


Abstract
It is now widely accepted that G protein-coupled receptors (GPCRs) are highly dynamic proteins that adopt multiple active states linked to distinct functional outcomes. Furthermore, these states can be differentially stabilized not only by orthosteric ligands but also by allosteric ligands acting at spatially distinct binding sites. The key pharmacologic characteristics of GPCR allostery include improved selectivity due to either greater sequence divergence between receptor subtypes and/or subtype-selective cooperativity, a ceiling level to the effect, probe dependence (whereby the magnitude and direction of the allosteric effect change with the nature of the interacting ligands), and the potential for biased signaling. Recent chemical biology developments are beginning to demonstrate how the incorporation of analytical pharmacology and operational modeling into the experimental workflow can enrich structure-activity studies of allostery and bias, and have also led to the discovery of a new class of hybrid orthosteric/allosteric (bitopic) molecules. The potential for endogenous allosteric modulators to play a role in physiology and disease remains to be fully appreciated but will likely represent an important area for future studies. Finally, breakthroughs in structural and computational biology are beginning to unravel the mechanistic basis of GPCR allosteric modulation at the molecular level.
Introduction
G protein-coupled receptors (GPCRs) are the largest family of cell-surface receptors encoded by the human genome (Lagerstrom and Schioth, 2008), and they participate in virtually all aspects of (patho)physiologic control. GPCR mutations can cause disease, and they remain one of the largest target classes for drug discovery (Overington et al., 2006; Thompson et al., 2008). However, and in common with many other drug target classes, the development of new GPCR-based therapeutics is hampered by an unacceptable attrition rate (Allison, 2012). Foremost among the possible reasons for this is the fact that most major illnesses are polygenic in nature, which suggests that a single-target drug may not be the optimal therapeutic choice (Roth et al., 2004). In addition, there remains an inadequate understanding of various aspects of GPCR biology that may contribute to the pathogenesis or modulation of disease in such instances, which makes it very difficult to link preclinical indices of cellular efficacy with desired therapeutic efficacy.
At the preclinical target and hit selection/validation stage, another important consideration has been the means by which a GPCR is exploited pharmacologically. Traditionally, this has been via targeting the primary GPCR-binding site recognized by the endogenous agonist(s)—that is, the orthosteric site (Christopoulos et al., 2014; Neubig et al., 2003); however, since the turn of the millennium, it has become increasingly appreciated that GPCRs can also be targeted via spatially distinct allosteric sites (Christopoulos, 2002; Christopoulos and Kenakin, 2002; May et al., 2007; Wootten et al., 2013). Ligands that bind to these sites are referred to as allosteric modulators because they can alter the conformation of the receptor to modulate its concomitant interaction with other ligands and/or intracellular signal transducing molecules, such as G proteins or β-arrestins. GPCRs are also highly dynamic and promiscuous proteins that can be directed in a ligand-specific manner to couple to certain pathways to the relative exclusion of others, a phenomenon that has been termed biased agonism, functional selectivity, pluridimensional efficacy, or collateral efficacy (Urban et al., 2007; Stallaert et al., 2011; Reiter et al., 2012; Kenakin and Christopoulos, 2013; Shonberg et al., 2014).
Allosteric modulation thus introduces further texture into GPCR pharmacology that can vary in a ligand-, receptor-, species-, or cell-dependent manner. It is possible that a failure to appreciate, detect, quantify, and validate allosteric drug effects at the preclinical stage has contributed to a number of translational failures in the past in programs that were “orthocentric” (i.e., orthosteric focused), although this remains speculative. Irrespective, there is now a growing appreciation that many (Fig. 1), if not all, GPCRs possess targetable allosteric sites that provide both challenges and opportunities for drug discovery.
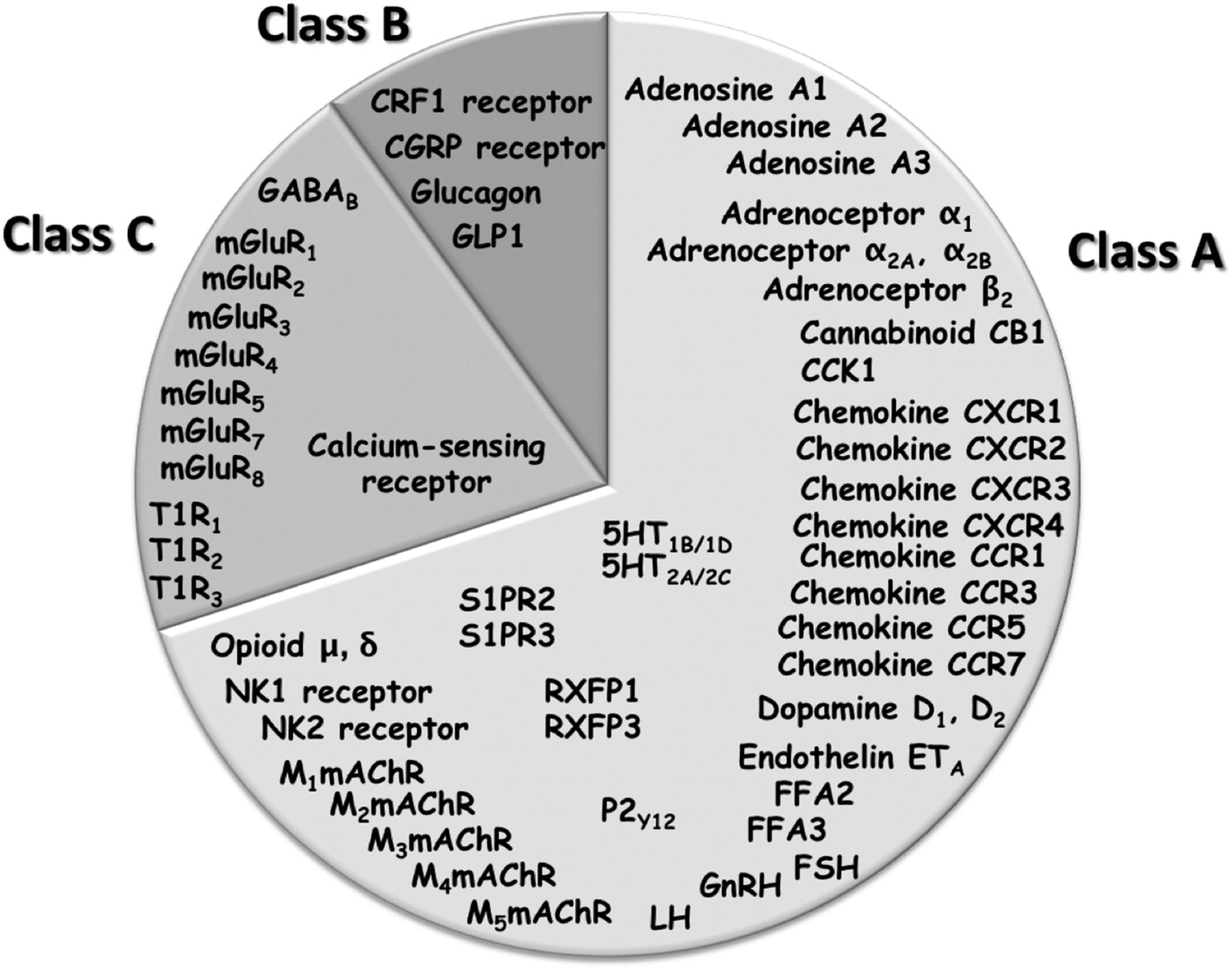
GPCRs for which allosteric modulators have been disclosed.
Much of the current interest in GPCR allosteric modulators as a true avenue for drug discovery can be traced to the turn of the millennium and the supplanting of (orthosteric) radioligand binding by cell-based signaling in the screening assays of choice; studies of the class C metabotropic glutamate receptors were particularly instrumental in this regard (Litschig et al., 1999; Varney et al., 1999). However, the phenomenon had actually been demonstrated (though largely unappreciated) many decades earlier. For instance, Lüllmann et al. (1969) were the first to propose a putative allosteric mechanism of action for a novel series of synthetic antimuscarinic agents based on tissue bioassay studies. A seminal report by Clark and Mitchelson (1976) described and quantified the mechanism of action of gallamine as a negative allosteric modulator of the M2 muscarinic acetylcholine receptor (mAChR) in an isolated atria bioassay, and this was confirmed in radioligand binding studies by Stockton et al. (1983). The first small molecule positive allosteric modulators of a GPCR agonist were described for the adenosine A1 receptor by Bruns and Fergus (1990), and that decade witnessed a progressive increase in the awareness of GPCR allostery that culminated in substantial activity after the turn of the millennium. This brief review will focus on developments and lessons learned in the study and exploitation of GPCR allosteric sites, with a particular emphasis on the last decade and a half.
Classification of Allosteric Modulators
In addition to a plethora of studies in the field of enzymology, where the phenomenon of allostery was first described (Monod et al., 1963, 1965; Koshland et al., 1966; Changeux, 2013), allosteric modulators have now been identified for all receptor superfamilies, including ligand-gated ion channels (Olsen et al., 2004; Taly et al., 2009; Traynelis et al., 2010), voltage-gated ion channels (Spedding et al., 1995; Catterall et al., 2007), nuclear hormone receptors (Estebanez-Perpina et al., 2007; Hughes et al., 2014), receptor tyrosine kinases (Bono et al., 2013; De Smet et al., 2014), and GPCRs. As a consequence, there exists a large body of literature in which the definition of the term “allosteric” and the description/classification of allosteric ligands can be quite varied and, in many instances, confusing (Colquhoun, 1998; Fenton, 2008; Nussinov and Tsai, 2013). Although a unified classification scheme based on molecular mechanisms would be desirable, this is currently not feasible for many systems because such mechanisms are not routinely obtainable from the common cell-based assays used to assess drug behavior. Furthermore, the assay conditions themselves can have a profound effect on the manifestation of an allosteric effect (Spedding and Mir, 1987).
Some fields use ligand-binding locus as a means for classifying allosteric modulators, such as ATP-site versus non-ATP site binders for kinases, but this approach is not readily transferable between protein families. Thus, at this point in time, allosteric modulators can be described operationally by ascribing a minimal set of properties expressed by allosteric ligand at a given receptor. These properties are 1) modulation of the affinity of a reference probe (typically an orthosteric ligand, but it can also be another allosteric ligand acting at a different allosteric site); 2) modulation of the signaling efficacy of a reference probe; 3) any direct agonist/inverse agonist effects of the modulator in the absence of other ligands. With regards to the effects on orthosteric ligands, therefore, allosteric compounds can be classified as positive allosteric modulators (PAMs), negative allosteric modulators (NAMs), or neutral allosteric ligands (NALs); the latter is preferable to terms such as “silent (or neutral) allosteric modulator” because if the interaction with an orthosteric ligand is neutral, then the allosteric compound is not a modulator per se. Superimposed on these classifications is the additional property of allosteric agonism/inverse agonism in terms of what the allosteric ligand does to the receptor system on its own (Christopoulos et al., 2014).
Ideally, each of these properties should be explicitly addressed when trying to classify the actions of an allosteric ligand, as should the reference ligand and assay, because the classification of an allosteric ligand is conditional on the nature of the reference ligand and the assay/pathway under investigation (Kenakin, 2005). For example, the mAChR allosteric modulator LY2033298 (3-amino-5-chloro-6-methoxy-4-methyl-thieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide) is a PAM of acetylcholine-mediated extracellular-regulated protein kinase 1/2 (ERK1/2) phosphorylation at the M4 mAChR, but essentially is a NAL of acetylcholine acetylcholine-mediated ERK1/2 phosphorylation at the M2 mAChR, despite having some direct allosteric agonism in its own right (Leach et al., 2010; Valant et al., 2012b).
Although it is not ideal, it is common to see shorthand terms used to describe allosteric ligands, such as “ago-PAM” for compounds that display both agonistic effects and positive allosteric modulation. These terms can be problematic if used as stand-alone descriptors because they do not take the conditional nature of allosteric interactions into account. If a shorthand term is to be used, it is more useful when the modulator action (PAM, NAM, or NAL) of the ligand is emphasized first as well as the interacting orthosteric ligand and, ideally, the assay or pathway involved. Using this approach, for instance, one can say that LY2033298 is a PAM-agonist at M2 mAChR-mediated ERK1/2 phosphorylation when tested against oxotremorine-M, but a NAL-agonist when tested against acetylcholine (Valant et al., 2012b). For further details, readers are referred to the guide for the classification of receptor allostery and allosteric ligands that has recently been published by the nomenclature committee of the International Union of Basic and Clinical Pharmacology (Christopoulos et al., 2014).
Pharmacologic Characteristics of GPCR Allostery
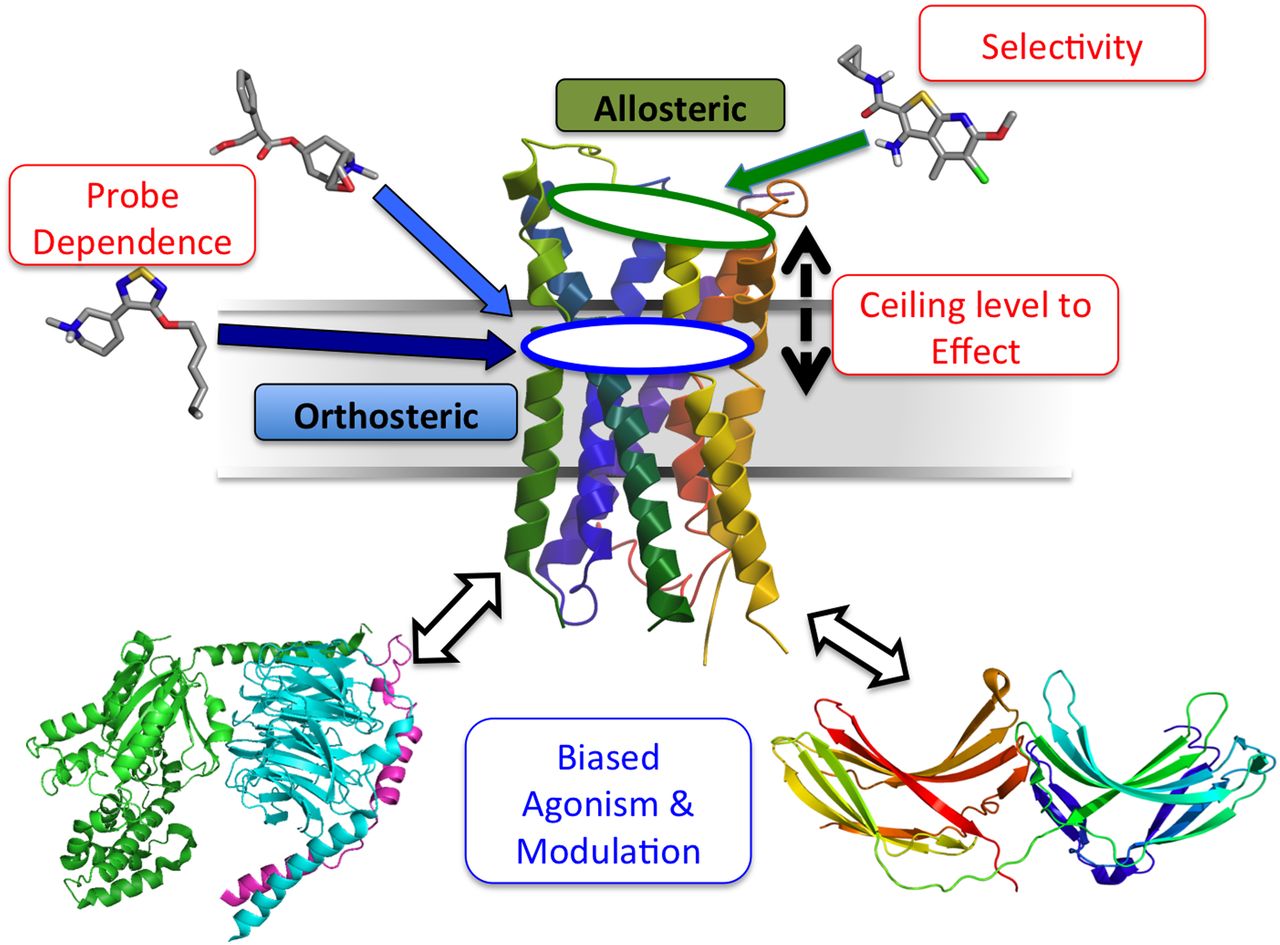
There are a number of key characteristics associated with the pharmacology of GPCR allosteric modulators, and these are summarized schematically in Fig. 2. The most obvious of these is the property of selectivity, which can be attained in two ways. First, unlike orthosteric sites, allosteric sites need not have evolved to accommodate an endogenous ligand (but see below) and thus can show greater divergence in their amino acid sequences between subtypes (and species). This is most pertinent to targeting GPCRs that show extremely high sequence similarity within the orthosteric pocket, such as the mAChRs (Conn et al., 2009; Kruse et al., 2014); despite substantial clinical potential for selectively targeting different mAChR subtypes in disorders such as Alzheimer’s disease, schizophrenia, and drug addiction (Kruse et al., 2014), clinical progression of orthosteric mAChR ligands has been hindered due to insufficient subtype selectivity (e.g., Bodick et al., 1997).

Pharmacologic characteristics of GPCR allostery.
The second mechanism by which selectivity can be obtained is not via targeting a less conserved allosteric site but via selective cooperativity with the orthosteric ligand, even if the allosteric site is shared between subtypes (Lazareno et al., 2004). As will be outlined here, the mAChRs are another good example of this. They all possess an extracellular vestibule that contains a so-called common allosteric site (Ellis and Seidenberg, 1992; Tränkle et al., 1998), but substantial selectivity is still attained in this region due to a combination of variability in amino acid sequence and pronounced differences in the magnitude of cooperativity between this site and the orthosteric site across subtypes. Indeed, the latter is the major mechanism by which allosteric compounds such as thiochrome and LY2033298 show high selectivity for the M4 mAChR over the other subtypes (Lazareno et al., 2004; Chan et al., 2008; Suratman et al., 2011).
A second key characteristic of GPCR allostery is that the allosteric effect approaches a ceiling, or limit, over and above which no further activity is observed, irrespective of the dose of the modulator. This is also referred to as saturability of effect, and it reflects the cooperativity between the orthosteric and allosteric sites, which determines the magnitude and direction of the allosteric effect. Depending on the GPCR type and its role in physiology/disease, markedly different ranges of saturability can be observed. For example, at the M2 mAChR, the NAM, gallamine, reduces the affinity of acetylcholine by approximately 200-fold before the ceiling is reached (Christopoulos, 2000), whereas the mGluR5 NAM VU0366249 [N-(3-chloro-4-fluorophenyl)-3-cyano-5-fluorobenzamide) maximally reduces glutamate signaling by a factor of only 3-fold (Gregory et al., 2012).
Similar effects can be observed for PAMs. For instance, LY2033298 and benzyl quinolone carboxylic acid (BQCA) maximally potentiate acetylcholine signaling by a factor of 100-fold or greater at the M4 and M1 mAChRs, respectively (Chan et al., 2008; Canals et al., 2012), and this magnitude of effect is likely required to see in vivo efficacy for these receptors (Ma et al., 2009; Shirey et al., 2009; Suratman et al., 2011). In contrast, the marketed PAM of the calcium-sensing receptor (CaSR) cinacalcet only potentiates CaSR signaling to the Gq/11-intracellular calcium mobilization by a factor of approximately 3 (Davey et al., 2012; Leach et al., 2013), yet this is clearly sufficient for in vivo efficacy in humans; the compound is successfully used to manage secondary hyperparathyroidism by normalizing levels of parathyroid hormone via potentiating the actions of serum calcium at the CaSR expressed in the parathyroid glands (Block et al., 2004). It is logical that certain neurotransmitter or hormone systems requiring tight regulation, such as glutamate, γ-amino butyric acid (GABA), and Ca++, equally require only subtle degrees of modulation; too high a cooperativity in such instances may actually contribute to on-target toxicity (Parmentier-Batteur et al., 2014).
The ceiling level of allosteric modulator effects is relevant both to the potential therapeutic advantages of allosteric modulators and to the design of screening assays. Specifically, the saturability of the allosteric effect means that such ligands have a greater potential to fine-tune physiologic responses in either a positive or negative direction, akin to the way a dimmer switch can control a lamp. Limited degrees of positive or negative cooperativity are thus associated with a lower risk of target-based overdose, as no further pharmacologic effect will be observed at saturating modulator concentrations over and above that determined by the cooperativity. This is not just a theoretical consideration: the best clinically validated example of allosteric medicines that exploit this property are the benzodiazepine PAMs of GABA at the GABAA ligand-gated ion channel, exemplified by compounds such as diazepam (Christopoulos et al., 2014). These molecules produce anxiolytic, sedative, anticonvulsant, and muscle relaxant effects by potentiating the central actions of GABA by only small degrees of positive cooperativity (e.g., 3-fold to 5-fold; Braestrup et al., 1979; Ehlert et al., 1982). This ceiling level to the benzodiazepine effect explains why they are relatively safe in overdose situations (when taken alone), and their greater selectivity arises dues to their targeting of an allosteric site at the interface of the receptor’s variable α/γ subunits (Christopoulos, 2002). In the absence of any direct allosteric agonism, pure PAMs and NAMs will also only act where and when endogenous tone is present, thus maintaining the spatiotemporal aspects of physiologic signaling. With regards to screening, however, limited degrees of positive or negative cooperativity may not be detected if the assay is not appropriately designed for small effect windows (Christopoulos, 2002), even though compounds with such properties could represent scaffolds with which structure-activity programs can be initiated.
A third unique characteristic of allosteric interactions is referred to as probe dependence (Kenakin, 2005), whereby the magnitude and direction of the allosteric effect mediated by the same modulator acting at the same receptor can vary depending on the orthosteric ligand that is used to probe receptor function. There are at least two (related) mechanisms that can contribute to the observation of probe dependence. The first is one of state dependence, whereby the modulator uniformly moves the equilibrium of receptor states either in more a positive (active) or a negative (inactive) direction. This is the simplest mechanism of allostery within a two-state receptor system (Monod et al., 1965; Canals et al., 2011) and predicts that the degree of modulation will track with the intrinsic efficacy of the interacting ligands; a PAM of orthosteric agonists will be a NAM of orthosteric inverse agonists, and vice versa. Furthermore, the higher the efficacy of the orthosteric agonist, the greater the PAM effect. Both of these effects have been observed at the M1 mAChR with the allosteric modulator BQCA, which shows PAM activity when tested against agonists but NAM activity when tested against inverse agonists (Canals et al., 2012). Figure 3 illustrates the positive cooperativity (αβ values) mediated by BQCA on cAMP signaling in the presence of various agonists; the high efficacy agonists acetylcholine and carbachol are potentiated to a greater extent than the low efficacy agonists pilocarpine and xanomeline. It is known, of course, that GPCRs can adopt more than two states in the presence of various ligands, but the above mechanism may still manifest if the modulator uniformly changes the abundance but not the quality/nature of the different microstates that govern receptor activity (Fig. 3, top). An overall change in the abundance of active microstates in one direction relative to inactive microstates would still appear, at a macroscopic level, as a two-state system (Abdul-Ridha et al., 2013).

Conformational mechanisms underlying probe dependence at GPCRs. GPCRs adopt multiple active and inactive states that can be represented schematically as a continuum of states that vary in abundance (black). At the macroscopic level these can be further averaged (red). Addition of ligand into this system changes the abundance of the states via conformational selection. If an allosteric modulator (e.g., BQCA at the M1 mAChR) generally enriches the abundance of active states at the expense of inactive states to a similar extent, then the system would behave as a simple two-state scheme macroscopically. Probe dependence would then be correlated with the intrinsic efficacy of the interacting ligands, as shown in the top panel where the high-efficacy agonists acetylcholine (ACh) and carbachol (CCh) are potentiated to a greater extent than the low-efficacy agonists pilocaprine and xanomeline. Cooperativity factors (αβ values) for M1 mAChR-mediated cAMP accumulation are calculated from data presented in Canals et al. (2012). However, if the allosteric modulator (e.g., LY2033298 at the M2 mAChR) nonuniformly changes the abundance across different active and inactive states, even at the macroscopic level clear differences would be observed in the apparent distribution of states, and the probe dependence will vary in a uniquely ligand- and pathway-dependent manner. This is shown in the bottom panel for the cooperativity between LY2033298 and various mAChR agonists in mediating ERK1/2 phosphorylation. Data replotted from Valant et al. (2012b).
In contrast, the other mechanism underlying probe dependence is also related to the fourth key characteristic associated with GPCR allostery: biased agonism and/or modulation. This refers to the ability of different ligands to preferentially stabilize a subset of functionally relevant GPCR conformations such that different signaling outputs are emphasized to the relative exclusion of others (Rajagopal et al., 2011; Stallaert et al., 2011; Kenakin and Christopoulos, 2013). Biased agonism can be observed directly with either orthosteric or allosteric agonists, but it can also be imposed on the signaling of an orthosteric agonist by an allosteric modulator. For instance, Fig. 4A shows the effects of the CaSR modulator cinacalcet on Ca++-mediated mobilization of intracellular calcium and ERK1/2 phosphorylation. Although cinacalcet is clearly a PAM for intracellular calcium mobilization, it is a NAL at the ERK1/2 pathway (Davey et al., 2012; Leach et al., 2013). Similar pathway-dependent changes in that nature of the modulatory effect for a given pair of allosteric/orthosteric ligands have been observed at other GPCRs, including the NK2 (Maillet et al., 2007), A1 (Valant et al., 2010, 2012a), M1 (Marlo et al., 2009), M2 (Valant et al., 2012b), M3 (Stewart et al., 2010), CRTH2 (Mathiesen et al., 2005), mGluR5 (Zhang et al., 2005; Bradley et al., 2011), PGF2α (Goupil et al., 2010), and CB1 (Ahn et al., 2012) receptors. A particularly striking example of probe dependence that is indicative of biased modulation has been observed with LY2033298 at the M2 mAChR (Valant et al., 2012b). As shown in Fig. 3, this modulator is a PAM to various extents for some agonists at mediating ERK1/2-phosphorylation, but a NAM of other agonists signaling to the same pathway. This suggests that LY2033298 is enriching some and/or deemphasizing other microstates in a manner that changes both the quality and the abundance of the states “seen” by the orthosteric agonist (Fig. 3, bottom).

Biased allosteric modulation. (A) Cinacalcet, the allosteric modulator of the CaSR, is a PAM of Ca2+-mediated mobilization of intracellular calcium but a NAL of Ca2+-mediated ERK1/2 phosphorylation in the same cell background. Data replotted from Leach et al. (2013). (B) “Cooperativity bias plots” showing the change in Ca2+ potency (ΔpEC50) for signaling to intracellular calcium mobilization or ERK1/2 phosphorylation in the presence of increasing concentrations of the PAM cinacalcet or NAM NPS2143 at the CaSR. If the cooperativity was the same between pathways, the plots would fall on the line of identity (dotted line). It can be seen, however, that at the wild-type CaSR (solid black line), the modulation is biased toward calcium mobilization (especially with the PAM). Interestingly, the indicated mutations (in the CaSR transmembrane domain) are all naturally occurring and can themselves change the bias of the receptor. Data replotted from Leach et al. (2013).
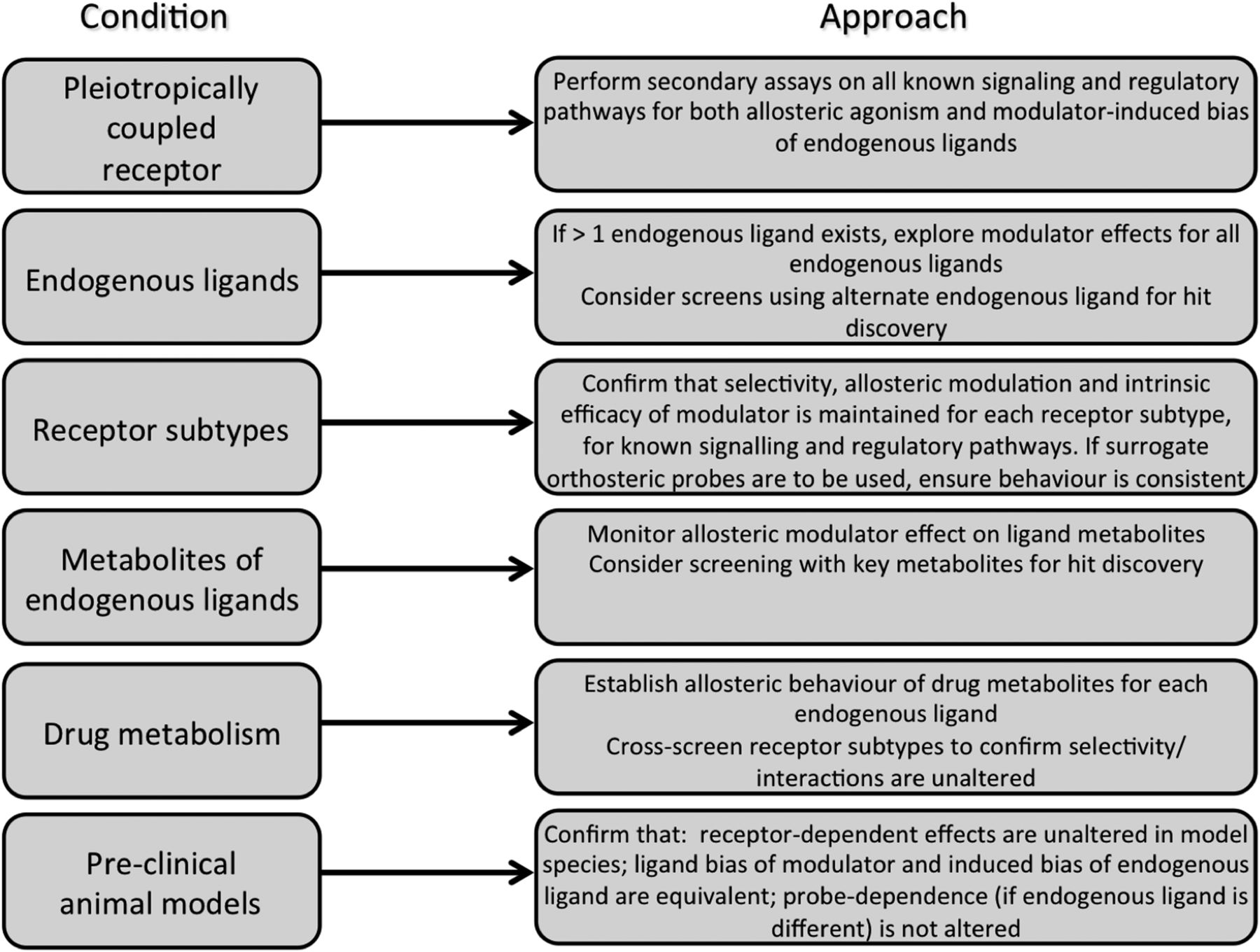
Probe dependence and biased modulation have substantial implications for drug discovery and for understanding GPCR allosteric ligand biology in general. For example, many GPCRs possess multiple endogenous agonists, and this requires screening of putative allosteric compounds against all of them (Koole et al., 2010; Keov et al., 2011; Wootten et al., 2013). Allosteric sites may show species variability, which requires counterscreening against receptors from species that will subsequently be used for in vivo efficacy studies. Even if the allosteric site is relatively conserved between species, differences may still arise from changes in the cooperativity/probe dependence between species (Suratman et al., 2011). If surrogate agonists need to be used in allosteric screening—resulting from metabolic instability of the endogenous agonist or accompanying the study of orphan GPCRs—the observed activity may not be retained when tested against the cognate agonist. Similar considerations apply for the potential of combination therapies involving a given orthosteric agonist-allosteric modulator pair. For single-agonist receptors, it is still possible that the metabolites of that agonist may be prone to different types of modulation. Indeed, at the M2 mAChR, the acetylcholine metabolite choline is potentiated by LY2033298 to a far greater extent than acetylcholine itself. Similarly, the otherwise inactive glucagon-like peptide-1 (GLP1) metabolite GLP1(9–36)NH2 is markedly potentiated by the modulator BETP [4-(3-benzyloxyphenyl)-2-ethylsulfinyl-6-(trifluoromethyl)pyrimidine] at the GLP1 receptor compared with the modest effect of the same modulator on the endogenous agonist GLP1(7–36)NH2 (Wootten et al., 2012). Figure 5 summarizes some key considerations when developing screening strategies for allosteric GPCR drug development.

Strategies for enhancing preclinical translation of allosteric modulators. The left column highlights potential issues that arise during drug screening/development, and the right describes potential approaches for overcoming these issues. Adapted from Wootten et al. (2013).
A major consequence of biased agonism/modulation is the potential to recruit pathways that are linked to a desired therapeutic outcome while not recruiting or even blocking those linked to adverse effects; there are now some biased agonists in clinical development (Soergel et al., 2014; Violin et al., 2014). However, as indicated in the introduction, the link between cellular signaling and pathophysiology is not well established in many cases, and thus allosteric drug candidates need to be profiled broadly as well as against the endogenous orthosteric agonist(s), where possible. Biased allosteric modulation may also play a direct role in disease. For instance, a patient with acquired hypocalciuric hypercalcemia was identified as possessing an allosteric autoantibody that enhances Gq/11-mediated phosphoinositide accumulation but allosterically inhibits Gi/o-mediated ERK1/2 phosphorylation via the CaSR (Makita et al., 2007); the bacterium Neisseria meningitidis possess filamentous structures, known as pili, that act as biased allosteric agonists of the β2 adrenergic receptor to facilitate meningeal colonization (Coureuil et al., 2010). Even where an allosteric modulator has been identified as a possible drug candidate, its properties may change due to naturally occurring mutations, which may themselves be associated with biased signaling. As shown in Fig. 4B, the ability of the PAM cinacalcet or the NAM NPS2143 (2-chloro-6-[(2R)-3-[[1,1-dimethyl-2-(2-naphthalenyl)ethyl]amino-2-hydroxypropoxy]benzonitrile hydrochloride) to modulate intracellular calcium mobilization or ERK1/2 phosphorylation mediated by Ca++ at the CaSR can change as a consequence of naturally occurring CaSR mutations (Leach et al., 2013).
In addition to GPCRs, there are now numerous allosteric ligands either marketed or in different stages of preclinical or clinical development for therapeutic targets such as ion channels, kinases, caspases, and phospholipases (Christopoulos et al., 2014; Wenthur et al., 2014). This highlights the fact that the paradigm is very broad and has gained substantial traction in modern drug discovery. A working knowledge of the key pharmacologic characteristics of allostery outlined here can thus prove very useful in assisting preclinical workflow decisions during the progression of allosteric drug candidates.
Enriching Structure-Activity Studies of GPCR Allostery and Bias
Given the marked interest in allosteric modulators and biased agonists as an avenue for drug discovery, the attendant growth in medicinal chemistry programs in this arena, and the introduction of new high-throughput methodologies for interrogating cellular responses, it has become apparent that analytical approaches are required for guiding chemical biology and preclinical drug discovery programs focused on allostery and bias. This is particularly so with regards to enriching structure-activity relationships (SAR) in a manner that can inform the design of probe compounds and, ideally, even be incorporated into drug candidate selection matrices. At a minimum, allosteric modulator SAR needs to differentiate modifications on modulator affinity from the cooperativity with the orthosteric ligand, as the two properties are not correlated. Furthermore, the increasing prevalence of direct allosteric and biased agonism must be addressed.
To meet these challenges, analytical models based on the classic operational model of agonism of Black and Leff (1983) have been recently developed (Ehlert, 2005; Kenakin, 2005, 2012; Price et al., 2005; Leach et al., 2007; Figueroa et al., 2009; Koole et al., 2010; Evans et al., 2011; Rajagopal et al., 2011). The operational model treats the link between an agonist-occupied receptor and its entire signal transduction chain as a virtual Michaelis-Menten system (Kenakin and Christopoulos, 2013). Within this model, the relevant parameters (Fig. 6) are the orthosteric and allosteric ligand affinities for the free receptor (quantified as equilibrium dissociation constants KA and KB, respectively), the coupling efficiency of each ligand with regards to the signal transduction chain (quantified by the equilibrium coupling dissociation constants KE(A) and KE(B), respectively), the allosteric effect that each ligand exerts on the other ligand’s equilibrium binding affinity (quantified by the cooperativity factor α), and the effect of the allosteric modulator on the orthosteric ligand’s signaling efficacy (quantified by the scaling factor β). The ratio of receptor density ([R]T) to the coupling efficiency (KE) is defined as τ, and it is an overall measure of the efficacy that a given agonist (orthosteric or allosteric) possesses for the signaling pathway under investigation.
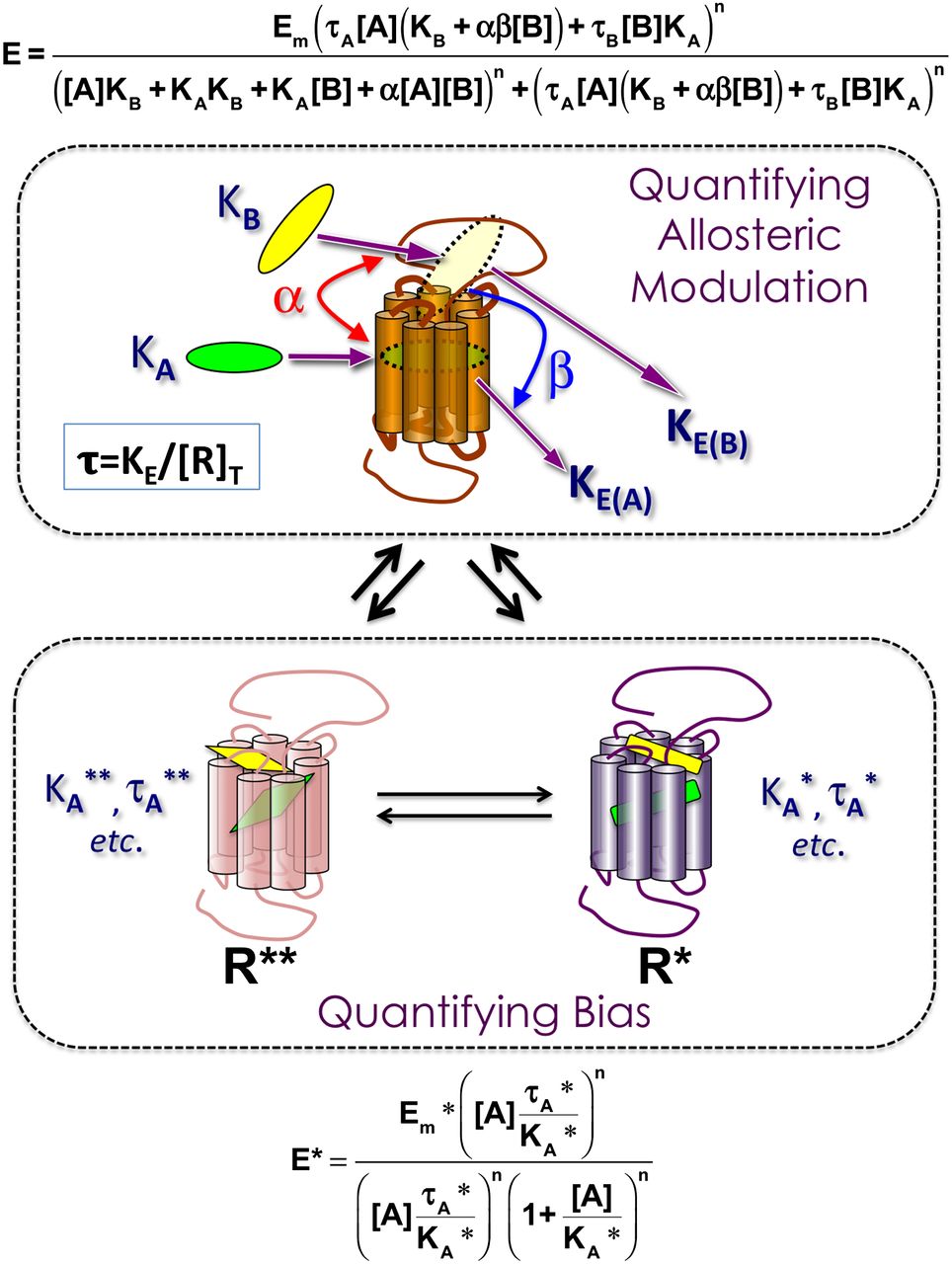
Operational parameters for quantifying allosteric modulation and biased agonism at GPCRs. Top: Allosteric effects can be described operationally in terms of orthosteric (KA) and allosteric (KB) ligand equilibrium dissociation constants, the coupling efficiency of each ligand with regards to a given signaling pathway (KE(A) and KE(B), respectively), the allosteric effect that each ligand exerts on the other ligand’s equilibrium binding affinity (cooperativity factor α), and the effect of the allosteric modulator on the orthosteric ligand’s signaling efficacy (scaling factor β). Values of αβ > 1 yield positive allosteric modulation, values of 0 < αβ < 1 yield negative modulation, whereas αβ = 1 defines a neutral allosteric ligand. The ratio of receptor density ([R]T) to coupling efficiency (KE) is defined as τ, and is an overall measure of agonist efficacy. Bottom: Biased agonism is characterized by different affinities and/or different efficacies of a given agonist for each active state (e.g., R*, R**), hence different KA and different KE (different τ) values. Also shown are the equations describing both phenomena that can be fitted to experimental data. Note that Em is the maximum effect of the system and n is the transducer slope for the function linking agonist-occupied receptor to the measured response.
The application of this model to quantify direct agonist bias is even simpler, and only requires the additional assumption that biased agonism is characterized by different affinities and/or different efficacies of a given agonist for each active state (R*, R**, etc.; Fig. 6, bottom), hence different KA and different KE (different τ) values. Although the individual values of these parameters are not easily obtainable from simple concentration-response data, the ratio of the two (τ/KA) can be obtained as a single parameter by fitting of the operational model to a family of agonist curves at a given pathway (Kenakin et al., 2012; Kenakin and Christopoulos, 2013) and forms the quantitative basis for calculations of agonist bias (Shonberg et al., 2014; van der Westhuizen et al., 2014). With regards to biased allosteric modulation, this would manifest as pathway-dependent differences in estimated modulator affinity (KB) or cooperativity (αβ), provided that the latter is not tracking with orthosteric agonist intrinsic efficacy or stimulus-response coupling (Davey et al., 2012; Langmead and Christopoulos, 2014).
An advantage of the operational model as applied to SAR is that it can almost always be fitted to experimentally derived data to provide estimates of some, or all, of its parameters with regards both to allosteric modulators (Aurelio et al., 2009; Gregory et al., 2012; Valant et al., 2012a; Huynh et al., 2013; Mistry et al., 2013) and biased agonists (Tschammer et al., 2011b; Shonberg et al., 2013; Szabo et al., 2014). Table 1 illustrates an example of allosteric modulator SAR determined through analysis of the effects of various thienopyridine modulators on acetylcholine-mediated ERK1/2 phosphorylation at the M4 mAChR. Four measures of compound activity are indicated. The first, EC50, is an empirical potency derived from a simple titration of test compound against a fixed concentration (and hence, effect) of orthosteric agonist. The other three parameters, KB, αβ, and τB, are derived from operational model analysis of the entire acetylcholine concentration-response relationship in the presence of increasing modulator concentrations (Huynh et al., 2013).
Empirical potency (EC50) versus operational model parameters describing the functional affinity (KB), global cooperativity (αβ), and direct agonism (τB) of thienopyridine allosteric ligands for modulating acetylcholine-mediated ERK1/2 phosphorylation at the M4 mAChR
Adapted from Huynh et al. (2013).
The advantage of the titration method, which represents the most common screening paradigm, is that it is simpler, uses fewer points, and can thus be applied to a very large number of compounds. The limitation, however, is that the derived potency from this method is an amalgam of all the operational parameters governing the observed effects (i.e., modulator and agonist affinities, efficacies, and cooperativities). As seen from Table 1, an inspection of the EC50 values shows very little apparent effect of the substitutions in this chemical series. Indeed, a common observation in the field of allosteric modulator chemistry is that the SAR is often “flat” (Melancon et al., 2012).
The operational model analysis, however, paints a different picture: modulator affinity and cooperativity can change in different directions with the same chemical substitution (compare KB and αβ values for compounds 1a and 1b), thus accounting for the apparent lack of effect on overall potency. Indeed, a more “enriched” conclusion from the operational analysis suggests that modification of this particular scaffold will have minimal impact on KB (the affinity of the modulator for the allosteric pocket on the free receptor) but substantial effects on the cooperativity (αβ). Thus, it is the latter parameter that can be chemically dialed up or down in this series, not the former. Determination of operational efficacies (τB) also allows one to look for correlations between the ability of the modulator to activate the receptor and its ability to modulate the orthosteric agonist (αβ), which appears to be the case in this instance (Huynh et al., 2013).
Such applications of operational modeling to allosteric (and biased agonist) SAR have the potential to link chemistry to key biologic parameters and thus facilitate the iterative generation of chemical probes for hypothesis testing and for a more informed pursuit of a therapeutic leads. One can use this to address questions around how much (and what type of) cooperativity, bias, and allosteric agonism are required to achieve in vivo efficacy and/or avoid on-target side effects. Similar analytical methods can be applied to mutational analysis of receptor allostery and bias to enrich structure-function analyses, in addition to SAR (Gregory et al., 2010; Nawaratne et al., 2010; Tschammer et al., 2011a; Koole et al., 2012; Leach et al., 2012, 2013; Valant et al., 2012b; Abdul-Ridha et al., 2014).
Bitopic GPCR Ligands
A more recent breakthrough in chemical biology studies of GPCR allostery has been the discovery, rational design, and exploitation of “bitopic” or “dualsteric” ligands, namely, hybrid bifunctional molecules that are composed of two pharmacophores, each known to independently interact with an orthosteric and allosteric site (Disingrini et al., 2006; Steinfeld et al., 2007; Valant et al., 2008, 2009, 2012c, 2014; Antony et al., 2009; Mohr et al., 2010, 2013; Narlawar et al., 2010; Bock et al., 2012; Lane et al., 2013). Some examples of rationally designed bitopic ligands are shown in Fig. 7A. All such molecules should be viewed as a special case of the “bivalent” ligand, which is composed of two distinct pharmacophores but whose sites of interaction (orthosteric or allosteric) are not explicitly defined (Valant et al., 2012c; Lane et al., 2013).

Bitopic GPCR ligands and biased agonism. (A) Examples of rationally designed bitopic ligands for the M2 mAChR (THRX-198321 [biphenyl-2-yl-carbamic acid 1-{9-[(R)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-piperidin-4-yl ester], Hybrid 2) and the adenosine A1 receptor (LUF6258 [N6-[2-amino-3-(3,4-dichlorobenzoyl)-4,5,6,7-tetrahydrothieno[2,3-c]pyridin-6-yl-9-nonyloxy-4-phenyl]adenosine], VCP746). (B) The bitopic agonist, VCP746, is biased toward native A1 receptor-mediated cytoprotection in rat cardiomyoblast cells (as measured by reduction in cell death due to simulated ischemia, SI), and away from A1-mediated bradycardia, in contrast to the canonical orthosteric agonist, CPA. Data replotted from Valant et al. (2014).
The advantages in the pursuit of bitopic ligands include the potential for greater receptor selectivity by virtue of targeting an allosteric site and greater affinity due to concomitant engagement with the orthosteric site. This was most strikingly observed when the negative allosteric modulator 4-aminobenzylpiperidine was linked to the orthosteric antagonist 3-benzhydryl pyrrolidine to yield the bitopic M2 mAChR-selective antagonist THRX-160209 [4-{N-[7-(3-(S)-(1-carbamoyl-1,1-diphenylmethyl)pyrrolidin-1-yl)hept-1-yl]-N-(n-propyl)amino}-1-(2,6-dimethoxybenzyl)piperidine)] (Steinfeld et al., 2007). This form of selectivity is also particularly pertinent to situations where pure allosteric modulators may lose effectiveness, such as in the context of neurodegenerative disease where endogenous tone is progressively lost while the receptor target remains functional. Another major advantage of bitopic ligands is the potential to engender biased agonism (Antony et al., 2009; Kebig et al., 2009; Valant et al., 2014) because concomitant binding to two different sites may promote unique receptor conformations. A recent successful example of this approach is shown in Fig. 7B, where the orthosteric agonist adenosine was linked to the A1 receptor-selective PAM VCP171 [2-amino-4-(3-(trifluoromethyl)phenyl)thiophen-3-yl)(phenyl)methanone] to yield the A1 biased agonist VCP746 [4-(5-amino-4-benzoyl-3-(3-(trifluoromethyl)phenyl)thiophen-2-yl)-N-(6-(9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-ylamino)hexyl)benzamide] (Fig. 7A). Importantly, this bias was (patho)physiologically relevant because VCP746 promoted cytoprotection without any significant effects on heart rate; the latter is an on-target side effect that has previously limited the pursuit of adenosine A1 agonists as cardioprotective agents in the clinic (Valant et al., 2014).
There are also design challenges and potential downsides to bitopic ligands. For example, mismatches between the nature of the orthosteric and allosteric pharmacophores (e.g., an agonist linked to a NAM) may not yield improved affinity, although selectivity can still be achieved (Mohr et al., 2010, 2013). Importantly, the linker between the two pharmacophores may itself have a profound effect on the potency or pharmacology of the designed bitopic ligand (Valant et al., 2012c; Lane et al., 2013): an inappropriate linker can fail to orient the two moieties toward their respective binding sites and thus not yield the expected affinity gains. Another consequence of large linkers (and multivalent compounds in general) is an increase in the size of the molecule and attendant decrease in its “druggability”: bitopic ligands can make very useful chemical probes, but some questions remain about whether they can yield true drug candidates (but see the discussion later). Finally, the actual validation of a bitopic mode of action can prove quite difficult and requires a number of complementary approaches (Lane et al., 2013). No GPCR crystal structures have been published to date with bitopic ligands bound, so the conclusion that a given ligand is truly bitopic remains largely inferential.
Despite the aforementioned caveats, the discovery of bitopic ligands has led to a reevaluation of the mechanism of action of other functionally selective ligands. Specifically, it is possible that some existing molecules may achieve their selectivity via a hitherto unappreciated bitopic mechanism. This was first demonstrated by reverse engineering of the M2 mAChR partial and biased agonist McN-A-343 [4-(m-chlorophenylcarbamoyloxy)-2-butynyl)trimethylammonium chloride] into two distinct pharmacophores: tetramethylammonium and 3-chlorophenylcarbamate, the former a high efficacy orthosteric agonist and the latter a negative allosteric modulator; combination of the two recapitulated the pharmacology of the parent molecule (Valant et al., 2008). There are two important consequences of this finding. The first is that it has led to a re-examination of compounds previously classified as “allosteric agonists”, such as AC-42 [(4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine hydrogen chloride)], 77-LH-28-1 [1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone)], and TBPB [(1-[1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one)] at the M1 mAChR (Spalding et al., 2002; Langmead and Christopoulos, 2006; Langmead et al., 2008; Jacobson et al., 2010), and resulted in the conclusion that they are actually bitopic agonists (Avlani et al., 2010; Gregory et al., 2010; Keov et al., 2013). The second is to highlight the fact that bitopic ligands can indeed display drug-like characteristics. AC-42, TBPB, and related compounds are small molecules arising out of drug discovery programs. If the orthosteric and allosteric sites are in close enough apposition, it should be possible to bridge them without the need for very large linkers. It would thus be very interesting to see if known drugs that have the potential to adopt extended binding poses, such as salmeterol at the β2 adrenergic receptor or aripiprazole at the dopamine D2 receptor, engage their targets in a bitopic manner to display biased signaling.
Finally, it should be noted that virtually all studies of bitopic ligands to date have focused on targeting monomeric GPCRs with such molecules. Under this condition, a successful bitopic mode of engagement should involve a single ligand occupying both orthosteric and allosteric sites at the same time on the (single) GPCR, and thus would still exhibit competitive behavior (because one of the pharmacophores occupies the orthosteric site)—essentially behaving like a more selective orthosteric ligand. Only at very high (supraphysiologic) concentrations and/or in the presence of a different (smaller) orthosteric ligand, it might be possible for the bitopic ligand to adopt a second, lower-affinity conformation via the allosteric site to exhibit noncompetitive behavior. It is of interest, therefore, to consider how this behavior could change in the context of GPCR dimers or oligomers, which remains an ongoing area of research and debate in the field (Smith and Milligan, 2010; Ferre et al., 2014).
A very recent study of the dopamine D2 receptor has revealed that the “pure” drug-like allosteric modulator SB269652 [trans-1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide] (Silvano et al., 2010) appears to mediate its allostery by binding to one D2 receptor protomer to allosterically modulate the binding of dopamine at an associated protomer (Lane et al., 2014). What distinguishes the molecule’s behavior from other prior studies of cooperativity across dimers is the specific requirement for SB269652 to engage both the orthosteric site and a secondary pocket on the first D2 receptor protomer to transmit its allosteric effect to the second protomer (Fig. 8). The synthesis of the cis isomer of this compound (MIPS1217, cis-1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide), which cannot access the secondary pocket, converts the molecule from allosteric to competitive (Fig. 8). Mutational impairment of the secondary pocket, or preventing protomer-protomer crosstalk at the D2 receptor, has the same effect (Lane et al., 2014). This example highlights a means by which a bitopic ligand can behave either allosterically or competitively, depending on the receptor context (dimer versus monomer), and illustrates how the study of such molecules continues to yield new insights into GPCR functionality.
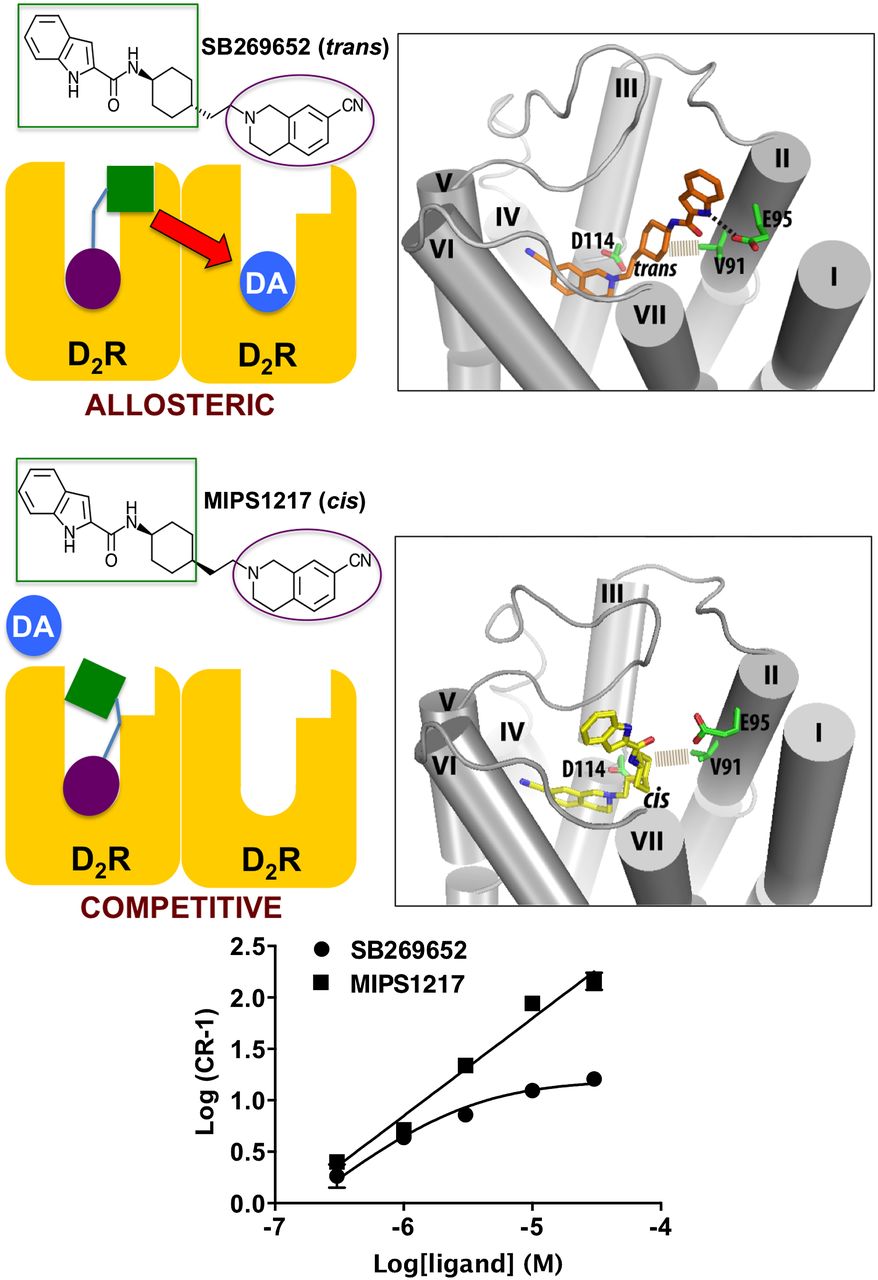
Allostery mediated by the bitopic ligand SB269652 across a GPCR dimer. The trans isomer SB269652 binds in a bitopic pose on one D2 receptor protomer to act as a negative allosteric modulator of dopamine (DA) binding and signaling via a second protomer. This negative allosteric effect manifests as a curvilinear Schild regression (for the ERK1/2 phosphorylation pathway), shown at the bottom, and requires association of the indole moiety of the molecule with a secondary pocket near the top of TM domains 2 and 7. Key residues for engagement with the orthosteric site (D114) and secondary site (E95 and V91) are indicated in the molecular model. In contrast, the indole moiety of the cis isomer MIPS1217 cannot effectively engage the secondary pocket to transmit any allostery. Instead, the behavior of the entire molecule is consistent with simple competition. Data replotted from Lane et al. (2014).
Endogenous Allosteric Modulators
GPCRs are natural allosteric proteins, as their mode of activation requires the transmission of extracellular stimuli to intracellular transducers. The best-known endogenous allosteric modulator of these receptors, therefore, is the G protein itself; classic thermodynamic principles of allosteric reciprocity have been well documented for the interaction between GPCR ligands and G proteins (De Lean et al., 1980; Ehlert, 1985). Nonetheless, a common question remains as to the existence of other, more receptor-specific allosteric molecules that may be expressed endogenously. On the one hand, as outlined earlier, it seems reasonable that the allosteric sites commonly targeted by exogenous small molecules or proteins represent serendipitous sites because they have not evolved to accommodate an endogenous ligand. They may serve other roles in the receptor, for instance, as a consequence of folding, trafficking, expression, or signal transduction. On the other hand, it is possible that some of these instances reflect the existence of “orphan” allosteric sites that have not been fully appreciated. To date, this issue remains largely unaddressed, probably because the focus of the field thus far has been largely on drug discovery. However, this area is likely to become very important in the coming years, as more research is directed to addressing key aspects of endogenous allosteric modulators in physiology and in disease.
An obvious physiologic extension of the allostery paradigm is with respect to the interacting proteins other than the G protein that can modulate GPCR biology. This is a very broad research area in its own right and beyond the scope of the current review, but it is worthwhile to highlight a few key examples as illustrative of the concept. An important protein family in this regard are the β-arrestins, originally identified as scaffolding proteins involved in signal termination but since shown to mediate noncanonical signaling pathways in their own right (Reiter et al., 2012). Early work by Gurevich et al. (1997) found that a β-arrestin bound receptor showed a markedly higher affinity for agonists than the nonoccupied receptor, which is in accordance with the expectations of a ternary complex model. A more recent study using double-exponential fluorescent lifetime decay analysis showed distinct ghrelin receptor conformational changes upon addition of β-arrestin (or Gq protein) in reconstituted lipid nanodiscs (Mary et al., 2012). The receptor activity modifying proteins (RAMPs) are another striking example of a single-transmembrane-spanning protein family that can associate with certain GPCRs (predominantly class B receptors) to create new receptor phenotypes or to bias signaling (McLatchie et al., 1998; Christopoulos et al., 1999, 2003; Hay et al., 2006). Allosteric communication between GPCRs in dimeric or oligomeric arrays, already mentioned earlier, remains an area of much current research interest (Kenakin and Miller, 2010; Ferre et al., 2014).
Numerous amino acids and peptides have also been proposed as putative endogenous allosteric GPCR modulators. For example, aromatic amino acids, such as l-Phe and l-Trp, are well-validated allosteric modulators of signaling via the CaSR (Conigrave et al., 2000; Busque et al., 2005; Conigrave and Hampson, 2010; Liou et al., 2011), and a role for l-Leu as an endogenous modulator of the GABAB receptor has also been proposed (Kerr and Ong, 2003). Another endogenous allosteric modulator of the CaSR is the γ-glutamyl-peptide glutathione (Broadhead et al., 2011), whereas the tetrapeptide “5HT-moduline” (Leu-Ser-Ala-Leu) has been proposed as an endogenous modulator of the 5HT1B receptor in the central nervous system (Fillion et al., 1996; Massot et al., 1996; Rousselle et al., 1996). More recently, studies in mouse and hamster brain tissues identified a family of peptides, known as the pepcans, that have been proposed as endogenous allosteric modulators of the cannabinoid CB1 receptor (Bauer et al., 2012).
The role of various ions in regulating GPCR binding and activation has been known for decades. Zinc has been implicated as an allosteric modulator at least of dopamine (D1A, D2L, D4) (Schetz and Sibley, 1997; Schetz and Sibley, 2001), MC1 and MC4 (Holst et al., 2002), and α1A and β2 adrenergic (Swaminath et al., 2002; Swaminath et al., 2003; Ciolek et al., 2011). The role of sodium as a GPCR modulator has been even more studied, with numerous investigations linking this ion to the ability of GPCRs, particularly class A GPCRs, to transition between active and inactive states (Pert et al., 1973; Simon and Groth, 1975; Christopoulos and Kenakin, 2002; Katritch et al., 2014). Structural studies have begun to shed light on the atomistic basis of this effect. For example, the 1.8-Å resolution crystal structure of the adenosine A2A receptor bound to an antagonist identified three clusters of ordered water molecules, one of which harbored a sodium bound to a highly conserved aspartate residue in the second transmembrane domain (TM2), which had previously been implicated in sodium modulation of GPCRs (Liu et al., 2012). The more recent solution of the δ opioid receptor crystal structure revealed a key sodium-binding site that not only was involved in modulating the transition between inactive to active states but also was involved in biased signaling to the β-arrestin pathway (Fenalti et al., 2014). Other key ions implicated in allosteric control of GPCRs include magnesium (Pasternak et al., 1975; Williams et al., 1978; Rodriguez et al., 1992) and calcium (Galvez et al., 2000).
Another very topical area is the role of endogenous lipids in modulating GPCR functionality. Cholesterol is a very important constituent of membrane lipid rafts, and the affinities of ligands to GPCRs such as the cholecystokinin subtype 1 (CCK1) receptor, oxytocin receptor, mGluR1a and 5HT1A receptors have all been shown to be highly dependent on the cholesterol content of their environment (Gimpl et al., 1997; Eroglu et al., 2003; Prasad et al., 2009; Potter et al., 2012). Crystal structures have revealed distinct cholesterol binding sites in the β2 adrenergic receptor, adenosine A2A receptor, and μ opioid (Cherezov et al., 2007; Hanson et al., 2008; Liu et al., 2012). The amidated lipid oleamide was proposed as a modulator of multiple 5HT receptor subtypes (Thomas et al., 1997, 1998; Hedlund et al., 1999), and progesterone has been linked to modulation of the oxytocin receptor (Grazzini et al., 1998). The cannabinoid CB1 receptor has been the focus on a number of studies identifying putative endogenous modulators, including oleamide (Leggett et al., 2004), lipoxin A4 (Pamplona et al., 2012), and pregnenolone (Vallee et al., 2014). It should be noted, however, that many of these studies have yet to be replicated or confirmed by multiple laboratories, so further research into the exact nature of endogenous lipid allosteric modulators of GPCRs is required.
Finally, a generally unappreciated but important area of inquiry relates to the potential of endogenous allosteric modulators in disease. As mentioned earlier in the example of bias for the CaSR (Makita et al., 2007), autoantibodies can act allosterically to mediate pathophysiology. Other examples where this has been seen include Chagas disease, a parasitic infection by Trypanosoma cruzi that leads to the generation of allosteric antibodies targeting β1 adrenergic and M2 mAChRs in the heart (Leiros et al., 1997; Sterin-Borda et al., 1999; Hernandez et al., 2003), and Sjogren syndrome, an autoimmune disorder associated with dysfunction of secretory glands that has been proposed to be mediated, at least in part, by autoantibodies against various mAChR subtypes (Borda et al., 1996). Interestingly, the polycationic human eosinophil major basic protein (MBP), a peptide that constitutes ∼50% of the inflammatory peptides released after infiltration and degranulation of eosinophils (Verbout et al., 2007), has been shown to specifically bind to the M2 mAChR, potentially in an allosteric fashion (Jacoby et al., 1993). It is well established that antigen-challenged animal models and humans with asthma are characterized by a hyperresponsiveness of the vagal nerves, leading to hyperreactivity and bronchoconstriction. Concomitantly, degranulation of eosinophils accumulated within the airway wall releases a variety of inflammatory peptides, including major basic protein (MBP). This finding suggests an unappreciated mechanism by which inflammatory mediators may contribute to pathophysiology—that is, by targeting allosteric sites on GPCRs. Indeed, the impact of the inflammatory milieu on GPCR regulation remains an area that warrants further investigation and may prove a rich source of novel allosteric biology.
Understanding the Structural Basis of GPCR Allostery
Given the increasing prevalence of allosteric modulators for GPCRs, recent studies have turned toward a deeper understanding of the molecular mechanisms underlying allostery. The simplest mechanism to account for allosteric behavior is the classic Monod-Wyman-Changeux model (Monod et al., 1965), which postulates that receptors exist in an equilibrium between different states that are preferentially stabilized by orthosteric or allosteric ligands binding to spatially distinct sites (Canals et al., 2011). The observed allosteric interaction thus reflects how one ligand “senses” the receptor conformation that has been selected by the other. The simplest expression of this model is restricted to two states, and its predicted macroscopic behaviors were discussed earlier (Fig. 3). Extension of this model to multiple active states accommodates ligand bias, but there remains ongoing debate as to the minimal molecular unit that determines the ultimate biologic manifestation of allostery and bias: the ligand-receptor complex or the ligand-receptor-transducer complex (Onaran and Costa, 2012). Recent breakthroughs in GPCR structural and computational biology are beginning to provide some insights into these questions.
Undoubtedly, the crystal structure solution of the “classic” ternary complex of an activated GPCR (β2 adrenergic receptor) bound to an agonist and nucleotide free G protein (Gs) was a landmark for the field that provided the first molecular snapshot of a GPCR as an allosteric machine (Rasmussen et al., 2011). Complementary studies focusing on the agonist-receptor interaction in the absence or presence of either G protein or G protein-mimicking nanobodies have also highlighted the vital role that the transducer plays in stabilizing the active state; a binary complex composed of a very high efficacy agonist and receptor alone is insufficient to maintain a fully active state (Rosenbaum et al., 2011). Other structural complexes that have provided molecular insights into allosteric mechanisms at GPCRs include the previously mentioned studies on sodium ion binding as well as the first GPCR structures bound to small molecules with a presumed or validated allosteric pharmacology. These include the class A chemokine CCR5 receptor [chemokine (C-C motif) receptor 5] bound to maraviroc (Tan et al., 2013), the TM-spanning region of the class B CRF1 receptor [corticotropin-releasing factor receptor 1] bound to CP-376395 [N-(1-ethylpropyl)-3,6-dimethyl-2-(2,4,6-trimethylphenoxy)-4-pyridinamine hydrochloride] (Hollenstein et al., 2013), and the TM-spanning region of the class C metabotropic glutamate mGluR1 receptor bound to FITM [4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazol-2-yl)-N-methylbenzamide] (Wu et al., 2014). The recent solution of the smoothened receptor crystal structures, each bound to a different ligand, revealed a long narrow cavity in the TM domain that can comprise multiple binding sites (Wang et al., 2014). In addition to providing new insights into prior pharmacologic studies of this receptor that suggested a potential for allosteric modulation (Rominger et al., 2009), it is possible that the long smoothened receptor TM cavity can be actively exploited to design bitopic ligands.
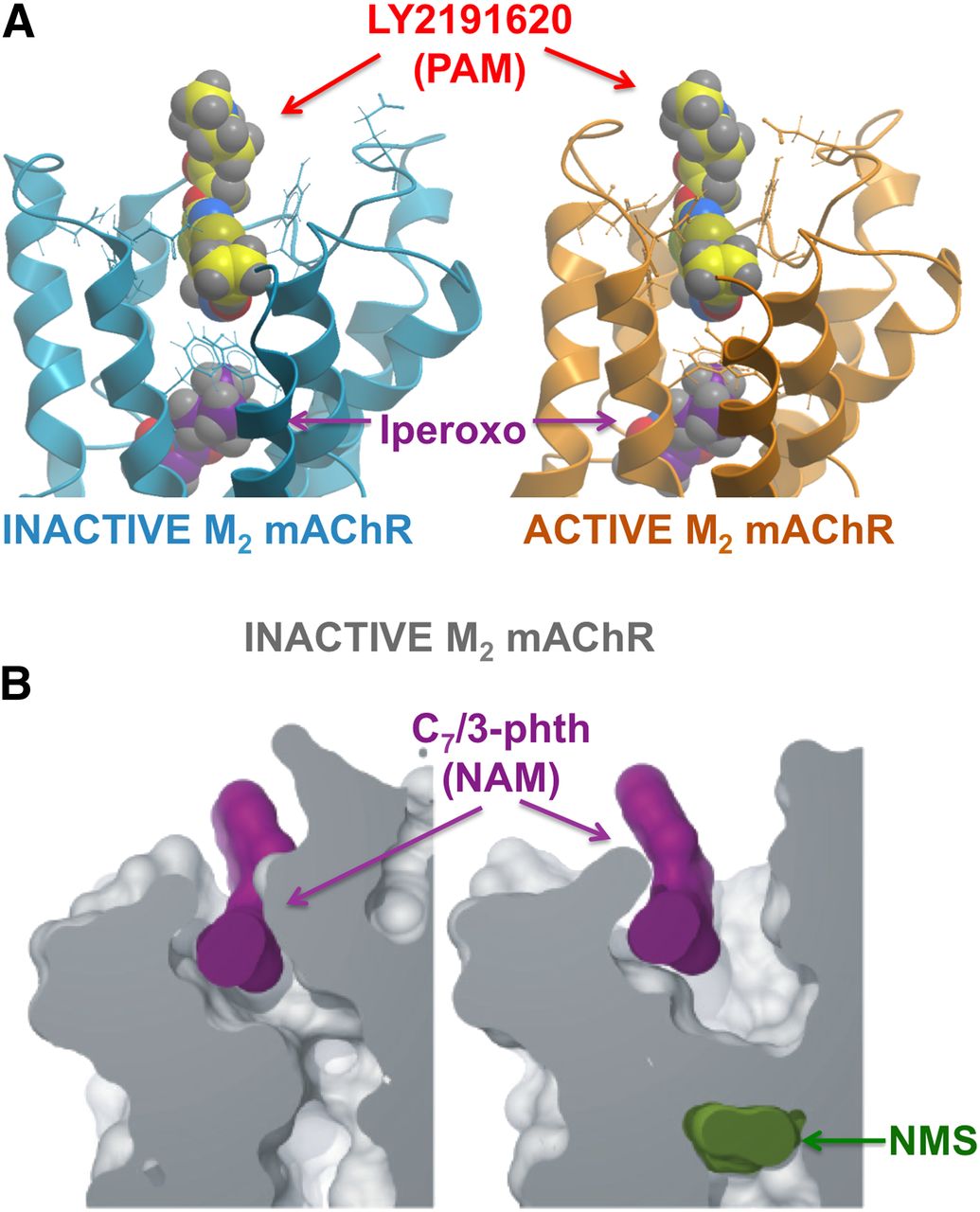
To truly understand the atomistic basis of small molecule allostery at GPCRs via structural biology, the solution of multiple structures is required, including binary and ternary receptor-ligand complexes in both active and inactive states. This feat has yet to be achieved, but an important first step has been the recent solution of a nanobody-bound active M2 mAChR bound to a high efficacy agonist agonist, iperoxo, in the absence or presence of the small-molecule PAM LY2119620 [(3-amino-5-chloro-N-cyclopropyl-4-methyl-6-[2-(4-methylpiperazin-1-yl)-2-oxoethoxy] thieno[2,3-b]pyridine-2-carboxamide)] (Kruse et al., 2013). Figure 9A compares the agonist-receptor-PAM crystal structure to that of the two compounds modeled into the earlier inactive M2 mAChR structure (Haga et al., 2012). As can be seen, the transition of the receptor to the active state involves a contraction in the extracellular vestibule that houses the allosteric site; in the absence of this contraction, the vestibule is too wide to accommodate the PAM. Receptor activation is also accompanied by a contraction within the orthosteric site around the agonist and a large opening on the intracellular end of the receptor, all these mediated in a large part due to an outward movement of the intracellular end and inward movement of the extracellular end of TM6. Interestingly, the active receptor bound to agonist in the absence of PAM looks very similar to the PAM-bound ternary complex, suggesting that the PAM pocket is almost preformed as part of the allosteric transition (Kruse et al., 2013), in agreement with the classic conformational selection mechanism of the Monod-Wyman-Changeux model. This finding can also explain why many PAMs, including LY2119620, show agonist activity because they preferentially stabilize active states.

Structural and computational insights into GPCR allostery. (A) X-ray crystal structures of the M2 mAChR in active (Protein Data Bank: 4MQS) and inactive (Protein Data Bank: 2RH1) receptor states bound to the high-efficacy agonist iperoxo and the PAM LY2119620. The compounds were solved bound in the active state, and have been modeled into the inactive state. (B) Models based on long time-scale molecular dynamics simulations of the inactive M2 mAChR where the NAM C7/3-phth is bound to a receptor in which the orthosteric site is empty (left) or occupied by the antagonist NMS (right). It can be seen that antagonist binding is conformationally linked to the extracellular vestibule, promoting a more open conformation that disfavors NAM binding. Reproduced from Dror et al. (2013).
In contrast, no NAM-receptor-orthosteric ligand ternary complex structure has been solved to date. This, in part, reflects the fact that a NAM by its very nature has lower affinity for a GPCR occupied by orthosteric ligand, and vice versa. A possible strategy for obtaining a NAM-orthosteric agonist-receptor ternary complex would thus be to use a NAM with low rather than high negative cooperativity such that it still retains sufficient affinity to bind to a receptor that already contains the orthosteric ligand.
Nonetheless, molecular level insights into the nature of NAM interactions with GPCRs can be obtained using alternative approaches, such as computational biology. For example, Dror et al. (2013) used the inactive state crystal structure of the M2 mAChR as a template for performing unbiased, long time-scale molecular dynamic simulations using multiple, structurally diverse, allosteric modulators in both the absence and presence of an orthosteric antagonist. One important outcome of this study was the identification of two core “centers” that contribute to the so-called common allosteric site in the wide extracellular vestibule. Another important finding was the contribution of multiple mechanisms that govern cooperativity with the orthosteric antagonist N-methylscopolamine (NMS), including electrostatic interactions and conformational coupling. As shown in Fig. 9B, the NAM C7/3-phth [heptane-1,7-bis-(dimethyl-39-phthalimidopropyl) ammonium bromide] forms better contacts in the extracellular vestibule of the M2 mAChR in the absence of orthosteric site occupancy; binding of the antagonist, NMS, results in a further widening of the extracellular vestibule and loss of favorable contacts for the modulator. Thus, C7/3-phth has a higher affinity for the unoccupied receptor relative to the NMS-occupied receptor and is thus a NAM for NMS binding. Ongoing structural and computational biology breakthroughs likely will continue to shed new light on both common and receptor-specific mechanisms underlying GPCR allostery.
Concluding Remarks
Like many receptor paradigms in the past, the concepts of GPCR allostery and bias have gone from pharmacologic curiosities to universal mechanisms underlying both the function of the receptors and a means of targeting them with novel therapeutic agents. Much of the impetus has come from changes in the way drugs are discovered, and thus many of the challenges and opportunities remain in this space. For instance, how successful will be the translation of the theoretical properties of allosteric (and biased) drugs as more of these compounds progress into clinical trials and onto the market? What are the long-term effects of allosteric ligands on receptor trafficking, regulation, and homeostatic mechanisms, especially given that many drug therapies are chronic? To what extent do endogenous allosteric modulators play a role in physiology and disease? How many different allosteric sites, including intracellular pockets, do GPCRs possess that can be pharmacologically exploited? Will the new era of GPCR structural and computational biology lead to a renaissance in structure-based GPCR drug design? At the very least, the answers to these questions will undoubtedly continue to yield exciting biologic insights, and in this regard the future remains bright.
Acknowledgments
The author thanks the numerous investigators who have contributed to the field of GPCR allostery and apologizes for not citing many of their studies due to space limitations. This review was commissioned to coincide with the 2014 IUPHAR Analytical Pharmacology Lecture, delivered by the author at the 17th World Congress of Basic and Clinical Pharmacology, Cape Town, South Africa, and thus borrows from many examples of the author’s work and that of his collaborators.
Footnotes
- Received June 21, 2014.
- Accepted July 24, 2014.
Portions of the work from the author’s laboratory were funded by the National Health and Medical Research Council of Australia (NHMRC) [Program Grant APP1055134 and Project Grant APP102696]. The author is a Principal Research Fellow of the NHMRC.
Abbreviations
- AC-42
- 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl]-piperidine hydrogen chloride
- BETP
- 4-(3-benzyloxyphenyl)-2-ethylsulfinyl-6-(trifluoromethyl)pyrimidine
- BQCA
- benzyl quinolone carboxylic acid
- C7/3-phth
- heptane-1,7-bis-(dimethyl-39-phthalimidopropyl) ammonium bromide
- CaSR
- calcium-sensing receptor
- CB1
- cannabinoid receptor 1
- CP-376395
- N-(1-ethylpropyl)-3,6-dimethyl-2-(2,4,6-trimethylphenoxy)-4-pyridinamine hydrochloride
- ERK1/2
- extracellular-regulated protein kinase 1/2
- FITM
- 4-fluoro-N-(4-(6-(isopropylamino)pyrimidin-4-yl)thiazol-2-yl)-N-methylbenzamide
- GLP1
- glucagon-like peptide-1
- GPCR
- G protein-coupled receptor
- 5HT
- serotonin
- 77-LH-28-1
- 1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone
- LUF6258
- N6-[2-amino-3-(3,4-dichlorobenzoyl)-4,5,6,7-tetrahydrothieno[2,3-c]pyridin-6-yl-9-nonyloxy-4-phenyl]adenosine
- LY2033298
- 3-amino-5-chloro-6-methoxy-4-methyl-thieno[2,3-b]pyridine-2-carboxylic acid cyclopropylamide
- LY2119620
- 3-amino-5-chloro-N-cyclopropyl-4-methyl-6-[2-(4-methylpiperazin-1-yl)-2-oxoethoxy] thieno[2,3-b]pyridine-2-carboxamide
- mAChR
- muscarinic acetylcholine receptor
- McN-A-343
- 4-(m-chlorophenylcarbamoyloxy)-2-butynyl)trimethylammonium chloride
- MIPS1217
- cis-1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide
- NAL
- neutral allosteric ligand
- NAM
- negative allosteric modulator
- NMS
- N-methylscopolamine
- NPS2143
- 2-chloro-6-[(2R)-3-[[1,1-dimethyl-2-(2-naphthalenyl)ethyl]amino-2-hydroxypropoxy]benzonitrile hydrochloride
- PAM
- positive allosteric modulator
- SAR
- structure-activity relationships
- SB269652
- trans-1H-indole-2-carboxylic acid {4-[2-(cyano-3,4-dihydro-1H-isoquinolin-2-yl)-ethyl]-cyclohexyl}-amide
- TBPB
- 1-[1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one
- THRX-160209
- 4-{N-[7-(3-(S)-(1-carbamoyl-1,1-diphenylmethyl)pyrrolidin-1-yl)hept-1-yl]-N-(n-propyl)amino}-1-(2,6-dimethoxybenzyl)piperidine
- THRX-198321
- biphenyl-2-yl-carbamic acid 1-{9-[(R)-2-hydroxy-2-(8-hydroxy-2-oxo-1,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-piperidin-4-yl ester
- TM
- transmembrane
- VCP171
- (2-amino-4-(3-(trifluoromethyl)phenyl)thiophen-3-yl)(phenyl)methanone
- VCP746
- 4-(5-amino-4-benzoyl-3-(3-(trifluoromethyl)phenyl)thiophen-2-yl)-N-(6-(9-((2R,3R,4S,5R)-3,4-dihydroxy-5-(hydroxymethyl)tetrahydrofuran-2-yl)-9H-purin-6-ylamino)hexyl)benzamide
- VU0366249
- N-(3-chloro-4-fluorophenyl)-3-cyano-5-fluorobenzamide
- Copyright © 2014 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Classification of Allosteric Modulators
- Pharmacologic Characteristics of GPCR Allostery
- Enriching Structure-Activity Studies of GPCR Allostery and Bias
- Bitopic GPCR Ligands
- Endogenous Allosteric Modulators
- Understanding the Structural Basis of GPCR Allostery
- Concluding Remarks
- Acknowledgments
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters