazgan and Pfarr, 2002; Tsuji, 2005). This phosphorylation site also conserved in cJun (Ser73) is detected using phospho-Ser73 antibody in Western blot experiments. Although both ERK and JNK pathways were activated by tBHQ, we showed that increased phosphorylation of JunD by tBHQ was mediated by JNK. Previous experiments demonstrate increased cFos expression is mediated by activated ERK, which phosphorylates and activates ternary complex factors (Eferl and Wagner, 2003). Activated ternary complex factor binds to the serum response element in the promoter of fos, resulting in increased expression. We showed that tBHQ-induced expression of cFos was regulated by ERK activation. To assess the relevance of JNK and ERK signaling pathways in tBHQ-mediated increase in CYP2C9 expression, selective kinase inhibitors were used. PD98059, a selective inhibitor of MEK1, inhibits the phosphorylation of ERK1/2, whereas SP600125, a specific inhibitor of JNK, inhibits the phosphorylation and activation of JunD. The incubation of cells with these inhibitors attenuates cFos expression and JunD phosphorylation by tBHQ and resulted in significant attenuation of tBHQ induction of CYP2C9 mRNA, suggesting major roles of ERK and JNK in induction of CYP2C9 by tBHQ.
DNA looping is a mechanism that can bring cis-response elements with their bound proteins in close proximity to each other or even distal regulatory elements into close proximity with the transcription start site to activate transcription by direct or indirect interaction between transcription factors. In the present study, we demonstrate that the distal and proximal AP-1 sites in CYP2C9 promoter are brought in close proximity by DNA looping, possibly allowing formation of a complex containing cFos at the distal AP-1 site and JunD at the proximal AP-1 site.
Although this is the first report to describe the formation of a DNA loop mediated by two AP-1 sites on a target cytochrome P450 gene promoter, previous studies described the formation of short DNA looping of other gene promoters mediated by AP-1 and other transcription factors such as nuclear factor κB (NF-κB). DNA looping created by interactions between the NF-κB and AP-1 binding site of osteopontin promoter synergistically transactivates the osteopontin gene in murine macrophages (Zhao et al., 2011). The interaction between cJun at −1069 and NF-κB p65 at −86 mediated by DNA looping on the inducible nitric oxide synthase promoter was shown to be responsible for initiation of iNOS transcription in lipopolysaccharide-stimulated ANA-1 murine macrophages (Guo et al., 2008). It is known that interaction between factors bound at nonadjacent cis-acting elements can induce bending or looping of the DNA to form higher order structures (Matthews, 1992; Kerppola and Curran, 1993; Natesan and Gilman, 1993). Indeed, the transcription factors ATF and NF-κB have been shown to induce bending or looping of DNA on the E-selectin promoter (Meacock et al., 1994). We have demonstrated the pivotal role of cFos and JunD in looping of the CYP2C9 promoter to possibly allow formation of a complex containing these proteins. Overexpression of cFos enhanced looping, whereas knockdown of JunD decreased the formation of loop product. We also show that both the distal and proximal AP-1 sites are involved in CYP2C9 looping. DNA loop formation was confirmed by the absence of looped product after EcoRV digestion. We propose that DNA looping brings the distal and proximal AP-1 sites into close proximity, possibly leading to interaction between cFos and JunD for induction of CYP2C9 gene.
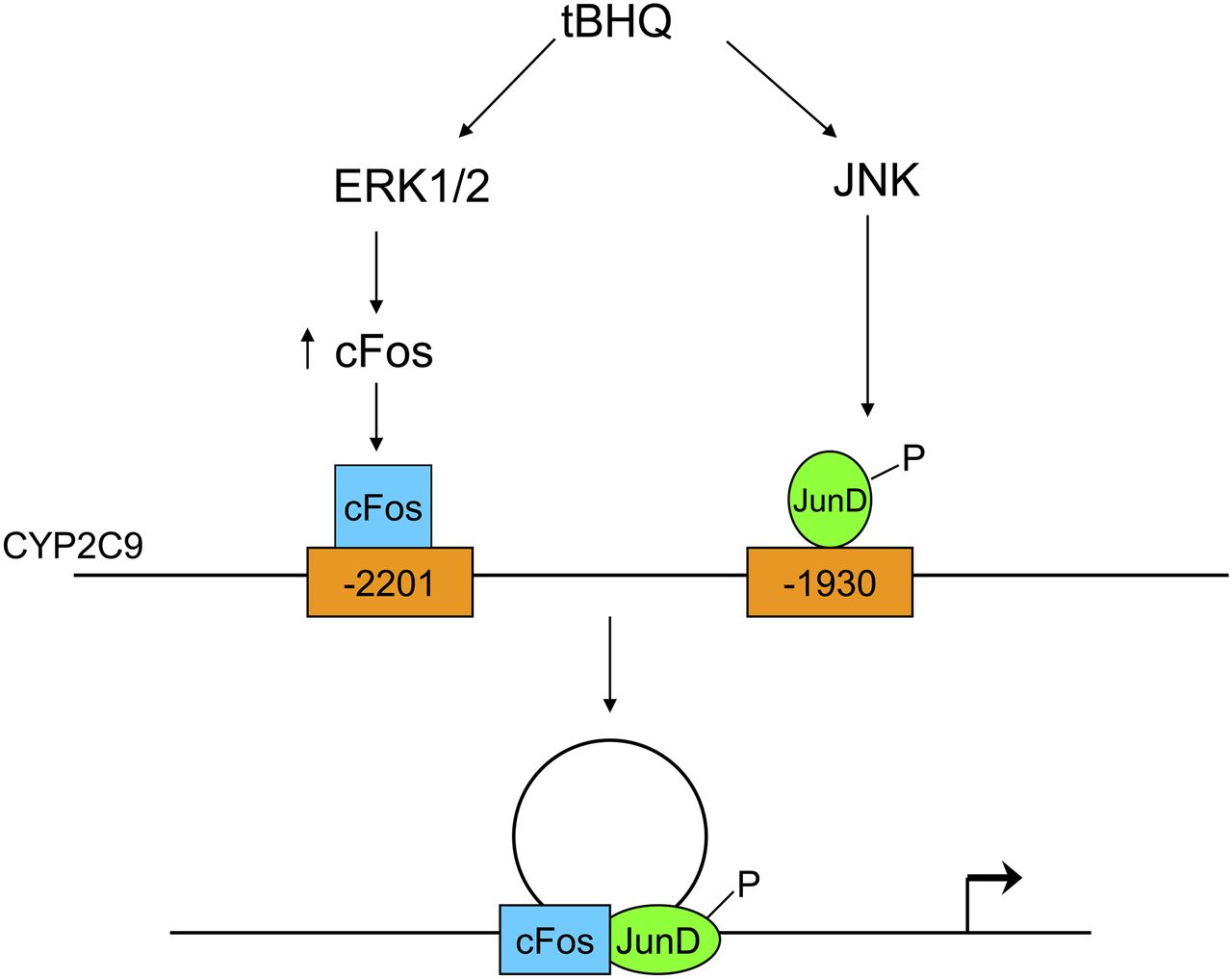
A proposed model for the regulation of CYP2C9 by tBHQ is illustrated in Fig. 9. The induction of CYP2C9 expression by the pro-oxidant, tBHQ, is mediated by cFos/JunD heterodimers. ERK and JNK are essential for tBHQ-mediated induction of CYP2C9 expression. Activation of ERK by tBHQ induces cFos expression, whereas JNK activation stimulates JunD phosphorylation. The cFos and JunD distinctly bind to the distal (−2201) and proximal (−1930) AP-1 sites, respectively, in EMSAs. Both AP-1 sites are brought together by DNA looping, as shown by 3C assay, for successful induction of CYP2C9 by tBHQ. These results provide strong evidence that compounds capable of inducing oxidative stress enhance the expression of CYP2C9 and represent an additional mechanism for induction of CYP2C9 and CYP2C19, thus altering the clearance of substrates metabolized by these enzymes.

Schematic model for the regulation of CYP2C9 expression by the electrophile, tBHQ. tBHQ stimulates the activation and phosphorylation of ERK and JNK. Activation of ERK by tBHQ induces cFos expression, whereas JNK activation stimulates JunD phosphorylation. cFos and JunD distinctly bind to the distal (−2201) and proximal (−1930) AP-1 sites, respectively. The two AP-1 sites are brought together by DNA looping for induction of CYP2C9 gene expression.
Acknowledgments
The authors thank Joyce Blaisdell, National Institute of Environmental Health Sciences, for assistance with this work. We also thank Drs. Masahiko Negishi and Tatsuya Sueyoshi of the Laboratory of Reproductive and Developmental Toxicology at the National Institute of Environmental Health Sciences for helpful conversations concerning the appropriate controls for 3C assays to exclude end-on-end ligation and for constructive criticism of the manuscript.
Authorship Contributions
Participated in research design: Makia, Monostory, Surapureddi, Prough, Goldstein.
Conducted experiments: Makia, Monostory, Surapureddi.
Performed data analysis: Makia, Monostory, Surapureddi, Prough, Goldstein.
Wrote or contributed to the writing of the manuscript: Makia, Monostory, Surapureddi, Prough, Goldstein.
Footnotes
- Received March 7, 2014.
- Accepted May 14, 2014.
This work was supported in part by the Intramural Research Program of the National Institutes of Health National Institute of Environmental Health Sciences [Project ES021024]; National Institutes of Health National Institute of Environmental Health Sciences [Grant ES11860]; National Development Agency [GOP-1.3.1-11/B-2011-0042]; and Hungarian Scientific Research Fund [OTKA K104459].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- ANOVA
- analysis of variance
- AP-1
- activator protein 1
- ATF
- activating transcription factor
- 3C
- chromosome conformation capture
- ChIP
- chromatin immunoprecipitation assay
- DMSO
- dimethylsulfoxide
- EMSA
- electrophoretic mobility shift assay
- ERK
- extracellular signal-regulated kinase
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- JNK
- Jun N-terminal kinase
- MAPK
- mitogen-activated protein kinase
- MEK
- mitogen-activated protein kinase kinase
- NF-κB
- nuclear factor κB
- Nrf2
- nuclear factor E2-related factor 2
- P450
- cytochrome P450
- PCR
- polymerase chain reaction
- PXR
- pregnane X receptor
- siRNA
- small interfering RNA
- tBHQ
- tert-butylhydroquinone
- U.S. Government work not protected by U.S. copyright





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}