


Abstract
The small heterodimer partner (SHP) (NR0B2) is an atypical nuclear receptor that lacks a DNA-binding domain. It interacts with and inhibits many transcription factors, affecting key metabolic processes, including bile acid, cholesterol, fatty acid, and drug metabolism. Our aim was to determine the influence of steatotic drugs and nonalcoholic fatty liver disease (NAFLD) on SHP expression and investigate the potential mechanisms. SHP was found to be repressed by steatotic drugs (valproate, doxycycline, tetracycline, and cyclosporin A) in cultured hepatic cells and the livers of different animal models of NAFLD: iatrogenic (tetracycline-treated rats), genetic (glycine N-methyltransferase–deficient mice), and nutritional (mice fed a methionine- and choline-deficient diet). Among the different transcription factors investigated, CCAAT-enhancer-binding protein α (C/EBPα) showed the strongest dominant-repressive effect on SHP expression in HepG2 and human hepatocytes. Reporter assays revealed that the inhibitory effect of C/EBPα and steatotic drugs colocalize between −340 and −509 base pair of the SHP promoter, and mutation of a predicted C/EBPα response element at −473 base pair abolished SHP repression by both C/EBPα and drugs. Moreover, inhibition of major stress signaling pathways demonstrated that the mitogen-activated protein kinase kinase 1/2 pathway activates, while the phosphatidylinositol 3 kinase pathway represses SHP in a C/EBP-dependent manner. We conclude that SHP is downregulated by several steatotic drugs and in advanced NAFLD. These conditions can activate signals that target C/EBPα and consequently repress SHP, thus favoring the progression and severity of NAFLD.
Introduction
The small heterodimer partner (SHP) (NR0B2) is a unique orphan nuclear receptor that contains a dimerization and ligand-binding domain but lacks the conserved DNA-binding domain (Zhang et al., 2011; Garruti et al., 2012). SHP interacts with a wide variety of conventional nuclear receptors and transcription factors and negatively regulates their transcriptional activity (Bavner et al., 2005). SHP expression is predominantly observed in the liver (Seol et al., 1996; Lee et al., 1998), where a large number of direct and indirect SHP target genes have been identified, including bile acid (BA), cholesterol, fatty acid, glucose, and drug metabolism genes (Boulias et al., 2005; Zhang et al., 2011). Moreover, the involvement of SHP in key hepatic regulatory networks denotes its relevance and central role in liver homeostasis (Zhang et al., 2011; Garruti et al., 2012). Therefore, chronic deregulation of SHP expression may lead to pathologic consequences and favor liver disease. The potential negative consequences of SHP deregulation can be extrapolated from studies with liver-specific SHP-transgenic and SHP-deficient mice (Wang et al., 2003; Boulias et al., 2005; Park et al., 2008; Zhang et al., 2008, 2010, 2014; Chanda et al., 2009).
BAs are potent agonists of FXRα, which induces the expression of SHP. Induced SHP in turn leads to transcriptional repression of the rate-limiting CYP7A1 and CYP8B1 enzymes of the BA biosynthetic pathway (Chiang, 2003). In line with this, transgenic mice with sustained overexpression of SHP in the liver show depletion of the hepatic BA pool. Moreover, these mice show an indirect activation of the lipogenic factors PPARγ and SREBP-1c, which trigger triglyceride (TG) accumulation and fatty liver (Boulias et al., 2005).
SHP also represses genes with an important role in liver fibrosis (Zhang et al., 2011). Consequently, the lack of expression of SHP in SHP−/− mice results in increased sensitivity to liver damage and liver fibrosis induced by bile duct ligation (Park et al., 2008; Zhang et al., 2014). In agreement, progressive fibrosing steatohepatitis was reversed in wild-type mice after AMPK-mediated induction of SHP, but this was not observed in SHP−/− mice (Chanda et al., 2009). SHP deficiency could have even more severe consequences as spontaneous hepatocellular carcinoma (HCC) is observed in SHP−/− mice due to hepatocyte hyperproliferation and increased cyclin D1 expression (Zhang et al., 2008). Moreover, SHP targets mitochondria, induces apoptosis, and inhibits tumor growth (Zhang et al., 2010), but this protective role may be lost when SHP is deficient.
All these studies reinforce the notion that SHP controls major liver functions; therefore, sustained changes in SHP expression may significantly trigger or worsen liver injury and disease progression. Induction of SHP may exacerbate diet-induced dyslipidemia and fatty liver (Boulias et al., 2005; Hartman et al., 2009). On the contrary, repression of SHP may promote fibrosis and HCC (Park et al., 2008; Zhang et al., 2008, 2010, 2014; Chanda et al., 2009). A key question then is whether hepatic SHP expression is altered in pathophysiological conditions.
It has been proven that repression of SHP by epigenetic silencing is a common denominator of HCC (He et al., 2008). However, the regulation of SHP in other conditions, such as hazardous xenobiotic exposure or fatty liver disease, is not so well characterized or is controversial. The response of SHP to endogenous factors, such as BAs, fatty acids, cholesterol, glucose, insulin, cytokines, and hormones, has been well characterized (Zhang et al., 2011; Garruti et al., 2012), but the effect of drugs and xenobiotics on SHP has not been investigated. On the other hand, SHP seems to be upregulated in genetic (leptin-deficient ob/ob mice) and dietary [high-sucrose or high-fat diet (HFD)] mouse models of fatty liver (Huang et al., 2007; Trauner, 2007). However, SHP expression appears diminished in human nonalcoholic steatohepatitis (NASH) (Zhang et al., 2014).
Our aim in the present study was to determine whether SHP is regulated by steatotic drugs and nonalcoholic fatty liver disease (NAFLD) and investigate the potential mechanisms. We demonstrate that SHP is repressed by several steatotic drugs and that SHP is downregulated in models of severe NAFLD. We identify CCAAT/enhancer-binding protein α (C/EBPα) as a SHP repressor and demonstrate that several steatotic drugs and stress pathways modulate SHP by signaling via a C/EBP response element at −473 bp in the human SHP promoter.
This study uncovers new mechanisms of transcription regulation of the human SHP gene and reveals novel pathways that target SHP expression that could favor the severity and progression of liver disease.
Materials and Methods
Animals.
Male mice (3 and 9 months of age) deficient in methionine adenosyltransferase 1A (MAT1A) (Lu et al., 2001; Martinez-Chantar et al., 2002) and glycine N-methyltransferase (GNMT) (Martinez-Chantar et al., 2008) and their wild-type C57BL/6 littermates were obtained from the animal facility of CIC bioGUNE (Derio, Bizkaia, Spain). Mice were housed at 22°C with a 12-hour light-dark cycle and allowed food (Teklad Global 18% Protein Rodent Diet 2018S; Madison, WI) and water ad libitum. To induce NAFLD, male C57BL/6J mice (10–12 weeks old) were fed a methionine and choline deficient (MCD) diet for up to 5 weeks (ICN, Aurora, OH). Control mice received the same MCD diet supplemented with DL-methionine (3 g/kg) and choline chloride (2 g/kg) (ICN). At the end of the experiments, mice were anesthetized (sodium pentobarbital, 60 mg/kg, i.p.) and livers were collected. The presence of steatohepatitis was proven histologically. Male Sprague-Dawley rats (125 g) were purchased from Charles River (Barcelona, Spain), maintained for 4 weeks in an air-conditioned animal room (21–25°C, 30–70% humidity, 12-hour light-dark cycle), and fed ad libitum with a standard diet (Research Diets, Gentofte, Denmark). Rats were randomly placed into two groups (n = 6), and tetracycline dissolved in methylcellulose 0.5% (vehicle) was administered by oral gavage (2 g/kg per day) during 4 consecutive days. Control rats were treated with vehicle. Animals were euthanized 24 hours after the last administration, and livers were collected. The Institutional Animal Care and Use Committee that specifically approved this study was the Bioethical and Animal Welfare Committee of CIC bioGUNE (IACUC: P-CBG-CBBA 02-10) in accordance with the guidelines of the European Research Council for animal care and use.
Culture of Human HepG2 and HepaRG Cells.
HepG2 cells (American Type Culture Collection, Rockville, MD) were cultured in Ham’s F-12/Leibovitz L-15 (1:1, v/v) medium (Gibco BRL/Invitrogen, Barcelona, Spain) supplemented with 7% newborn calf serum and 2 mM l-glutamine. In addition, culture media were supplemented with 50 U/ml penicillin and 50 μg/ml streptomycin. HepaRG cells were obtained from Biopredic International (Rennes, France). Undifferentiated cells were cultured in a growth medium (Biopredic) for 2 weeks, and cell differentiation was induced with a differentiation medium (Biopredic) for 2 more weeks. At that stage, HepaRG cells reached a differentiated hepatocyte-like morphology. Cells were further maintained in a HepaRG differentiation medium for the duration of the experiments.
Isolation and Culture of Hepatocytes.
Human hepatocytes were isolated from nonsteatotic liver biopsies (1–4 g) using a two-step perfusion technique (Gomez-Lechon et al., 1990). Liver samples were obtained in conformity with the rules of the Hospital’s Ethics Committee. None of the donors (four men and one woman aged between 37 and 74) were regular consumers of alcohol or other drugs and were not suspected of harboring any infectious disease. Rat hepatocytes were obtained from Sprague-Dawley male rats (180–250 g) by perfusion of the liver with collagenase. Freshly isolated hepatocytes (150 × 103 viable cells/cm2) were plated on a six-well plate precoated with a collagen gel layer. Sandwich cultures were established by the deposition of a second layer of collagen 24 hours later, as described elsewhere (Gomez-Lechon et al., 1998). Cells were cultured in serum-free Williams/Ham’s F-12 (1:1, v/v) medium (Gibco BRL/Invitrogen) supplemented with 5 mM glucose, 0.2% bovine serum albumin, 0.01 µM insulin, and 0.01 µM dexamethasone.
Chemicals and Treatment of Cultured Cells.
Amitriptyline, cyclosporin A, doxycycline, fluoxetine, flutamide, ketotifen, simvastatin, tamoxifen, tetracycline, tianeptine, tilorone, valproate, U0126 [1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene], and LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride] were acquired from Sigma-Aldrich (Madrid, Spain). Steatotic drugs and nonsteatotic compounds were chosen on the basis of previous information on their steatotic, phospholipidosic, hepatotoxic, or nonhepatotoxic effect (Benet et al., 2014). Stock solutions were prepared in dimethylsulfoxide (DMSO) or water and were diluted in a culture medium. The kinase inhibitors U0126 and LY294002 were prepared in DMSO and added 24 hours before analysis. The final concentration of DMSO never exceeded 0.5% (v/v), and control cultures were treated with the same amount of solvent.
Adenoviral Infection.
The recombinant adenoviruses encoding CAR, PXR, C/EBPα, C/EBPβ, FOXA1, PGC1α, RXRα, and PPARα were developed in our laboratory as described (Guzman et al., 2013). Cultured cells were infected with recombinant adenoviruses encoding transcription factors or with an insertless adenoviral vector (Ad-CTRL) for 24 hours at a multiplicity of infection ranging from 2 to 45 plaque-forming units/cell. Thereafter, cells were washed and a fresh medium was added. To activate the expressed nuclear receptors (CAR, PXR, RXRα, and PPARα), selective ligand agonists were added at 24 hours postinfection, and at 48 hours, postinfection cells were harvested and analyzed. Ligands were dissolved in DMSO and added to culture media at the following concentrations: 500 nM CITCO [6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime] (CAR), 1 μM GW7647 [ 2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-2-methylpropanoic acid] (PPARα), 50 μM rifampicin (PXR), and 1 μM 9-cis retinoic acid (RXRα). Control cultures were treated with 0.5% DMSO.
RNA Purification, Reverse Transcription, and Quantitative Real-Time Polymerase Chain Reaction.
Total RNA was extracted from cultured cells with the RNeasy Plus Mini Kit from Qiagen (Madrid, Spain). To extract RNA from tissue, frozen mouse and rat liver samples (25–50 mg) were placed in 2-ml tubes containing CK14 ceramic beads (Precellys, Saint Quentin en Yvelines, France) and 800 μl of RLT buffer (Qiagen). Then, liver tissues were homogenized twice for 10 seconds at 6000 rpm at 4°C in a Precellys 24 dual system equipped with a Criolys cooler. Tubes were centrifuged at 3000g for 5 minutes at 4°C, and total RNA was extracted from supernatants and reverse transcribed as described (Benet et al., 2014). Diluted cDNA was amplified in a LightCycler 480 Instrument (Roche Applied Science, Barcelona, Spain) using LightCycler DNA Master SYBR Green I (Roche Applied Science). In parallel, mouse and rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) or human porphobilinogen deaminase (PBGD) were analyzed for normalization. In each amplification, a reference calibrator cDNA made from a pool of human livers was included. The relative mRNA was calculated with the LightCycler Relative Quantification Analysis software (Roche Applied Science). The efficiency of each polymerase chain reaction (PCR) was estimated from a serially diluted human liver cDNA curve. Based on these PCR efficiencies and the cycle thresholds (Ct), the relative concentration of the target and reference (GAPDH or PBGD) cDNAs and their ratio were determined.
Specific primers for quantitative real-time PCR were as follows: human PBGD, forward, 5′- CGGAAGAAAACAGCCCAAAGA, and reverse, 5′- TGAAGCCAGGAGGAAGCACAGT; human SHP, forward, 5′- CAGCAGCAGTGGAGGCAGTG, and reverse, 5′- CGGGGTTGAAGAGGATGGTC; human SREBP1c, forward, 5′- GGAGGGGTAGGGCCAACGGCCT, and reverse, 5′- CATGTCTTCGAAAGTGCAATCC; mouse and rat GAPDH, forward, 5′- AGGGCTCATGACCACAGTCCAT, and reverse, 5′- GCCAGTGAGCTTCCCGTTCAG; mouse SHP, forward, 5′- AGGGCACGATCCTCTTCAAC, and reverse, 5′- AGGGCTCCAAGACTTCACAC; rat SHP, forward, 5′- GGCACTATCCTCTTCAACCCA, and reverse, 5′- TCCAGGACTTCACACAATGCC.
Protein Samples and Immunoblotting Analysis.
Cultured cells or liver tissue were homogenized in an M-PER or T-PER reagent from Pierce (Fisher Scientific, Madrid, Spain) and a protease inhibitor cocktail. Protein concentration was determined using the Protein Assay Kit from Bio-Rad Laboratories (Madrid, Spain). Proteins were resolved by 12% SDS-PAGE, transferred to Immobilion membranes (Millipore Iberica, Madrid, Spain), and incubated with anti-SHP, antitubulin (Santa Cruz Biotechnology, Dallas, TX), or anti–β-actin (Sigma-Aldrich) antibodies. Blots were developed with horseradish peroxidase–labeled IgG (DAKO, Glostrup, Denmark) using an ECL kit from Amersham Biosciences/GE Healthcare (Buckinghamshire, UK). Equal loading was verified by Coomassie Brilliant Blue or Ponceau S staining.
Plasmid Constructs.
Chimeric luciferase reporter constructs with different lengths of the 5′ flanking region of the human SHP gene were obtained by cloning SHP-5′ flanking sequences into the enhancerless, promoterless pGL3-Basic vector (Promega Biotech Iberica, Madrid, Spain). The −2500 to +27 bp SHP-5′ flanking sequence was synthesized by GenScript Technologies (Piscataway, NJ) and cloned in a pUC57 plasmid. Fragments corresponding to −2500, −1362, and −612 were excised from the pUC57 plasmid by restriction enzymes to be subcloned in pGL3-Basic. Three DNA fragments spanning nucleotides −509, −340, and −150 to +10 bp of the SHP-5′ flanking region were PCR cloned from the −612-SHP-pGL3 plasmid using the Expand High Fidelity PCR System (Roche Applied Science). The PCR primers used are SHP-UP-150-MluI, 5′-CCTACGCGTACCACTTCCCCACCAT, SHP-UP-340-MluI, 5′-AATACGCGTACACCTGCTGATTGTG, SHP-UP-509-MluI, 5′-CTGACGCGTGCTTCTGGCTGACAACA, and SHP-DN-HindIII, 5′-CTCAAGCTTCCAGCTCTCTGGC. The resulting amplicons were digested with the restriction and ligated into the pGL3-Basic vector previously digested with the same enzymes.
Chromatin Immunoprecipitation Assay.
A chromatin immunoprecipitation (ChIP) assay was done as previously reported (Benet et al., 2010). Briefly, HepG2 cells were infected with Ad-C/EBPα or with a control (insertless) adenovirus (Ad-pACC) and 48 hours after infection, cells were treated with 1% formaldehyde for 10 minutes. Thereafter, cells were lysed and sonicated on ice for 8 × 15 second steps at a 20% output in a Branson Sonicator (VWR International Eurolab, Barcelona, Spain). DNA content was quantified and properly diluted to maintain an equivalent amount of DNA in all samples (input DNA). For immunoprecipitation of C/EBPα-DNA complexes, 4 µg of a specific antibody (Santa Cruz Biotechnology) was added (bound DNA fraction). Mock immunoprecipitation with rabbit preimmune IgG (Santa Cruz Biotechnology) was performed in parallel (background DNA fraction). Samples were incubated overnight at 4°C on a 360° rotator. The immunocomplexes were affinity absorbed with protein G agarose/salmon sperm DNA (Millipore) and collected by centrifugation. DNA fractions were washed, and the crosslinks were reversed using 100 µl of 10% Chelex (Bio-Rad Laboratories) (Benet et al., 2010). Amplification was real-time monitored, stopped in the exponential phase of amplification, and analyzed by agarose gel electrophoresis. Amplification of a SHP 5′-flanking sequence (−329/−678 bp) among the pull of DNA was performed with specific primers flanking these regions: forward, 5′- CAGACCTGGCCTTTCAGGAG, and reverse, 5′- ATCAGCAGGTGTCCCCATTG. An exonic region in the ribosomal protein, large, P0 (RPLP0) gene devoid of C/EBPα consensus binding sites and expected not to be enriched in antibody-bound DNA fractions was PCR amplified as a negative control (Guzman et al., 2013).
Statistical Analysis.
Differences among groups were analyzed by one-way analysis of variance and Tukey’s pairwise comparisons. Differences between groups were evaluated by the nonparametric Mann-Whitney test as indicated. A P value of less than 0.05 was considered to be statistically significant. Values are expressed as mean ± S.D.
Results
Regulation of SHP by Drugs.
Human hepatoma HepG2 cells were incubated for 24 hours with different drugs previously characterized and classified as nonhepatotoxic, hepatotoxic, phospholipidosic, and steatotic. The concentrations selected were always subcytotoxic (≤IC10), thus avoiding acute necrotic or apoptotic side effects (Benet et al., 2014). Moreover, we have previously shown that in these conditions, the selected steatotic drugs trigger lipid accumulation in HepG2 cells (Benet et al., 2014). We observed that most steatotic drugs (valproate, doxycycline, tetracycline, cyclosporine A, and tianeptine) negatively affected SHP expression (Fig. 1A). However, this was not a common effect observed with all known steatotic compounds, as tamoxifen, amiodarone, and zidovudine did not repress SHP (Fig. 1A and data not shown). Hepatotoxic and phospholipidosic drugs did not influence or consistently affect SHP expression.
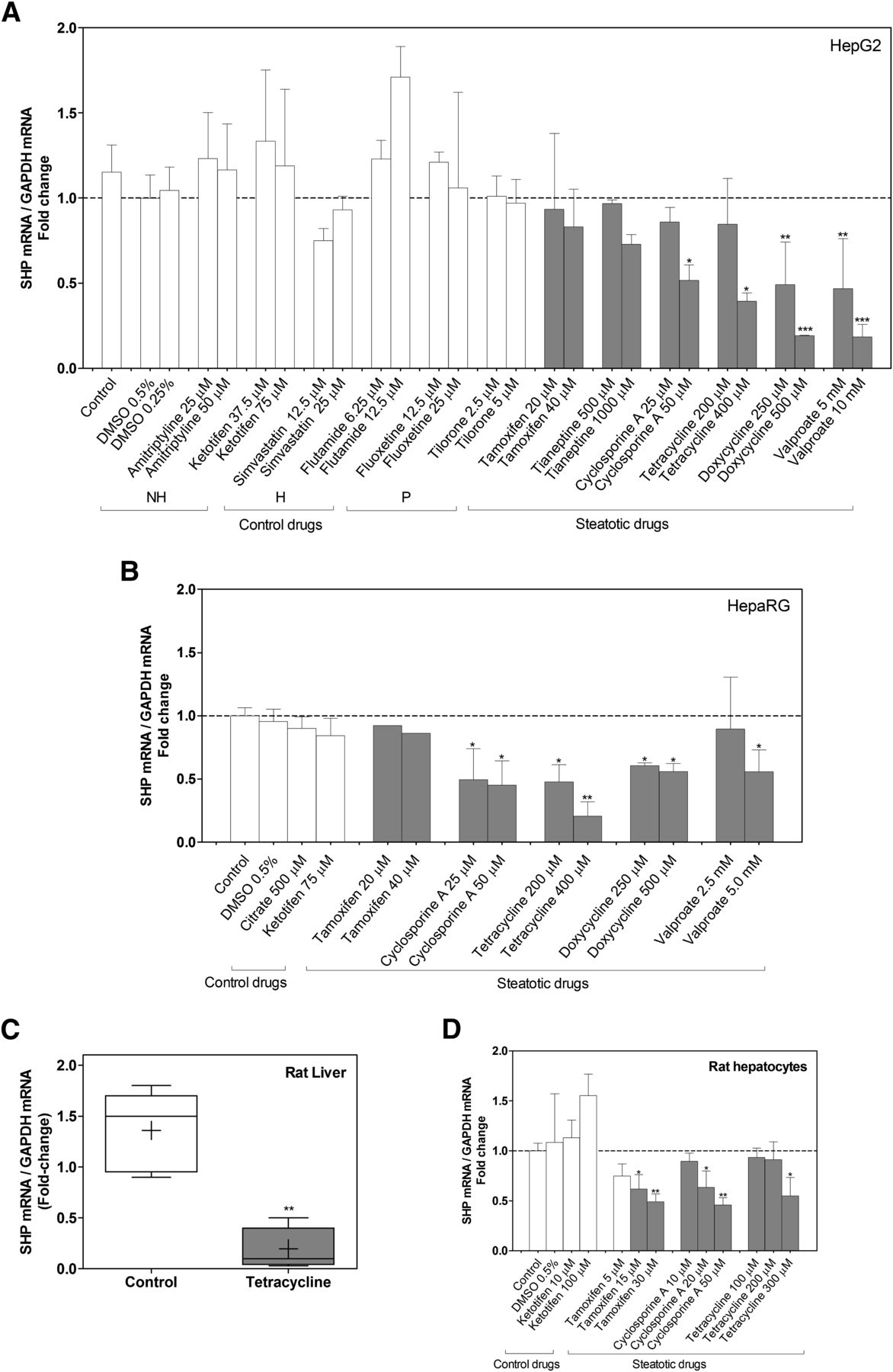
Regulation of SHP by drugs. (A) HepG2 cells, (B) HepaRG cells, and (D) cultured rat hepatocytes were incubated with steatotic and nonsteatotic control compounds for 24 hours, and SHP mRNA was measured and normalized to GAPDH. H, hepatotoxic; NH, nonhepatotoxic; P, phospholipidosic. Results are expressed as a fold change versus DMSO-treated cells and represent the mean ± S.D. of three to five independent experiments. *P < 0.05; **P < 0.01; and ***P < 0.001 versus control cells (one-way analysis of variance and Tukey’s test). (C) Liver SHP expression on rats treated with tetracycline (n = 5) or vehicle (n = 5) was determined by quantitative real-time PCR and normalized to GAPDH. Results are represented in box plots with the median (black line) and the mean (cross), and expressed as a fold change versus a control rat liver pool. **P < 0.01 (Mann-Whitney test).
To better support the relevance of these findings, we investigated the response of SHP to steatotic drugs in HepaRG cells, a more differentiated human hepatic cell model, which is much more similar to primary human hepatocytes and liver tissue than HepG2 cells (Hart et al., 2010). Results in Fig. 1B demonstrate that cyclosporine A tetracycline, doxycycline, and valproate also caused a significant repression of SHP in differentiated HepaRG cells.
To find out if the repressive effect of steatotic drugs occurs in vivo, we treated rats with tetracycline or vehicle during 4 consecutive days. Results demonstrate that tetracycline strongly repressed SHP in the rat liver (Fig. 1C). Finally, we incubated cultured rat hepatocytes in a collagen-sandwich configuration with some selected steatotic drugs to search for potential interspecies differences between human and rat models. We found that some steatotic drugs, such as cyclosporine A and tetracycline, decreased SHP expression as in human hepatic cultures; however, tamoxifen only significantly repressed SHP in rat hepatocytes (Fig. 1D).
Regulation of SHP in Mouse Models of NAFLD.
Liver SHP expression was first analyzed in two genetic models of NAFLD: MAT1A−/− and GNMT−/− mice. These two models spontaneously develop fatty liver, where GNMT−/− is a more severe NAFLD model than MAT1A−/−.
The 3-month-old MAT1A−/− mouse does not present hepatosteatosis but shows increased expression of lipid synthesis genes (Martinez-Chantar et al., 2002) and decreased TG output in very low-density lipoprotein (Cano et al., 2011), whereas the 9-month-old MAT1A−/− exhibits macrovesicular steatosis (25–50% of hepatocytes) and focal areas of inflammation (Lu et al., 2001). Only the 9-month-old mouse shows increased serum aminotransferases, but fibrosis is not observed (Cano et al., 2011). Intriguingly, SHP was significantly induced in 3-month-old MAT1A−/− mice, but no differences were observed in 9-month-old deficient mice with established fatty liver (Fig. 2A).

Liver SHP expression is decreased in mouse models of advanced NAFLD. Total RNA from 3- and 9-month old MAT1A−/− (A) and GNMT−/− (B) male mouse livers was purified, and the mRNA level of SHP was determined by quantitative real-time PCR (Q-RT–PCR) and normalized to GAPDH mRNA. WT, wild-type. (C) SHP expression level was analyzed in mice livers fed with an MCD or control diet for up to 5 weeks by both Q-RT–PCR and Western blot (SHP molecular mass: ∼28 kDa). Data represent the mean ± S.D. (n = 5–6) expressed as a fold change versus a control mouse liver pool. *P < 0.05 and **P < 0.01 by the nonparametric Mann-Whitney test.
The GNMT−/− mouse shows elevated serum aminotransferases at both 3 and 9 months of age. The livers of the 3-month-old mutant mouse already shows steatosis and fibrosis, which are more pronounced in the livers of 9-month-old animals. Moreover, apoptosis increases and genes involved in inflammation are induced. At 9 months, the GNMT−/− mouse also spontaneously develops multifocal HCC (Martinez-Chantar et al., 2008; Varela-Rey et al., 2010). SHP was found repressed in the liver of GNMT−/− mice, particularly 9-month-old deficient mice that exhibit more manifest liver disease (Fig. 2B).
We next investigated the regulation of SHP in the MCD diet model of NAFLD. Mice fed the MCD diet for more than 2 weeks exhibit elevated serum aminotransferases, inflammation, and hepatic fibrosis in addition to simple steatosis (Anstee and Goldin, 2006; Itagaki et al., 2013). Liver SHP was also found repressed in mice on an MCD diet. The repression began after 1 week and was very significant at 5 weeks of diet (Fig. 2C). The reduced expression at the mRNA level correlated with progressive, reduced expression at the protein level, as determined by immunoblotting (Fig. 2C).
Altogether, these data support that both steatotic drugs and advanced NAFLD (NASH/fibrosis) trigger SHP downregulation in the liver, and therefore, a common mechanism may exist that causes SHP gene repression in these conditions.
Screening for SHP Gene Repressors: Identification of C/EBPα as an SHP Dominant Negative Factor.
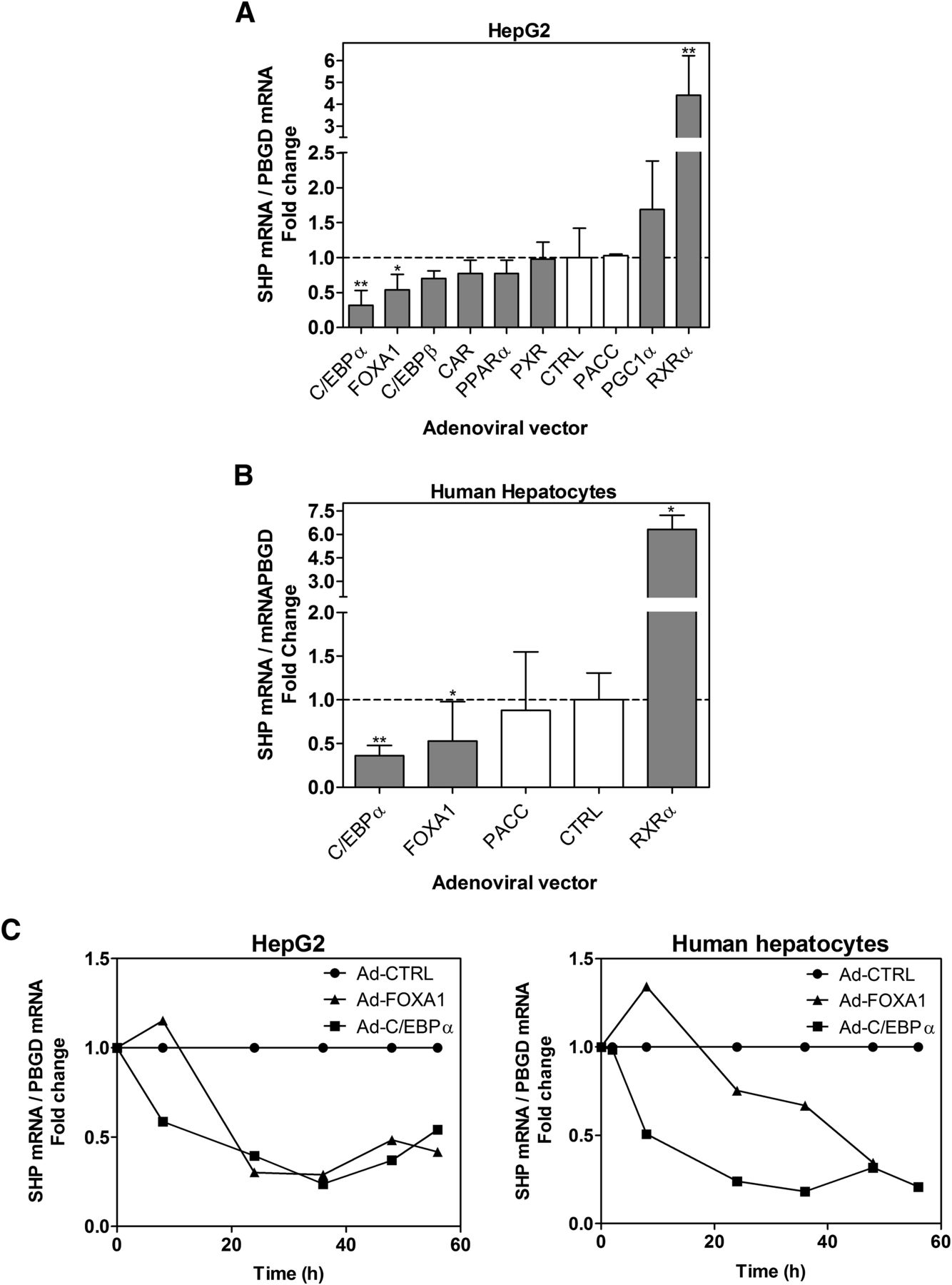
SHP is activated by multiple transcription factors, but no SHP gene repressor has been identified. We aimed to investigate the potential repressive role of new transcription factors whose effects on the SHP gene have not been investigated yet: C/EBPα, C/EBPβ, FOXA1, CAR, PXR, PPARα, RXRα, and the coactivator PGC1α. These factors are associated with liver lipid/energy metabolism and the hepatic phenotype. Previous studies from our laboratory have shown that all these factors are expressed in HepG2 at a lower level than in human liver (Donato et al., 2013; Guzman et al., 2013), which discourages the use of knockdown approaches to study their role. Therefore, we decided to force their re-expression and transfected HepG2 cells with adenoviral vectors encoding these factors. The experimental conditions selected induced expression in HepG2 to levels within the wide range found in human liver (Guzman et al., 2013).
Results demonstrate that C/EBPα and FOXA1 downregulated SHP, whereas PGC1α and RXRα caused gene activation (Fig. 3A). Moreover, C/EBPα, FOXA1, and RXRα caused the same effect on SHP in cultured human hepatocytes (Fig. 3B).

C/EBPα and FOXA1 repress SHP, whereas RXRα [(+)-9-cis-RA] induces SHP transcription. (A) HepG2 cells and (B) cultured human hepatocytes were infected with adenoviruses encoding eight transcription factors for 48 hours. pACC, insertless control adenovirus. Selective ligand agonists for CAR, PXR, RXRα, and PPARα (see Materials and Methods) were added during the last 24 hours. The SHP mRNA level was determined by quantitative real-time PCR and normalized to PBGD. Data represent mean ± S.D. (n = 3–5) expressed as a fold change versus control noninfected cells. (A) *P < 0.05 and **P < 0.01 versus control cells (one-way analysis of variance and Tukey’s test). (B) *P < 0.05 and **P < 0.01 versus control cells (Mann-Whitney test). (C) Time-dependent alterations in SHP mRNA after transduction of HepG2 and human hepatocyte cells with Ad-FOXA1 and Ad-C/EBPα. Data represent the mean of two experiments expressed versus control adenovirus-infected cells.
Time-course analysis demonstrated that Ad-C/EBPα causes a fast downregulation, which reaches ∼50% by 8 hours after adenoviral infection, both in HepG2 and cultured human hepatocytes. Ad-FOXA1 required ≥24 hours to trigger SHP gene repression (Fig. 3C).
A combination of these transcription factors demonstrated that C/EBPα acts on SHP as a dominant negative factor, which is able to substantially repress both basal and RXRα-induced SHP levels. The repressive effect of FOXA1 was less significant when combined with RXRα, and no further repression was observed when combined with C/EBPα (Fig. 4).

SHP response to combined transcription factors. HepG2 cells were infected with Ad-C/EBPα, Ad-FOXA1, and Ad-RXRα either individually or combined. Twenty-four hours later, 1 μM 9-cis-retinoic acid or solvent (DMSO) was incorporated, and at 48 hours postinfection, cells were harvested and processed for quantitative real-time PCR and Western blot. Data represent the mean ± S.D. (n = 3) expressed as a fold change versus control noninfected cells. *P < 0.05 and **P < 0.01 versus Ad-RXRα infected cells (one-way analysis of variance and Tukey’s test). ns, not significant.
Analysis of SREBP1c, one of the few positive targets of SHP (Boulias et al., 2005), demonstrated a very similar response. C/EBPα repressed both basal and induced SREBP1c mRNA levels, whereas FOXA1 had no significant effect on this gene (Fig. 4). These results indicate that repression of SHP by C/EBPα translates into downstream SHP-dependent functions.
Altogether, our results demonstrate that C/EBPα is an effective dominant repressor of SHP both in HepG2 and cultured human hepatocytes.
C/EBPα Negatively Regulates the Human SHP Gene by Binding to a Response Element at −473 bp in the SHP Promoter.
We cloned 2500 bp of the 5′-flanking region of the human SHP gene and generated serially deleted luciferase (LUC) reporter constructs. Basal reporter analysis of the different fragments in HepG2 cells indicated that the activity of the proximal promoter between +10 and −340 bp is very low (Fig. 5). Transcription increased notably and stepwise between −340 and −612 bp, and no significant further increase was observed with longer sequences up to −2500 bp. When the basal response was assayed in the nonhepatic HeLa cells (Fig. 5), a very low activity with all SHP promoter fragments was detected and no major increase in activity with the sequence beyond −340 bp was achieved. Results demonstrate that most of the constitutive transcription of the SHP gene in HepG2 depends on response elements (REs) located between −340 and −612 bp, and that the transcription factors acting on these regions are likely liver-enriched factors that are only present in the hepatic cell line. The significant increase in reporter activity observed in HepG2 with the sequence between −509 and −612 bp can be elicited by HNF4α (not present in HeLa), as an HNF4α-RE at −580 bp in the human SHP gene has been previously reported (Shih et al., 2001).

Basal activity of SHP promoter deletion constructs in HepG2 and HeLa cells. HepG2 (A) or HeLa (B) cells were transfected with sequential deletion fragments of SHP-firefly-LUC vectors and the normalization pRL-SV40 plasmid. Luciferase activities were determined 48 hours post-transfection. Bars represent the mean ± S.D. of four to five independent experiments expressed as firefly/Renilla luciferase activity ratios. Statistical differences were evaluated by one-way analysis of variance and Tukey’s pairwise comparisons. ***P < 0.001.
We next investigated the response of the SHP promoter to C/EBPα. Results in Fig. 6A demonstrate that C/EBPα represses SHP-LUC activity in HepG2 in consonance with the negative effects of this factor on SHP expression (mRNA and protein). The repressive effect of C/EBPα is located between −340 and −509 bp.

Transactivation of SHP reporter constructs by C/EBPα. (A) Repressing effect of C/EBPα on SHP promoter. HepG2 cells were infected with Ad-control or Ad-C/EBPα. The next day, cells were transfected with the different SHP promoter vectors and the normalization pRL-SV40 plasmid. At 48 hours post-transfection, cells were lysed, and both firefly and Renilla reniformis luciferase activities were determined. Bars represent the mean ± S.D. of three to five independent experiments expressed as a fold change versus Ad-control.*P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant (one-way analysis of variance and Tukey’s test). (B) Response of the SHP promoter with mutated C/EBP-RE to C/EBPα. Cells were infected with Ad-control or Ad-CEBPα, and the next day, they were transfected with wild-type (509) or C/EBP-RE mutated (509-mut) promoter reporter vectors. Luciferase activities were measured 48 hours post-transfection. Bars represent the mean ± S.D. of three to five independent experiments expressed as fold change versus Ad-control. *P < 0.05 (Mann-Whitney test). (C) ChIP assay demonstrates binding of C/EBPα to the SHP promoter. Formaldehyde crosslinked chromatin from adenovirus-infected HepG2 cells was incubated with antibody against C/EBPα or with preimmune IgG. Immunoprecipitated purified DNA was PCR amplified with primers specific for the SHP 5′-flanking region (−329/−678 bp, 34 cycles) or for an exonic region of the human RPLP0 (negative binding control, 39 cycles). Aliquots (10 µl) of PCR amplifications from a representative experiment were subjected to electrophoresis on 2% agarose gel and stained with SYBR Green Safe. M, 100-bp DNA ladder. (D) Recovery of DNA in the immunoprecipitates is represented as the percentage of input DNA (100%).
Sequence analyses with MatInspector software (Genomatix Software GmbH, Munich, Germany) and defined C/EBP frequency matrices from the PAZAR database lead to the identification of a potential C/EBP-RE at −473 bp (GTTAGGCAAA).
To prove that this is the RE for C/EBPα in the SHP promoter, we constructed a −509 bp reporter vector with 3-bp substitution in the predicted C/EBP-RE at −473 bp (509-mut, TCAAGGCAAA, Fig. 6B). We found that mutation of this element completely abolished the repressive effect of C/EBPα on SHP (Fig. 6B). Indeed, the 509-mut-SHP-LUC construct showed a very similar response to that of the 340-SHP-LUC.
ChIP assays were performed to assess binding of the transcription factor to the native SHP promoter. The DNA sequence responsive to C/EBPα was PCR amplified after immunoprecipitation. Infection of HepG2 cells with Ad-C/EBPα caused a noticeable increase in the amount of immunoprecipitated SHP DNA by anti-C/EBPα antibody, suggesting actual binding of this transcription factor in vivo (Fig. 6, C and D). C/EBPα is expressed at a low level in human hepatoma cells (Jover et al., 1998), and consequently forced re-expression by adenoviral vector is needed to observe significant binding by a ChIP assay.
We conclude that C/EBPα represses the human SHP gene via direct binding to a newly identified C/EBP-RE located at −473 bp.
Steatotic Drugs Modulate SHP Expression by Signaling via the −473 bp C/EBP-RE.
We have demonstrated that steatotic drugs repress SHP mRNA in cultured hepatic cells and in vivo. Therefore, our next step was to analyze the response of the SHP promoter to these drugs. We found that the repressive effect of valproate (1 and 2.5 mM), doxycycline (250 and 500 µM), and cyclosporine A (25 and 50 µM) localized between −340 and −509 bp of the SHP promoter (Fig. 7A), the same fragment through which C/EBPα causes SHP repression, thus suggesting that steatotic drugs may repress SHP by signaling via C/EBPα.
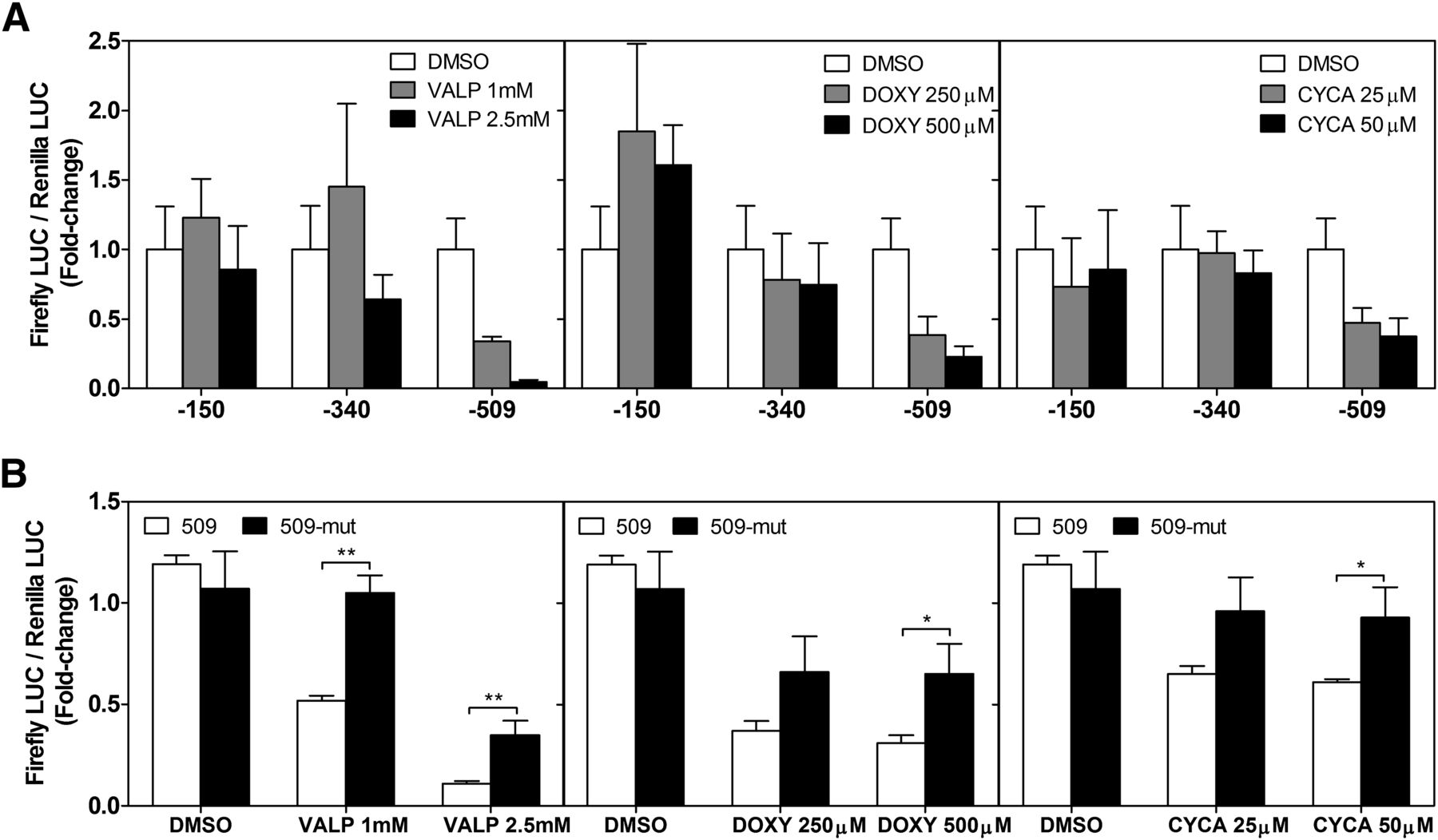
Steatotic drugs negatively modulate SHP by a C/EBPα-dependent mechanism. (A) HepG2 cells were transfected with three different SHP-promoter vectors (−150, −340, and −509). The next day, cells were treated with steatotic drugs or vehicle (DMSO). Twenty-four hours later, cell extracts were prepared and assayed for firefly and Renilla luciferase activities. (B) Response of the SHP promoter with mutated C/EBP-RE to steatotic drugs. Cells transfected with reporter vectors with mutated CEBP-RE (509-mut) or wild-type (509) SHP promoters were lysed at 48 hours post-transfection. Steatotic drugs were added 24 hours before processing. Bars represent the mean ± S.D. (n = 3–5) expressed as a fold change versus DMSO. *P < 0.05; **P < 0.01 (one-way analysis of variance and Tukey’s test).
To evaluate this possibility, we analyzed the effect of steatotic drugs on the −509 bp reporter vector with the mutated C/EBP-RE (509-mut) and found that the repressive effects of these drugs were abolished or significantly reduced (Fig. 7B). Results demonstrate that steatotic drugs repress SHP at least partially by a C/EBP-dependent mechanism.
Mitogen-Activated Protein Kinase Kinase 1/2 and Phosphatidylinositol 3 Kinase Kinases Modulate SHP Expression by Signaling via the −473 bp C/EBP-RE.
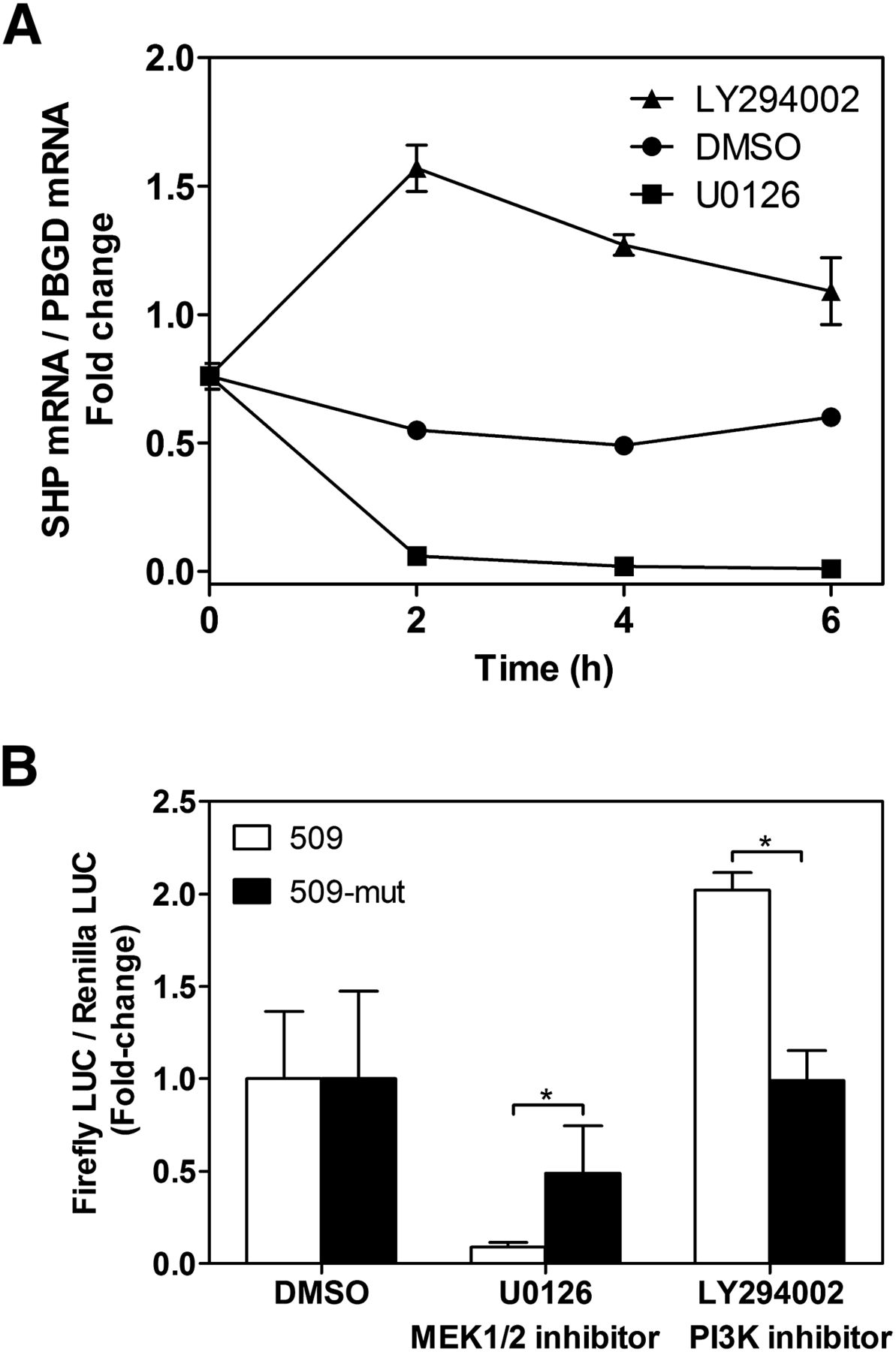
Because SHP repression occurs both by steatotic drugs and in advanced steatosis, it is tempting to suggest that a common mechanism may be involved. Many different pathways may be involved in metabolic and iatrogenic NAFLD, including insulin, lipid intermediate, mitochondrial dysfunction, oxidative stress, and inflammatory pathways (Larrain and Rinella, 2012). To preliminarily investigate the involvement of major cell stress pathways on SHP repression, we incubated HepG2 cells with well characterized inhibitors of phosphatidylinositol 3 kinase (PI3K) (LY294002), p38 (SB203580 [4-(4′-fluorophenyl)-2-(4′-methylsulfinylphenyl)-5- (4′-pyridyl)-imidazole]), JNK (SP600125 [anthra[1-9-cd]pyrazol-6(2H)-one]), and MEK1/2 (U0126) kinases. Results demonstrated that inhibition of PI3K resulted in upregulation of SHP mRNA, whereas inhibition of MEK1/2 caused SHP repression. Maximal effects were observed by 2-hour incubation (Fig. 8A). Inhibition of p38 and JNK did not significantly modify SHP mRNA levels in HepG2 (data not shown).

SHP mRNA expression and promoter activity after inhibition of MEK1/2 and PI3K pathways. (A) Time-course analysis of SHP mRNA level in HepG2 cells after treatment with 20 μM LY294002 (PI3K inhibitor), 10 μM U0126 (MEK1/2 inhibitor), or vehicle. (B) HepG2 cells were transfected with the −509 and 509-mut reporter vectors and the next day were exposed to kinase inhibitors for 24 hours. Data represent firefly activities normalized to Renilla luciferase. Bars represent the mean ± S.D. of three to four independent experiments. *P < 0.05 (Mann-Whitney test).
We next investigated the effects of PI3K and MEK1/2 inhibitors on the 509-SHP-reporter construct. Inhibition of PI3K induced, whereas inhibition of MEK1/2 repressed the human SHP promoter (Fig. 8B), which is in agreement with their effects on SHP mRNA. This suggests that these pathways modulate factors that control SHP gene transcription.
MEK1/2 activates ERK1/2 kinases, whereas PI3K represses the GSK3 kinase. Both ERK1/2 and GSK3 can phosphorylate C/EBP factors and modulate C/EBPα activity (Nakajima et al., 1993; Ross et al., 1999, 2004). It is then feasible to think that the effects of these signaling pathways on SHP could be C/EBP dependent. To examine this possibility, we compared the effects of the MEK1/2 and PI3K inhibitors on the wild-type and C/EBP-RE-mut SHP promoters. Results demonstrate that the repression of 509-SHP-LUC by U0126 (MEK/ERK inhibitor) was partially but significantly abrogated in 509-mut-SHP-LUC, whereas the induction of 509-SHP-LUC by LY294002 (PI3K inhibitor, GSK3 activator) was completely abolished in the mutated construct (Fig. 8B). This experimental evidence supports the notion that phosphorylated C/EBP forms (by ERK1/2 and/or GSK3) activate SHP, whereas nonphosphorylated C/EBPs cause SHP repression.
Discussion
One important finding of this study is that several steatotic drugs repressed SHP in human HepG2 and HepaRG cells. However, other steatotic drugs failed to significantly repress SHP in these cell models. There are several possible explanations for this discrepancy. First, not all steatotic drugs share the same mechanism of action: inhibition of β-oxidation, increased TG synthesis, alterations of mitochondria (respiratory chain, membrane potential, oxidative phosphorylation, and mitochondrial DNA), inhibition of lipoprotein synthesis, and secretion, etc. (Begriche et al., 2011). Secondly, only some steatotic drugs induce lipid peroxidation, lipotoxicity, and reactive oxygen species (ROS). Regarding the present study, some steatotic drugs with no effect on SHP induced only a low level of lipid accumulation in HepG2 (i.e., tamoxifen and amiodarone) (Benet et al., 2014), whereas others did not induce ROS (i.e., tianeptine and zidovudine) (Donato et al., 2012). Therefore, a threshold level of lipid accumulation and a particular subsequent toxic mechanism may be required for drug-induced SHP repression.
The drug concentrations selected in this in vitro study were above therapeutic plasma concentrations. However, the ratio of the effective concentration in vitro to the therapeutic peak plasma concentration (Cmax) is considered a risk index that provides an estimation of the safety margin. We found significant effects of steatotic drugs on SHP at ratios of 10- (tetracycline and valproate) and 25-fold (cyclosporine A and doxycycline) Cmax. These ratios are considered low safety margins (O’Brien et al., 2006; Xu et al., 2008), and suggest these four steatotic drugs are at risk of having negative effects on SHP under therapeutic doses in vivo.
These observations prompted us to study the regulation of SHP in different mouse modes of fatty liver (iatrogenic and NAFLD/metabolic) and found that SHP was repressed in tetracycline-treated rat livers and MCD diet and GNMT−/− mice livers, but was not repressed in the MAT1A−/− model. One major difference among these NAFLD models is that the livers of GNMT−/− and MCD diet mice show early symptoms of severe disease with fibrosis and even HCC in GNMT−/− (Anstee and Goldin, 2006; Martinez-Chantar et al., 2008; Varela-Rey et al., 2010; Itagaki et al., 2013), whereas MAT1A−/− express a less severe phenotype with delayed onset and no fibrosis or HCC (Lu et al., 2001; Martinez-Chantar et al., 2002).
In a previous study, Huang et al. (2007) reported that SHP is upregulated in fatty livers of genetic (ob/ob and KKAy) and dietary (high sucrose and high fat) mouse models. This apparent contradiction could also be due to differences in the disease phenotype of the models. Leptin-deficient (ob/ob) and KKAy mice are similar models of hyperphagia, type 2 diabetes, and obesity that do not develop steatohepatitis and are resistant to fibrosis. Similarly, HFD mice develop obesity, hyperinsulinemia, and hyperglycemia. However, severe fibrosis and HCC do not appear even after prolonged HFD (Takahashi et al., 2012; Imajo et al., 2013). On the contrary, GNMT−/− and MCD mice develop steatohepatitis with fibrosis, but without obesity and insulin resistance (Varela-Rey et al., 2010; Takahashi et al., 2012; Imajo et al., 2013). Moreover, the MCD diet induces greater ROS production, mitochondrial DNA damage, and apoptotic cell death than many other diet models of NAFLD (Gao et al., 2004; Anstee and Goldin, 2006).
Therefore, we can speculate that SHP is upregulated at the onset or in less severe NAFLD, but the progression of the disease, with inflammation, liver injury, and fibrosis, triggers signals that inhibit SHP expression. In agreement with this view, SHP expression is significantly decreased in human NASH cirrhotic livers (Zhang et al., 2014).
Overall, our results suggest that independently of their iatrogenic or metabolic origin, hepatosteatosis with associated liver injury triggers SHP downregulation. The potential clinical consequence of SHP repression could be extrapolated from studies with SHP−/− mice, which showed increased sensitivity to liver damage and liver fibrosis (Park et al., 2008; Chanda et al., 2009; Zhang et al., 2014) as well as hepatocyte hyperproliferation and HCC (Zhang et al., 2008). Moreover, the repression of SHP could affect the crosstalk of SHP with other nuclear receptors, particularly PXR, a well characterized lipogenic factor (Lee et al., 2008; Hoekstra et al., 2009). SHP suppresses PXR, which is also considered a master regulator of drug/xenobiotic metabolism (Krausova et al., 2011). On the contrary, PXR activation by rifampicin inhibits SHP (Li and Chiang, 2006). Therefore, the repression of SHP by steatotic drugs and in advanced NAFLD could lead to higher PXR activity and enhanced lipogenesis, which would also contribute to disease severity and progression.
However, the potential negative consequences mentioned above need to be demonstrated in future studies, as the data presented here do not demonstrate a causative link between the downregulation of SHP and the severity and progression of the disease. Our study has additional limitations because the different animal models are very complex systems and presumably many genes (not only SHP) show altered expression.
Because of the critical role of SHP in liver metabolism and disease, many different studies have addressed the regulation of the SHP gene. It has been found that SHP is activated by many transcription factors in response to multiple signaling pathways (Zhang et al., 2011; Garruti et al., 2012). In the present study, we demonstrate that RXRα [(+)-9-cis-retinoic acid] also induces SHP. This result is in line with the induction of SHP by all-transretinoic acid (Cai et al., 2010), and suggests that SHP can be regulated by retinoids via RXRα homodimers or RXRα-RAR heterodimers. Nevertheless, increased SHP expression by RXRα could also be mediated by other heterodimers, such as RXRα-FXR, RXRα-LXRα, or RXRα-PPARγ (Kim et al., 2007).
In the present study, we have also uncovered two new SHP repressors: FOXA1 and C/EBPα. However, comparative analysis showed that C/EBPα was faster and more effective and dominantly repressed SHP induction by RXRα. Further analyses demonstrated binding of C/EBPα to a negative response element at −473 bp in the human SHP gene. Mutation of this element evidenced its relevance for the regulation of SHP by C/EBPα, steatotic drugs, and stress-signaling pathways. Based on these results, it is feasible to suggest that lipid-dependent insults triggered by iatrogenic or metabolic steatosis can alter or modulate signaling pathways that modify C/EBPα phosphorylation and consequently cause SHP repression.
Our results demonstrate that inhibition of MEK1/2 kinases caused a very significant repression of both SHP mRNA and SHP-LUC activity. These results suggest that conditions causing MEK/ERK inhibition will cause SHP downregulation. ERK1/2 inhibition has already been reported in several models of NAFLD. For instance, mice on an MCD diet showed a remarkable suppression of hepatic ERK1/2 activation (Wang et al., 2010a). On the opposite side, an increase in the level of phosphorylated ERK1/2 improved liver steatosis (Aghazadeh and Yazdanparast, 2010). Chronic alcohol also caused a significant suppression of hepatic ERK1/2 activation and triggered fatty liver (Wang et al., 2010b). We hypothesize that one of the MEK/ERK targets could be C/EBPα or C/EBPβ. ERK can phosphorylate C/EBPα at Ser 21 (Ross et al., 2004). Moreover, phosphorylation of C/EBPβ at Thr235 by ERK2 is a key determinant of its capacity for transactivation (Nakajima et al., 1993). Based on these studies, we suggest that phosphorylated C/EBPs would activate SHP, whereas dephosphorylated forms would not activate or repress SHP.
Inhibition of PI3K induced both SHP mRNA and SHP-LUC activity in HepG2 cells. These results suggest that conditions causing PI3K activation will cause SHP repression. It has been shown that mitochondrial ROS induces hepatocyte steatosis by upregulating the PI3K pathway (Kohli et al., 2007) and that hepatocytes exposed to the MCD medium developed significant and progressive steatosis along with activation of PI3K/Akt (Sahai et al., 2006). Moreover, PI3K (p85) was significantly higher in NAFLD patients (Xu et al., 2011). Activation of PI3K leads to GSK3 inhibition, and GSK3 phosphorylates C/EBPα on an evolutionarily conserved consensus site (Thr222, Thr226, and Ser230) (Ross et al., 1999). Moreover, activation of the PI3K/Akt pathway in proliferating hepatocytes and liver tumor/hepatoma cells induced the protein phosphatase 2A-mediated dephosphorylation of C/EBPα on Ser193, leading to a failure of C/EBPα to cause growth arrest (Wang et al., 2004). Finally, insulin, a major activator of the PI3K pathway, causes a very significant repression of SHP in primary cultured hepatocytes (Van Rooyen et al., 2011). Therefore, the inhibitory activity of PI3K on SHP could be mediated through the reciprocal repression of GSK3 and/or activation of protein phosphatases, leading to both dephosphorylation of C/EBPα, which in turn would cause SHP repression. These results again suggest that dephosphorylated C/EBPα is the SHP repressor. Adenovirus-mediated overexpression of C/EBPα would mainly generate a nonphosphorylated (repressing) C/EBPα protein.
C/EBP proteins are a well known recipient of extracellular signals resulting in multiple phosphorylations, acetylations, and SUMOylations (Nerlov, 2008). Up to eight different phosphorylation sites have been described in C/EBPα (Tsukada et al., 2011), and therefore the investigation of the precise phosphorylated forms involved in SHP regulation is complex and out of the scope of this paper.
In summary, we hypothesize that the repression of SHP by C/EBPα is likely dependent on dephosphorylated C/EBPα. A similar scenario has been demonstrated for the hepatic phosphoenolpyruvate carboxykinase gene, which is activated (Qiao et al., 2006) or repressed (Pedersen et al., 2007) by C/EBPα, depending on its phosphorylation status. Our data also support that harmful xenobiotics or metabolic conditions causing inhibition of MEK1/2 or activation of PI3K pathways will trigger a C/EBPα-dependent repression of SHP, which could favor the progression and severity of NAFLD.
Authorship Contributions
Participated in research design: Benet, Castell, Jover.
Conducted experiments: Benet, Guzmán, Pisonero-Vaquero, García-Mediavilla, Sánchez-Campos, Martínez-Chantar.
Contributed new reagents or analytic tools: Donato, Castell, Jover.
Performed data analysis: Benet, Jover.
Wrote or contributed to the writing of the manuscript: Benet, Jover.
Footnotes
- Received October 8, 2014.
- Accepted January 9, 2015.
This work has been partly supported by Grants PI-13/01470 and PI-11/01588 from Fondo de Investigación Sanitaria (FIS) (Instituto de Salud Carlos III), Grant BFU2013-48141-R from Programa Estatal de Investigación (Ministerio de Economía y Competitividad), Grant LE135U13 from Consejería de Educación (Junta de Castilla y León), and Grant ETORTEK-2012/Sanidad Gobierno Vasco 2013. M.B. and M.V.G.M were recipients of CIBERehd contracts [EHD-10-DOC2 and EHD-24-DOC], respectively. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- BA
- bile acid
- ChIP
- chromatin immunoprecipitation
- CITCO
- 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime
- DMSO
- dimethylsulfoxide
- GAPDH
- glyceraldehyde 3-phosphate dehydrogenase
- GW7647
- 2-[[4-[2-[[(cyclohexylamino)carbonyl](4-cyclohexylbutyl)amino]ethyl]phenyl]thio]-2-methylpropanoic acid
- HCC
- hepatocellular carcinoma
- HFD
- high-fat diet
- LUC
- luciferase
- LY294002
- 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one hydrochloride
- MCD
- methionine and choline deficient
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- nonalcoholic steatohepatitis
- PBGD
- porphobilinogen deaminase
- PCR
- polymerase chain reaction
- RE
- response element
- ROS
- reactive oxygen species
- SB203580
- 4-(4′-fluorophenyl)-2-(4′-methylsulfinylphenyl)-5-(4′-pyridyl)-imidazole
- SHP
- small heterodimer partner
- SP600125
- anthra[1-9-cd]pyrazol-6(2H)-one
- TG
- triglyceride
- U0126
- 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio]butadiene
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}