


Visual Overview

Abstract
Pancreatic beta cells, unique cells that secrete insulin in response to an increase in glucose levels, play a significant role in glucose homeostasis. Glucose-stimulated insulin secretion (GSIS) in pancreatic beta cells has been extensively explored. In this mechanism, glucose enters the cells and subsequently the metabolic cycle. During this process, the ATP/ADP ratio increases, leading to ATP-sensitive potassium (KATP) channel closure, which initiates depolarization that is also dependent on the activity of TRP nonselective ion channels. Depolarization leads to the opening of voltage-gated Na+ channels (Nav) and subsequently voltage-dependent Ca2+ channels (Cav). The increase in intracellular Ca2+ triggers the exocytosis of insulin-containing vesicles. Thus, electrical activity of pancreatic beta cells plays a central role in GSIS. Moreover, many growth factors, incretins, neurotransmitters, and hormones can modulate GSIS, and the channels that participate in GSIS are highly regulated. In this review, we focus on the principal ionic channels (KATP, Nav, and Cav channels) involved in GSIS and how classic and new proteins, hormones, and drugs regulate it. Moreover, we also discuss advances on how metabolic disorders such as metabolic syndrome and diabetes mellitus change channel activity leading to changes in insulin secretion.
Introduction
Pancreatic beta cells, which act as glucose sensors, secrete insulin in response to elevated blood glucose levels. Insulin is an anabolic hormone that regulates the storage of nutrients in the liver, muscle, and adipose tissue and regulates the glucose uptake in muscle and adipose tissue. In this way, beta-cell glucose sensing and response are essential for life in mammals (Hiriart et al., 2014).
Several nutrients stimulate insulin secretion, although they must be metabolized, and glucose is the most potent and best-studied secretagogue (“glucose-stimulated insulin secretion,” GSIS). GSIS involves the interaction between metabolic events and ion channels activity. Beta cells also express different types of receptors that modulate GSIS through their activation by hormones, growth factors, neurotransmitters, incretins, and drugs (Hiriart et al., 2014).
Since 1968, Dean and Matthews (1968) demonstrated that high extracellular glucose depolarized the membrane of pancreatic beta cells and increased the action potential firing frequency in mouse islet beta cells. The activity of at least six different channels generated the electrical activity in rodent pancreatic beta cells: 1) ATP-sensitive potassium channels (KATP), which determine the resting potential and link glucose metabolism to electrical activity; 2) transient receptor potential channels (TRPs), which generate nonselective cationic currents; 3) voltage-gated sodium channels (Nav); 4) low- and high-voltage-activated calcium channels (Cav) that trigger action potentials to raise cytoplasmic calcium concentration and insulin secretion; 5) voltage-dependent potassium channels (Kv); and 6) calcium-sensitive voltage-dependent potassium channels that repolarize the membrane potential stopping insulin secretion.
Beta cells from different mammals express specific combinations of channels (Hiriart and Aguilar-Bryan, 2008; Drews et al., 2010; Hiriart et al., 2014). Despite the fact that the potential role of each type of ionic channel on insulin secretion has been discussed in the literature, there is agreement that they all participate in this process. Thus, in this review, we will focus only on extensively studied channels, such as KATP, Nav, and Cav. We examine recent information about the structure- function relationship of the channels, their role in insulin secretion, and their regulation by hormones, drugs, enzymes, receptors, and exocytotic proteins. Furthermore, we will consider some mutations localized in critical structural determinants and how they could impair channel gating, drug binding, or translocation to the membrane, which could contribute to different pathologies.
Glucose-Stimulated Insulin Secretion
The coupling between the rise in extracellular glucose concentration and insulin secretion involves metabolic and ionic events (Hiriart and Aguilar-Bryan, 2008). In the fasting state, the plasma glucose level is 4–5 mM and beta cells are electrically silent at their resting potential. Metabolic events start when glucose levels increase above 7 mM. Glucose enters the cells through glucose transporters, GLUT2 (Km = 11 mM) in rodents or Type-1 glucose transporter (Km = 6 mM) in humans (McCulloch et al., 2011). Glucose is then phosphorylated to glucose-6 phosphate by glucokinase, an essential step in glycolysis. Phosphorylated glucose enters the glycolytic pathway in the cytoplasm, the Krebs cycle, and oxidative phosphorylation in the mitochondria leading to a rise in intracellular ATP/ADP ratio (Hiriart and Aguilar-Bryan, 2008; Drews et al., 2010; Hiriart et al., 2014).
ATP inhibits KATP channel activity and reduces K+ efflux. In this condition, the basal activity of TRP nonselective cationic channels produces a slow depolarization until a threshold membrane potential is reached that increases the open probability of Na+ and T-type calcium channel, which further depolarizes the membrane. When the membrane potential reaches approximately −20 mV, high-voltage-activated calcium channels such as P/Q-type, N-type, and L-type calcium channels increase their conductance and raise intracellular calcium levels, leading to a fast depolarization and firing of superimposed action potentials. This triggers the exocytosis of insulin-containing granules (Hiriart and Aguilar-Bryan, 2008; Drews et al., 2010).
Finally, the activity of the delayed rectifier voltage-dependent potassium channels (Kv) and calcium-sensitive voltage-dependent potassium channels repolarize the membrane, acting as a brake for GSIS (Yang et al., 2014a).
High glucose concentration increases the cytosolic Ca2+, ATP, cAMP levels and triggers insulin secretion in a pulsatile manner, synchronized with calcium influxes during two phases (Jewell et al., 2010). The first phase occurs during 5–10 minutes after beta-cell stimulation and involves the exocytosis of plasma membrane-predocked granules, termed the readily releasable pool. The second phase is less robust and is sustained until the glucose stimulation stops. It has been proposed that it may involve the release of a deeper granule pool from within the cell named granule storage pool, which presumably replenishes the readily releasable pool (Barg et al., 2002; Jewell et al., 2010).
The molecular mechanism of exocytosis is partly understood and is mediated by soluble N-ethylmaleimide-sensible factor attachment protein receptors (SNAREs). SNARE proteins such as sintaxyn-1A and SNAP25/23 are located at the cell membrane, whereas vesicle-associated membrane protein or synaptobrevin is anchored to the granule membrane. Interaction among SNARE proteins and other regulatory proteins such as synaptotagmin, Munc 18-1, Munc 13-1, and GTPase and Rab3A allows the docking, tethering, priming, and fusion to the membrane of insulin-containing granules (Kasai et al., 2010).
ATP-sensitive Potassium Channels
ATP-sensitive potassium KATP channels are considered to be a metabolic sensor because they couple the metabolic status of the cell to the membrane potential and its electrical activity (Seino, 2003). KATP channels exist in several tissues, including brain, pancreas, and smooth and skeletal muscles, and their physiologic role has been mostly characterized in pancreatic beta cells. As mentioned above, the resting potential in beta cells is principally determined by KATP channels (Clark and Proks, 2010). In the absence of glucose, beta cells are electrically silent because the KATP activity is high (3 nS) and K+ efflux is maintained. In this condition, voltage-dependent channels are closed, and insulin secretion occurs only at basal levels. When plasma glucose levels increase above 6 mM, KATP channel activity is reduced by more than 75%, allowing the depolarization of the cell and increasing insulin secretion (Rorsman et al., 2011, 2014).
Molecular Structure of KATP Channel and SUR1 Nucleotide Binding Domains
The molecular structure of KATP channels consists of hetero-octameric complexes formed by four pore-forming inward rectifier potassium channel (Kir6.x) subunits and four sulfonylurea receptor (SURx) subunits (Aguilar-Bryan et al., 1995). There are two isoforms of inward rectifier potassium channel Kir6.1 and Kir6.2 (Clement et al., 1997). The sulfonylurea receptor is an enzymatic member of the ABC (ATP-binding cassette) superfamily, which uses the energy of ATP binding and hydrolysis to transport substrates across the cell membrane. This receptor has two nucleotide binding sites (Wilkens, 2015).
There are three isoforms of the sulfonylurea receptor: SUR1, SUR2A, and SUR2B (Devaraneni et al., 2015). The sulfonylurea receptor is a regulatory subunit because it: 1) confers sensitivity to Mg-nucleotides, 2) is activated by K+ channel openers such as diazoxide and pinacidil, and 3) is inhibited by sulfonylureas such as glibenclamide and tolbutamide (Aguilar-Bryan and Bryan, 1999). In addition, SUR1 increases the open probability of Kir6.2 via phosphatidylinositol-4, 5-biphosphate (PIP2) (Song and Ashcroft, 2001) and may increase the sensitivity to ATP (Tucker et al., 1997).
The combination of Kir6.2 (encoded by KCNJ11) and SUR1 (encoded by ABCC8) subunits form the KATP channel in beta cells (Fig. 1), and this combination is also present in alpha and delta cells from the islet (Clark and Proks, 2010; Ashcroft and Rorsman, 2013). Kir6.2 is a member of the inward rectifier potassium channels superfamily formed by two transmembrane domains, M1 and M2. These domains are linked by a sequence of amino acids that partially enters the membrane to form the pore (loop P) and a signature sequence of glycine-phenylalanine-glycine that corresponds to the K+ selectivity filter (see Fig. 1) (Xie et al., 2007). Furthermore, the N and C termini are intracellular in Kir6.2 channels and are structurally important in the regulation by ATP and sulfonylureas.
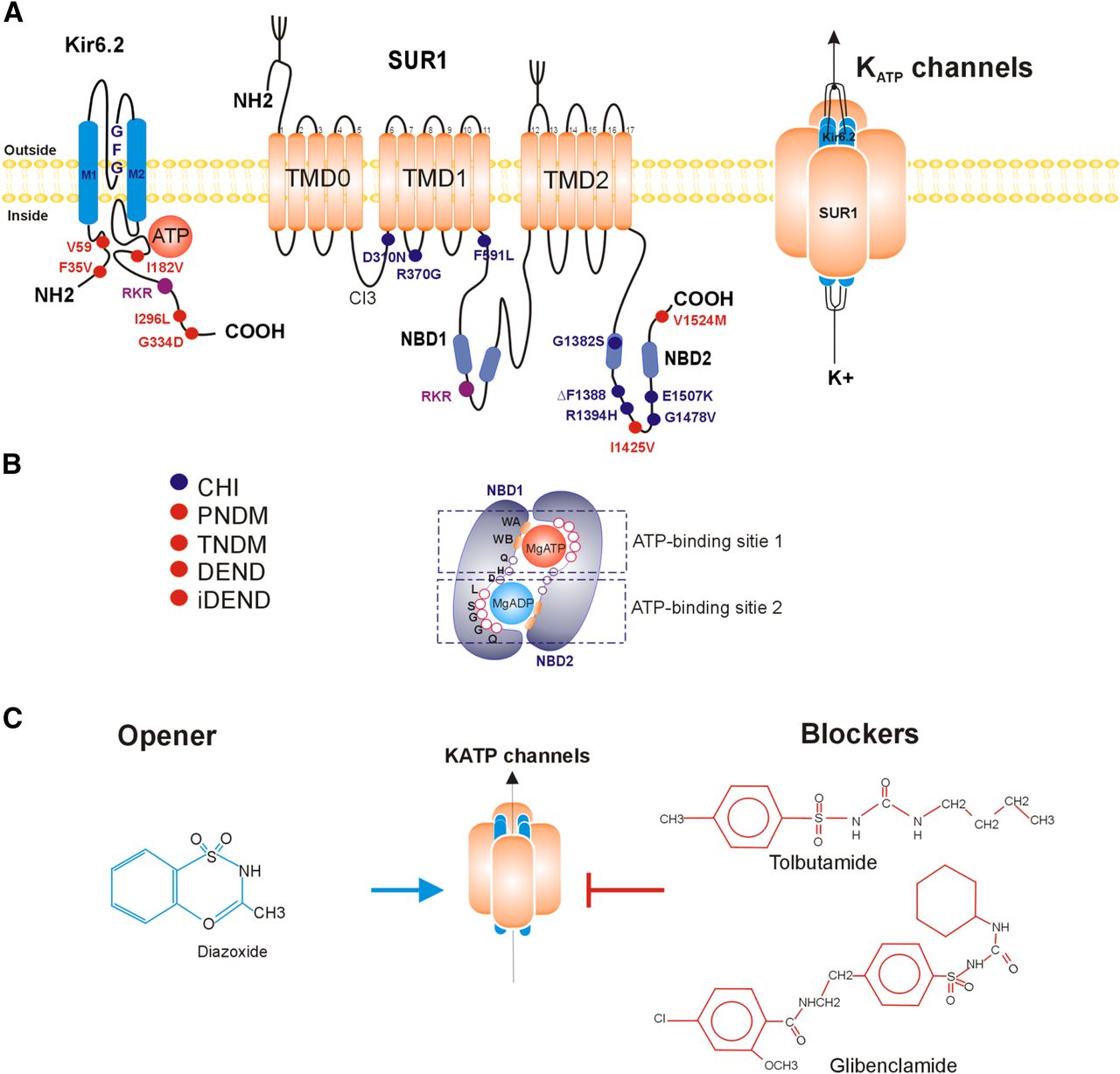
Pancreatic KATP channel structure: (A) Membrane topology of Kir6.2 channel and sulfonylurea receptor (SUR1) subunits. Functional KATP (right) is a 4:4 octamer. Transmembrane domains (TMD0, TMD1, and TMD2): Residues marked in red are associated with neonatal diabetes mellitus (NDM) and those marked in purple are associated with congenital hyperinsulinism (CHI). Retention motif (RKR) is marked in brown. (B) Cooperativity between nucleotide binding domains (NBD1 and NBD2) to form ATP-binding sites, walker A and B motif, loop of glutamine (loop-Q), histidine (loop-H) and aspartate (loop-D). NBS1 binds ATP or MgATP and NBS2 binds MgADP or MgATP. ABC signature sequence (LSGGQ): (C) Pharmacologic modulators of pancreatic KATP channels.
The SUR1 receptor is formed by two transmembrane domains (TMD1 and TMD2), each one with six transmembrane helices, and two cytosolic H (NBD1 and NBD2) subunits between the TMDs (Aguilar-Bryan and Bryan, 1999). Moreover, the N-terminal domain (TMD0) has five transmembrane helices connected to the TMD1 through a long cytosolic loop known as the L0 or CL3 linker (Fig. 1) (Devaraneni et al., 2015).
Both NBDs have a highly conserved structure, and each one is formed by a catalytic and an α-helical subdomain. The catalytic subdomain contains Walker A and B conserved motifs that play important roles in ATP binding and hydrolysis. The A motif (P-loop) contains a highly conserved lysine residue that interacts with the β and γ phosphates of the nucleotides (Devaraneni et al., 2015).
The B motif contains an aspartate residue that coordinates Mg2+ binding (ter Beek et al., 2014) and a basic glutamate residue that binds the γ-phosphate of ATP through a water molecule (Beis, 2015). Downstream the Walker B motif, the catalytic base consists on a glutamate residue that both hydrolyzes ATP and forms a dyad with the H-loop histidine of the antiparallel NBD (Jones and George, 2012). In addition, the catalytic domain contains functionally relevant residues, such as aspartate (D-loop), that allow the formation of the hydrolytic-competent state and enable the ATP hydrolysis.
The α-helical domain contains both the ABC signature sequence (LSGGQ), also known as C-loop, and a Q-loop (Beis, 2015). Both NBDs are grouped in an antiparallel manner to form two ATP binding sites (NBS). Thus, Walker A and B of NBD1 and the signature sequence of NBD2 form the first ATP-binding site, where nucleotides are located at the central part of the sandwich (Fig. 1). While in the second binding site, A and B motifs of NBD2 are associated with the signature sequence of NBD1(Vedovato et al., 2015).
Pharmacology of KATP Channels in Pancreatic Beta Cells
Nucleotide Regulation of KATP Channels.
The hallmark of KATP channels is the fine-tuned regulation by intracellular ATP that binds to the Kir6.2 channel and triggers its closure. Inversely, the Mg-ADP or Mg-ATP binding to the SUR1 promotes the opening of the KATP channels. The inhibition by ATP is the most predominant characteristic of KATP channels. The administration of ATP, with or without Mg2+, inhibits the KATP channels in excised patches with a half maximally inhibiting concentration (IC50) in the range of 5 to 25 µM (Tarasov et al., 2006; Schulze et al., 2007). Interestingly the intracellular ATP concentration is in a millimolar range (3–5 mM). This observation suggests that in intact cells, KATP channels should always be closed.
However, the fact that KATP channels are open during the resting potential could be due to several possible reasons: i) cytosolic proteins that regulate the ATP-sensitivity and the KATP channels are loosely bound in inside-out patches; ii) it is possible that ATP decreases with the activity of membrane-bound ATPases (like Ca2+/K+ or Na+/K+), building up an ATP gradient from below the membrane to the cytosol; iii) also, several studies have shown that MgADP opens KATP channels previously inhibited by ATP in inside-out patches, suggesting that the ATP/ADP ratio is more relevant than ATP alone to regulate the KATP channels (Aguilar-Bryan and Bryan, 1999; Drews et al., 2010). Furthermore, the ATP has a greater affinity to Mg2+ than to other cations; therefore, the ATP within the cells is attached to Mg2+ (Wilson and Chin, 1991). The ATP-inhibition of KATP channels occur through the Kir6.2 channel. Experimental data have shown that a truncated version of Kir6.2ΔC36 reaches the plasma membrane and lacks SUR1 interaction. In addition, these channels are inhibited by ATP, but with a lower affinity (Tucker et al., 1997).
The ATP binding is mediated by some residues such as the Arg50 in the N-terminal domain and the Lys185, Arg192, Arg201, and Gly334 in the carboxyl-terminal domain. Thus, both the N- and C-terminal domains of the Kir6.2 channel are involved in the ATP binding (Matsuo et al., 2005). Moreover, mutations at Arg192 and Arg201 decrease the sensitivity to ATP, ADP, and AMP, suggesting that they interact with their common α phosphate. On the other hand, mutations at Lys185 specifically diminish the sensitivity to ATP and ADP, due to an impaired interaction with their common β phosphate (Riedel and Light, 2005). The KATP channels have four ATP-binding sites, one in each Kir6.2 monomer; however, only one ATP molecule is sufficient to inhibit the channel (Markworth et al., 2000).
ATP hydrolysis is the canonical property of ABC proteins. The catalytic cycle starts with the “apo” ground state followed by the binding interaction of NBDs with the TMDs. The binding of each NBD to MgATP molecules allow their dimerization and a conformational change of the transmembrane domains. Finally, the ATP hydrolysis produces the dissociation of NBDs resetting the transporter to the ground state to start a new cycle (Wilkens, 2015).
The ATPase activity of SUR1 drives discrete conformational changes that modulate the KATP channels gating. Thus, the catalytic cycle of SUR1 in a prehydrolytic state favors the channel closing, whereas the posthydrolytic state promotes the channel opening (Zingman et al., 2001).
The SUR1 receptor contains a consensus (NBS2) and a degenerated (NBS1) ATP binding site. NBS1 binds ATP, but does not hydrolyze it (Inagaki et al., 1995; Matsuo et al., 2005). Although the KATP channels are inhibited by MgATP binding to Kir6.2 subunits, the channels are also activated when MgATP binds to SUR1 (Gribble et al., 1998a). The MgATP and MgADP interaction with NBDs of the SUR1 subunits results in the activation of KATP channels, an increase of the K+ efflux, and a diminished excitability of beta cells. This phenomenon occurs by cooperativity between NBD1 and NBD2 (Yang et al., 2014a).
Recently, the usage of a heterodimeric ABC protein (TM287/288) similar to SUR1 suggested that the cooperativity between NBDs may be mediated via two D-loops in the dimer interface. The D-loop at NBS1 ties the NBDs together even in the absence of nucleotides, while the D-loop in NBS2 is flexible (Hohl et al., 2014). ATP hydrolysis is necessary to switch SUR1 into a conformation that antagonizes nucleotide binding to the Kir6.2 pore and stimulates channel opening.
The ATP hydrolysis occurs in NBD1, which is a high-affinity site for ATP or MgATP, whereas NBD2 has low-affinity to MgATP or MgADP and intrinsic ATPase activity (Drews et al., 2010; Yang et al., 2014a). However, it has been demonstrated that MgATP does not activate the channel directly, but rather it must first be hydrolyzed to MgADP (Zingman et al., 2001). However, recent work demonstrated that nonhydrolyzable ATP analogs such as MgAMP-PNP [adenosine 5′-(β, γ-imino) triphosphate] and MgAMP-PCP [adenosine 5′-(β, γ, methylene-triphosphate)] fail to activate KATP channels because of selective binding to NBD1 that prevents NBD dimerization and the SUR1 conformational switching, rather than by enzymatic activity (Ortiz et al., 2013).
Regulation by Sulfonylureas.
Sulfonylureas are a group of drugs that decrease blood glucose levels, and for a long time, these compounds were the main treatment of type 2 diabetes mellitus. The sulfonylureas (SUs) were discovered serendipitously in the 1940s when hypoglycemia symptoms were observed after sulfonamide antibiotic treatment (Loubatieres, 1957). The first generation of SUs, including tolbutamide, acetohexamide, and chlorpropamide, was used in the 1960s (Deacon and Lebovitz, 2015). The second generation of SUs such as glibenclamide (glyburide), glipizide, gliclazide, and glimepiride appeared 10 years later. These drugs were produced by replacing an aliphatic side chain with a cyclohexyl group, which increases its molecular complexity and molecular affinity to the SUR1 subunit. With this changes, their adverse effects profile was also improved (Deacon and Lebovitz, 2015).
Sulfonylureas were used as an oral antidiabetic for 50 years, but their action mechanism was unknown. In the 1980, long before the cloning of SUR1 in 1995, the high-affinity sites for sulfonylureas were localized in the membrane of beta cells (Gaines et al., 1988; Aguilar-Bryan et al., 1995). According to the previously described model for the SUs and SUR interaction, there are two binding sites located on the eighth cytosolic loop between TM segments 15 and 16 and the third cytosolic loop between TM segments 5 and 6 (Ashfield et al., 1999), and it has been proposed that glibenclamide and glimepiride bind to both sites in the SUR1 receptor (Mikhailov et al., 2001), and recently suggested that the Kir6.2 N-terminus participates in this binding (Vila-Carriles et al., 2007; Kühner et al., 2012). On the other hand, tolbutamide binds only to the high-affinity site of SUR1 in the intracellular loop between TM 15 and 16. The substitution of tyrosine for serine 1237, localized between TM 15 and 16, blunts the channel inhibition by tolbutamide (Ashfield et al., 1999). Furthermore, it was demonstrated that tolbutamide also binds to a low-affinity site in the Kir6.2 channel (Gribble et al., 1997). SUs bind with different affinity to the three isoforms of SUR receptor (SUR1, SUR2A, and SUR2B), and, interestingly, the SUR1 receptor that forms the pancreatic KATP channels has the highest affinity to sulfonylureas with a dissociation constant in the nanomolar range (Aguilar-Bryan and Bryan, 1999).
KATP channels increase their opening time in response to several compounds known as potassium channel openers (see Fig. 1). The principal potassium channel openers are pinacidil, cromakalin, and diazoxide; the SUR1 receptor responds better to diazoxide than SUR2A cardiac receptor, but they do not respond to pinacidil (Aguilar-Bryan and Bryan, 1999).
KATP trafficking regulation.
Nucleotides are not the only protein that regulates KATP channels activity; a higher level of complexity in the structure-function relationship is added due to their regulation by other proteins. Particular attention has been paid to proteins that regulate the trafficking of KATP channels to the membrane. Biogenesis and assembly of KATP channels occur in the endoplasmic reticulum (ER); however, when Kir6.2 or SUR1 subunits are expressed alone, they are trapped in the ER and degraded, suggesting that assembled complexes are required for the forward trafficking and the surface expression (Zerangue et al., 1999). Both subunits are needed to form a fully functional KATP channel, and these subunits are assembled in the ER. The assembling occurs through a tripeptide (RKR) retention motif within the C terminus of Kir6.2 and between TMD1 and TMD2 of SUR1 (Tucker et al., 1997; Zerangue et al., 1999).
In 2004, an interaction between KATP channels and syntaxin 1A was demonstrated (Syn-1A; t-SNARE protein). Also, it was proposed that syntaxin 1A (Syn-1A) interacts with NBDs of SUR1 and inhibits the KATP channel activity (Pasyk et al., 2004). It has been observed that the levels of Syn-1A and cognate SNARE proteins are reduced in diabetic human and rodent beta cells (Nagamatsu et al., 1999; Ostenson et al., 2006). In addition, overexpression and downregulation of Syn-1A led to a decrease and an increase in the surface expression of KATP channels, respectively. It was suggested that the lower expression of KATP channels at the membrane is caused by an accelerated endocytosis and a decreased biogenesis, as well as altered traffic to the membrane of these channels. The authors even proposed that physiologic and pathologic changes in Syn-1A expression may control the KATP channel expression, thus modulating insulin secretion (Chen et al., 2011).
Leptin, a product of the LEP/ob gene, is an obesity-related hormone, which is predominantly secreted by adipocytes in direct proportion to body fat mass. Leptin inhibits insulin secretion by increasing the cell-surface expression and the open probability of KATP channels, resulting in a more hyperpolarized membrane potential (Holz et al., 2013). Leptin also induces an increased surface expression of Kv2.1 channels, leading to a rapid repolarization of membrane potential (Park et al., 2013b). These studies suggest that leptin exerts a coordinated trafficking regulation of KATP and Kv2.1 channels to inhibit insulin secretion (Wu et al., 2015). Furthermore, leptin activates the AMP-activated protein kinase (AMPK) through phosphorylation by the Ca2+/calmodulin-dependent protein kinase kinase beta and increases KATP channels trafficking (Park et al., 2013a).
The translocation of KATP channels to the plasma membrane could be promoted by AMPK activation, which regulates the actin cytoskeleton through the activation of Rac GTPase and phosphorylation of myosin regulatory light chain in pancreatic beta cells. The trafficking of K+ channels to cell surface could also be considered as a novel autocrine negative feedback mechanism for insulin secretion (Han et al., 2015). In this way in MIN6 cells, high glucose and exogenous insulin treatment increase KATP channels trafficking to the surface, stabilizing the membrane potential and avoiding insulin secretion. This increase in cell-surface KATP channels is mediated by vesicle-associated membrane protein 2 via the phosphoinositide 3 kinase (Xu et al., 2015). The hormonal and metabolic regulation of KATP channel trafficking constitutes an emerging field of potentially high significance to beta-cell physiology.
Pancreatic KATP channelopathies
Functional and structural defects in KATP channels impair insulin secretion, leading to the onset of diseases. In 1981, it was first described that glucose failed to promote insulin secretion in a concentration-dependent manner in isolated tissue from a child with hyperinsulinemia-induced hypoglycemia (Aynsley-Green, 1981; Dunne, 2000). The persistent hyperinsulinemia hypoglycemia of infancy, also named congenital hyperinsulinism (CHI), is a complex disorder composed of clinical, morphologic, and genetic changes (Shah et al., 2014). Excessive insulin secretion characterizes CHI, despite low blood glucose levels in the early neonatal period. This disorder has an incidence of around 1 in 50,000 live births.
Clinically, CHI is classified in mild and severe forms of the disease. The first one appears during the newborn period, but also in infancy and childhood. In the severe form the symptoms develop after the first hours or days after birth. Failure in the diagnosis and treatment of hypoglycemia could lead to severe brain damage and epilepsy (Kapoor et al., 2009).
Histologically, CHI is divided into diffuse, focal, and atypical forms. The focal form is sporadic in inheritance and only affects discrete areas of the pancreas. The diffuse disease is inherited in an autosomal recessive or dominant way. The latter is the most common of CHI and affects the entire pancreas. Finally, the atypical forms may be diffuse along the gland with normal or abnormal islet anatomy (Shah et al., 2014).
The etiology of CHI may be due to rare variants in nine different genes, such as: 1) ABCC8 (SUR1 receptor), 2) KCNJ11 (Kir6.2 channel), 3) GCK (glucokinase), 4) GLUD1 (mitochondrial enzyme glutamate dehydrogenase), 5) SLC16A1 [monocarboxylate transporter 1 (MCT1)], 6) HADH (mitochondrial L-3-hydroxyacyl-coenzyme A dehydrogenase), 7) UCP2 (uncoupling protein 2), and 8 and 9) HNF4A and HNF1A (hepatocyte nuclear factor 4alpha and 1alpha). All of these genes encode for proteins involved in beta-cell insulin secretion (Roženková et al., 2015). The most frequent causes for CHI are variants in KCNJ11 and ABCC8, which account for ∼70% of all cases with more than 200 mutations reported to date, some of them shown in Fig. 1. Both are inherited either by autosomal recessive or dominant form (Remedi and Koster, 2010).
KATP channel variants can also be grouped into two functional classes: those that reduce: i) the cellular membrane expression by affecting either channel biosynthesis, trafficking, assembly; or ii) reduce the intrinsic channel activity (Remedi and Koster, 2010). Genetic variants in ABCC8 and KCNJ11 along with HNF1A and GCK are associated not only with CHI but also with maturity onset of diabetes in young and neonatal diabetes mellitus (NDM), each of them are monogenic forms of glucose-regulation disorders in children.
Neonatal diabetes mellitus (NDM), caused either by Kir6.2 (KCNJ11) or SUR1 (ABCC8) mutations, is a rare disorder that appears during the first 6–9 months of life, with an incidence of 1:300,000 live births. The patients typically have low weight at birth, and only one-third of them develop severe ketoacidosis. NDM can be permanent or transient (TNDM) and requires lifetime glycemic control or until a remission period. Moreover, some patients present a more severe form of NDM with severe developmental delay and epilepsy, whereas other patients show a milder phenotype without epilepsy.
NDM-associated KATP channel mutations cause the opposite phenotype of CHI. Therefore, they have an overactive channel that remains opened, either increasing the KATP activity mediated by Mg nucleotides or altering the intrinsic gating (Edghill et al., 2010; Remedi and Koster, 2010).
Gloyn et al. (2004) reported genetic variants in Kir6.2 as the potential cause of NDM for the first time in 2004. To date, over 30 rare variants in Kir6.2 and approximately 15 in SUR1 have been identified and associated with NDM. These are the most common cause of permanent NDM (approximately 30–50%) and involved in 15% of the TNDM cases (Shimomura, 2009; Shimomura et al., 2009). TNDM also correlates with mutations in hepatocyte nuclear factor-1beta (HNF1b), which can also cause maturity-onset diabetes of the young 5. Furthermore, the majority of cases of TNDM are due to abnormalities of an imprinted locus on chromosome 6q24 that results in the overexpression of a paternally expressed gene (Aguilar-Bryan and Bryan, 2008).
Electrophysiological studies have observed that Kir6.2 variants at Arg50, Ile192, Arg201, and Phe333 residues involved in ATP binding decrease the KATP sensitivity. In addition, variants of Val59, Cys166, Ile197, and Ile296 residues involved in the channel gating impair the open probability of the channel (Proks et al., 2005; Edghill et al., 2010).
SUR1 mutations affect either the Mg-nucleotide-mediated activation or the intrinsic gating. These mutations could be located along the protein length, including many instances of the following amino acid substitutions V1523A, V1524M, I1425V, and R1531A in NBD2 (Edghill et al., 2010).
Mutations and polymorphism in Kir6.2 (KCNJ11) have already been linked to type 2 diabetes mellitus (Gloyn et al., 2003). Several genetic studies consistently demonstrated an association of the E23K polymorphism with a reduced insulin secretion in glucose-tolerant and -intolerant cohorts as a risk allele in the development of type 2 diabetes (Nielsen et al., 2003; Chistiakov et al., 2009; Villareal et al., 2009).
The E23K polymorphism is due to a substitution of a glutamic acid (E) by a lysine (K) at codon 23 of the KCNJ11. This residue substitution is located in the N-terminal domain of the Kir6.2 subunit (Bonfanti et al., 2015).
Electrophysiological recording of reconstituted KATP channels in mammalian expression consistently shows hyperactivity of E23K channels, which has been suggested to increase the intrinsic channel open probability with a decrease in the sensitivity to ATP inhibition (Villareal et al., 2009; Remedi and Koster, 2010) or enhanced activation by long-chain acyl coenzyme A (CoA) esters with minimal effects on ATP sensitivity (Riedel and Light, 2005).
Impairment of the KATP channel activity can alter beta-cell electrical activity and insulin secretion sufficiently to cause diabetes. The Kir6.2-G324R mutation reduces the channel ATP sensitivity in beta cells; however, the difference in ATP inhibition between homozygous and heterozygous patients was remarkably small. Nevertheless, the homozygous patient developed neonatal diabetes, whereas the heterozygous parents were unaffected (Vedovato et al., 2016).
Endogenous Regulation of KATP Channels in the Beta Cell
The regulation of KATP channels by endogenous ligands could be required for the appropriate function under physiologic conditions. ATP/ADP ratio is the principal physiologic regulator of these channels; however, they can also be regulated by lipids, such as long-chain acyl-CoA esters (LC-CoAs), phosphatidylinositol-4, 5-biphosphate (PIP2) (Tarasov et al., 2004), syntaxin-1A (Syn-1A) (Pasyk et al., 2004), and leptin (Gavello et al., 2016).
Acyl CoAs are products of free fatty acid (FFA) esterification by acyl-CoA synthetase. Larsson et al. (1996) demonstrated the activation of KATP channels by LC-CoAs at a physiologic concentration in mice and clonal pancreatic beta cells. LC-CoAs esters are also potent stimulators of KATP channels activity in human beta cells and do not require the presence of Mg2+ or nucleotides (Bränström et al., 2004). The increase in the activity of KATP channels occurs by interaction of LC-CoAs in the carboxyl-terminal domain of Kir6.2 subunit (Gribble et al., 1998b), where the Lys332 residue is a key structural determinant (Bränström et al., 2007). Moreover, the effects of LC-CoAs on the activity of KATP channels depend on both the length and the saturation degree of the acyl chain. Saturated acyl CoAs are the most potent activators of KATP channels, followed by monounsaturated and polyunsaturated acyl CoAs, respectively (Riedel and Light, 2005).
In addition, the KATP channels with E23K (0.6471 allele frequency) and I337V (0.6448 allele frequency) Kir6.2 polymorphisms, which are frequent in Caucasian type 2 diabetes populations, showed significantly increased activity in response to palmitoyl-CoA, compared with wild-type KATP channels. These observations suggest that the diabetogenic effects of E23K and I337V polymorphisms occur through an increased KATP channel activity mediated by high levels of LC-CoAs (Riedel et al., 2003).
To date, there is clear evidence supporting the regulating role of phosphoinositides in the activity of many ion channels (Hille et al., 2015). Fan and Makielski (1997) showed for the first time that the KATP channels are regulated by the phosphatidylinositol 4-5 biphosphate (PIP2). PIP2 is a second messenger that increases the activity of KATP channels. Electrostatic interaction occurs between negative phosphate head groups of PIP2 and positively charged residues in Kir6.2 (Schulze et al., 2003), including Arg54 in N terminus and Arg176, Arg177, and Arg206 in C terminus. All of these residues are localized above the ATP binding site and near to the plasma membrane (Ribalet et al., 2005).
Several studies demonstrated that SUR1 plays an important role in stabilizing the interaction of PIP2 with the Kir6.2 subunit. Recently, a variant (E128K) in the TMD0 of SUR1 showed diminished the open probability and the responses to PIP2 (Pratt et al., 2011), raising the possibility that TMD0 of SUR1 modulates the Kir6.2 gating by reducing the response to PIP2. Thus, PIP2 may play an important role in regulating the activity of K+ channels and, therefore, insulin secretion. Furthermore, PIP2 can interact with other proteins that also regulate the KATP channel activity, and these proteins appear to be involved with the exocytotic machinery [e.g., synaptotagmins, calcium-dependent activator protein for secretion, and syntaxin-1A (Syn-1A)]. Syntaxin-1A is a target protein of SNARE complex that regulates the KATP channel trafficking (Pasyk et al., 2004) and is a potent inhibitor endogenous of KATP channels (Chang et al., 2011). PIP2 directly and indirectly regulates the interaction of syntaxin-1A on SUR1. PIP2 also affects the Syn-1A binding on NBDs of SUR1, albeit PIP2 acts indirectly on syntaxin-1A clusters on the plasma membrane to sequester Syn-1A and reduce its availability and interaction with SUR1 at the plasma membrane (Liang et al., 2014).
Leptin is an adipocyte-derived hormone that regulates food intake, body weight, glucose homeostasis (Friedman and Halaas, 1998), and was recently demonstrated as a regulator of KATP trafficking within beta cells (Holz et al., 2013). Moreover, leptin can activate different ion channels, including KATP channels, both in central neurons and beta cells (Gavello et al., 2016). The mechanism by which leptin activates KATP channels involves phosphatase and tensin homologe inhibition mediated by phosphoinositide 3 kinase. phosphatase and tensin homologe inhibition induces F-actin depolymerization and, consequently, KATP channel opening (Park et al., 2013b; Wu et al., 2015).
The Role of KATP Channels in the Pathophysiology of MS and T2DM
In obesity and type 2 diabetes, acyl CoAs, PIP2, and Syn-1A levels change. Chronic plasma levels of FFA in obese (Golay et al., 1987) and diabetic patients (Reaven et al., 1989) can increase the cytosolic acyl CoAs and KATP activity (Riedel et al., 2003). Long-chain polyunsaturated fatty acids maintain cell membrane fluidity and facilitate the cellular signaling mechanisms. Long periods of high-fat diet, particularly saturated fat and trans fatty acids, alter cell membrane lipid composition, compromising transmembrane insulin receptor signaling and consequently glucose uptake peripheral tissues (Wilcox, 2005). Furthermore, in diabetic human and rodent beta cells, the levels of Syn-1A and cognate SNARE proteins are reduced (Nagamatsu et al., 1999; Ostenson et al., 2006). The increase in adipose mass will elevate plasma leptin levels and this, in turn, will feed back to pancreatic beta cells to inhibit insulin secretion via stimulation of KATP channels (Campfield et al., 1995). This suggests that during pathophysiological conditions such as obesity, insulin resistance, or metabolic syndrome, the regulation of KATP channels could be affected and may contribute to the disease. Recent studies have evaluated the KATP activity during the metabolic syndrome (MS), which is a group of symptoms including central obesity, hyperinsulinemia, insulin resistance, high plasma levels of triglycerides, and high arterial blood pressure that predispose the subject to develop type 2 diabetes mellitus, cardiovascular diseases, and certain types of cancer. In contrast to type 2 diabetes mellitus, where there is an irreversibly reduced insulin secretion and pancreatic beta-cell exhaustion, the MS condition may be reversible, allowing the study of channel modifications before type 2 diabetes mellitus develops. A metabolic syndrome model in Wistar rats, developed by adding 20% sucrose to the drinking water for 6 months, was used to study the KATP channel activity and ATP sensitivity in inside-out patches (Velasco et al., 2012). During the metabolic syndrome, the ATP sensitivity of the KATP channels is higher than in the control. The dissociation constant (Kd) and Hill coefficients for ATP interaction with KATP channels were 18.3 ± 0.01 and 1 ± 0.01 μM for control and 10.1 ± 0.9 and 0.9 ± 0.01 μM for MS cells, respectively. It will be important in the future to investigate the mechanisms of this change, considering that the intracellular environment modulates KATP channels activity and eventually the excess of ATP cause the channel dysfunction in type 2 diabetes mellitus (Velasco et al., 2012).
Voltage-Gated Na+ Channels
Voltage-gated Na+ channels (VGSC) have not been studied as exhaustively as other channels in beta cells. However, it is well known that modulators of these channels can influence insulin secretion (Diaz-Garcia et al., 2010), and several reports suggesting novel roles of Na+ channels in pancreatic beta cells have appeared. Therefore, we will address the roles for these channels in beta- cell physiopathology.
Structural Determinants of VGSC
Voltage-gated Na+ channels (VGSC) are multimeric proteins. The core of the channel is formed by the alpha subunit, which encodes four homologous domains (I–IV), each of them comprising six membrane-spanning alpha helices (S1–S6) (Ahern et al., 2016). Nine members (Nav1.1–1.9 or Scn1–9a) constitute the gene family of VGSC alpha subunits (Catterall et al., 2005). Among them, Nav1.7 and Nav1.3 are widely expressed in pancreatic beta cells of rodents (Vignali et al., 2006; Zhang et al., 2014), whereas Nav1.6 and Nav1.7 are expressed in humans (Braun et al., 2008).
Alpha subunits share a prototypical architecture (Fig. 2A), where the segments S5–S6 of each domain and the extracellular loop that connects them (called P, denoting pore), constitute the hydrophilic pore that allows the movement of ions and determines the particular selectivity of the channel for Na+. Furthermore, the S4 segments serve as voltage sensors due to positively charged residues in these regions (Catterall, 2000a; Ahern et al., 2016). These channels activate in response to membrane depolarization, but their activity decreases during a sustained stimulus, a phenomenon known as inactivation (Fig. 2B) (Ahern et al., 2016). The biophysical properties of alpha subunits, as well as their trafficking, are regulated by the interacting VGSC beta subunits, which comprise 4 genes and 5 members, as one of them presents two splicing variants (O'Malley and Isom, 2015). Among the latter, the predominant beta subunit expressed in pancreatic beta cells is Scn1b (Ernst et al., 2009; Zhang et al., 2014).

Structure and biophysical properties of voltage-gated Na+ channels. (A) The cartoon represents the structure of the pore-forming alpha subunit. In each domain (D-I to D-IV), S4 segments show positive charges and the S5–S6 segments with the P-loop in between are highlighted in red. (B) Schematic representation of the closed, open, and inactivated states of VGSC. At hyperpolarized membrane potentials (i.e., the resting membrane potential of −70 mV in pancreatic beta cells) Nav channels remain in the closed state. A depolarizing stimulus causes the transition to an open state in Nav channels, where Na+ ions permeate through the channel pore. A sustained depolarization induces the inactivation of the channel, represented as a second gate that prevents the flow of ions even when the activation gate is still open. The recovery from inactivation occurs at hyperpolarized potentials. (C) The classic role for Nav channels in beta-cell physiology involves their activation after the high glucose-dependent KATP channel closure, which in turn enhances depolarization and spiking activity, promoting Ca2+ entry and insulin secretion. The scheme also summarizes some of the pharmacological manipulations that potentiate (right panel, sharp arrowhead) or inhibit (let panel, blunted arrowhead) VGSC activity and consequently modulate glucose-stimulated insulin release
Historical Overview of Nav Channels and Insulin Secretion
Direct reports on functional VGSC in insulin-secreting cells first appeared in the 1980s, when a transient inward Na+ current was described in RINm5F cells, which could be blocked by 2.5 µg/ml of tetrodotoxin (TTX) (Rorsman and Trube, 1986). This finding was in contrast to a previous report from the same group on native pancreatic beta cells from NMRI mice, where the authors found no effect of TTX (20 µM) on inward currents elicited by membrane depolarization using the whole cell patch clamp recordings (Rorsman and Trube, 1986). At that time, a plausible explanation was that voltage-activated Na+ currents could reflect a dedifferentiated state of RINm5F cells (Rorsman et al., 1986). However, because the RINm5F cell line was derived from a rat insulinoma, species-related differences could also account for this discrepancy.
The report of a TTX-sensitive Na+ current in isolated pancreatic beta cells (Plant, 1988), helped advance an understanding of previous negative results in beta cells from NMRI mice. This report determined that the voltage for half-maximal steady-state inactivation (V0.5) was more negative than −100 mV at regular (2.6 mM) or low (0.2 mM) Ca2+ concentrations in the bath solution. The latter argued against a potential role of VGSC in insulin secretion in mice, because the resting membrane potential is around −70 mV at low glucose, a potential at which the channels of mice beta cells are expected to be fully inactivated. Consistently, the application of 1.5 µM TTX did not alter the electrical responses when glucose rose from 3 to 20 mM (Plant, 1988).
Almost at the same time, Hiriart and Matteson (1988) demonstrated that this was not the case for rat pancreatic beta cells. They consistently recorded voltage-activated Na+ currents with an almost complete steady-state inactivation around −40 mV, which was rightward shifted, compared with the study of Plant (1988) in mice. Accordingly, the average insulin release of individual beta cells was decreased by incubating with TTX (200 nM), as measured by a reverse hemolytic plaque assay at high glucose (15.6 mM) (Hiriart and Matteson, 1988).
Studies in other species (such as dogs and humans) confirmed the expression of VGSC (Philipson et al., 1993), as well as their contribution to the spiking activity of pancreatic beta cells. Similar results have been obtained in porcine beta cells (Pressel and Misler, 1990), where TTX (1 µM) blocks voltage-gated Na+ currents and action potentials elicited by 12 mM glucose (Silva et al., 2009). Even in mice, where VGSCs were supposed to marginally participate in beta-cell excitability because of inactivation at physiologic membrane potential (Plant, 1988), the genetic deletion of the ancillary beta 1 subunit (Scn1b) impairs the glucose-induced insulin secretion, causing glucose intolerance (Ernst et al., 2009).
Recently, Zhang et al. (2014) examined the role of VGSC in pancreatic beta cells of mice, combining several pharmacological and genetic tools and using state-of-the-art models for the assessment of beta-cell physiology (i.e., intact islet electrophysiology, insulin release measurements in isolated islets, and perfused pancreas). The authors found that although Scn9a/Nav1.7 is the isoform with the highest expression level in this model, the form Scn3a/Nav1.3 is also expressed and significantly contributes to VGSC-mediated currents in 30% of the beta-cell population (Zhang et al., 2014). Accordingly, glucose-induced insulin secretion was impaired in pancreatic islets from Scn3a−/− but not in Scn9a−/− mice (Zhang et al., 2014). These results support that a minor fraction of cells expressing higher relative amounts of Scn3a/Nav1.3 transcripts may exhibit greater excitability and mediate the recruitment through gap junction coupling of other beta cells with less sensitivity to rises in glucose concentration (Zhang et al., 2014).
Indeed, VGSC may be a clinically relevant target, because a decrease in their activity profoundly impacts beta-cell excitability and insulin release in humans. In an early paper, Pressel and Misler (1990) showed that replacing ∼94% of the extracellular Na+ with N-methylglucamine abrogated the action potentials elicited by 10 mM glucose in human pancreatic beta cells. Later, it was demonstrated that 1 µM TTX reduced the amplitude and broadened the width of the action potential of beta cells from human donors (Barnett et al., 1995). As expected, TTX application (as well as Na+ substitution) in the bath solution significantly reduced the voltage-activated Na+ currents in these cells, whereas the toxin impaired insulin secretion when islets were challenged from 3 to 6 mM glucose (Barnett et al., 1995). Braun and coworkers (2008) obtained similar results using TTX, which reduced insulin secretion in 6 and 20 mM glucose of isolated pancreatic islets from human donors. These authors also corroborated that TTX (0.1 µg/ml) decrease the amplitude of voltage spikes in pancreatic beta cells, as well as the voltage-gated Na+ whole cell currents (Braun et al., 2008).
The molecular identity of the channels mediating these currents has also been explored in human pancreatic islets, where the highest mRNA levels corresponded to Nav1.6 and Nav1.7 (Braun et al., 2008). However, only a minor component of the voltage-dependence inactivation of Na+ currents resembled that of beta cells of mice with an inactivation V0.5 of −105 mV (Zhang et al., 2014). The hyperpolarizing shift in the voltage of half-maximal inactivation for Nav1.7 in pancreatic beta cells seems to occur through the interaction with a beta-cell-specific intracellular factor (Zhang et al., 2014), yet to be determined. Changes in the activity of Nav1.7 channels may be linked to beta-cell dysfunction and the susceptibility of developing painful neuropathy, because this channel is also expressed in dorsal root ganglion neurons (Hoeijmakers et al., 2014). Recently, it was shown that Nav1.7 channels are linked to insulin production, because pancreatic islets from Scn9a−/− mice exhibit increased insulin content compared with their wild-type counterparts (Szabat et al., 2015). The latter also offers new perspectives for drugs like carbamazepine, which inhibits the channel and dose dependently increases the mRNA expression of both insulin genes (Ins1 and Ins 2), as well as Pdx1 (a marker of pancreatic beta cells) in isolated islets from mice (Szabat et al., 2015).
Pharmacology of Nav Channels in Pancreatic Beta Cells
From the previous section, it is evident that tetrodotoxin, a potent blocker of voltage-gated Na+ channels, stands out as the first-choice pharmacological agent to use in the study of these proteins. Toxins have been extensively used to explore the structure function of ion channels (Morales-Lazaro et al., 2015) and also because their high potency and selectivity can be harnessed in a more complex pathophysiological context (Diaz-Garcia et al., 2015). The chemical structure and the mechanisms of action of these bioactive molecules are diverse. The toolkit of toxins with proven effects on insulin secretion via VGSC includes compounds isolated from plants, snakes, and scorpions, which modulate the biophysical properties of these channels instead of blocking the pore, as TTX does. The use of toxins like veratridine, which prevents channel inactivation (Ulbricht, 2005) through the neurotoxin interaction site 2 (Cestele and Catterall, 2000), established the presence of VGSC in pancreatic beta cells years before the first direct electrophysiological recordings. An early paper from Donatsch et al. (1977) showed that veratridine (100 µM) increases insulin secretion in isolated islets from albino rats perfused with 5.6 mM glucose to a similar extent to that observed with a rise in glucose to 16.7 mM. Moreover, these authors demonstrated that the secretagogue activity of veratridine could be reversibly inhibited by 3 µM TTX (Donatsch et al., 1977).
The effect of veratridine (200 µM) and TTX (3 µM) on insulin secretion was later corroborated by Pace (1979) in pancreatic islets from Sprague-Dawley rats. The author also observed a veratridine-induced depolarization of pancreatic beta cells in culture, as well as an increase in their spiking activity (Pace, 1979). In agreement with these findings, the ability of veratridine (30 µM) to induce Na+ oscillations in islets cells from ob/ob mice was prevented by TTX (Grapengiesser, 1996). A concentration of 10 µM veratridine was not as effective as a 20-fold higher concentration, but increased insulin secretion when 20 nM of Leiurus toxin was applied simultaneously (Pace and Blaustein, 1979). The scorpion toxin from Leiurus quinquestriatus also delays the inactivation of Na+ currents (Gonoi et al., 1984), which is a common property of alpha-scorpion neurotoxins and certain spiders' toxins interacting with the neurotoxin receptor site 3 in the alpha subunit of VGSC (Cestele and Catterall, 2000). Another alpha-scorpion neurotoxin, TsTx-V (5.6 µg/ml) from Tityus serrulatus has been shown to reversibly increase the electrical activity of pancreatic beta cells from Swiss mice perfused with 11 mM glucose, in a similar fashion as 110 µM veratridine (Goncalves et al., 2003). Moreover, TsTx-V also increases the insulin secretion of pancreatic islets from Wistar albino rats when incubated with 2.8 or 8.3 mM glucose. Crotamine, a toxin from the rattlesnake Crotalus durissus terrificus, also promotes insulin release by preventing VGSC inactivation. An active fraction (F2) obtained from reversed-phase high-performance liquid chromatograph, enhanced insulin secretion of rat islets incubated with 16.7 mM glucose, exhibiting a threefold augment in potency with respect to the native crotamine (Toyama et al., 2000).
The effect of certain drugs has been studied on the late Na+ currents from insulin secreting cells, which increase after long-term exposure to 33 mM glucose, a condition that resembles hyperglycemia (Rizzetto et al., 2015). Notably, the blockers ranolazine (10 µM) and TTX (0.5 µM) decrease the spiking activity of INS-1E cells when stimulated by high glucose or tolbutamide, as well as the tolbutamide-induced Ca2+ transients (Rizzetto et al., 2015). Also, both blockers attenuated the potentiating effect of veratridine (40 µM) on glucose-stimulated insulin secretion (Rizzetto et al., 2015).
Recently, VGSC were identified as regulators of cytokine-induced beta-cell death. This finding arose from the discovery of carbamazepine as a protecting agent against a cocktail of cytotoxic cytokines (tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma) (Yang et al., 2014b). In the study, the authors demonstrated that carbamazepine (0.01–100 µM) dose dependently reduced the cytokine-induced apoptosis in beta cells and expanded these findings to other VGSC blockers such as lidocaine and TTX (Yang et al., 2014b). Interestingly, carbamazepine (0.3–60 µM) also prevents the lipotoxicity of palmitate (0.5 mM) on beta cells (Cnop et al., 2014). These results shed new light on unexpected roles for VGSC in inflammatory and metabolic events that may occur in diabetes mellitus.
Nav Channels Modulation by Endogenous Molecules
The activity of VGSC can also be modulated by endogenous signals, usually acting as downstream effectors of signaling cascades. This phenomenon was observed since the initial studies on VGSC in insulin-secreting cells, because the chronic (48 hours) application of phorbol ester (TPA, a PKC activator) was shown to decrease the Na+ current density in RINm5F cells (Rorsman et al., 1986). Nav channel activity may predispose to beta-cell death after an inflammatory insult (Yang et al., 2014b). On the contrary, nerve growth factor (NGF) favors beta-cell survival and insulin secretion (Vidaltamayo et al., 2002; Navarro-Tableros et al., 2004). It has been demonstrated that rat beta cells exhibited higher Na+ current densities when incubated with 50 ng/ml of 2.5S NGF for 5 days, which could be prevented by either actinomycin D or cycloheximide (Vidaltamayo et al., 2002). The latter suggested that the NGF-mediated increment in Na+ currents may be due to an increased transcription and protein synthesis, which is supported by elevated Nav1.3 transcripts in the NGF-treated cells (Vidaltamayo et al., 2002).
The ability of NGF to enhance the activity of voltage-gated ion channels in pancreatic beta cells is not exclusive to the Nav family, because it has also been demonstrated for Ca2+ channels (Navarro-Tableros et al., 2007a). In both cases, the effects are mediated by TrkA, a high-affinity receptor with intrinsic tyrosine kinase activity (Rosenbaum et al., 2001). Remarkably, the inhibition of protein tyrosine kinases leads to impaired insulin secretion (Persaud et al., 1999), which agrees with the role of these signaling molecules in maintaining proper beta-cell excitability.
Nav channels may also serve as downstream effectors in G-coupled receptor cascades. The muscarinic agonist carbachol (100 nM), for instance, promotes insulin release from isolated rat beta cells at low glucose concentrations (0–5.6 mM), which can be prevented by the coapplication of 319 nM TTX (Hiriart and Ramirez-Medeles, 1993). However, the role of Nav in the muscarinic signaling, at high glucose (20.6 mM), is not straightforward. Strikingly, carbachol decreases insulin secretion in this condition, as well as its inhibitory effect, and is not additive with that of TTX (Hiriart and Ramirez-Medeles, 1993).
A recent work from Salunkhe and coworkers (2015) demonstrated that the microRNA-375 also modulates the activity of Nav channels in insulin-secreting cells. The authors observed that the overexpression of microRNA-375 in INS-1 832/13 cells led to a decrease in the protein levels of Scn3a and Scn3b. Nevertheless, the relevance of this regulation is not completely understood in a more physiologic context, because glucose-induced insulin secretion was not affected by knocking down microRNA-375 (Salunkhe et al., 2015).
Interestingly, Nav channels are also connected to ATP levels in beta cells. It was proposed that Na+ currents may be enhanced by an increase in glucose levels and further ATP production in beta cells (Zou et al., 2013). This idea is supported by whole cell recordings of beta cells in slices from rat pancreas, where concentrations of ATP (2–8 mM) in the recording pipette shift the inactivation curve to more positive potentials and accelerate the recovery from inactivation of VGSC (Zou et al., 2013). In the other direction, the TTX-sensitive depolarizing Na+ influx contributes to the cytosolic Na+ transients (Nita et al., 2014), which, together with Ca2+ transients, propagate to the mitochondria and modulate ATP synthesis. Thus, a glucose-induced uptake of Na+ through the mitochondrial Na+/Ca2+ exchanger promotes Ca2+ extrusion and reverses the divalent cation uptake by the mitochondrial Ca2+ uniporter (Nita et al., 2014).
After four decades of study on VGSC in pancreatic beta cells, we have advanced in the understanding of how Na+ currents contribute to cell depolarization, voltage and Ca2+ oscillations in beta cells, and, finally, in the promotion of glucose-induced insulin secretion (Fig. 2C). However, the regulatory pathways involving VGSC, from endocrine signals to epigenetic mechanisms, are still an incipient field of research. The evidence suggesting that Nav channels interact with metabolism in a bidirectional way is fascinating, because the role of these channels is not yet entirely understood in metabolic diseases such as diabetes or the metabolic syndrome. Indeed, VGSC regulates the physiology of pancreatic beta cells through mechanisms other than just a depolarizing effect, a reason to consider them as potential drug targets for treating related pathologies.
Voltage-Dependent Calcium Channels
Voltage-dependent calcium channels (VDCCs) mediate calcium influx to the beta cells, coupling electrical signaling to Ca2+-dependent protein-protein interactions, and enzymatic responses, playing a crucial role in insulin secretion. Furthermore, Cav channels could also play roles in beta-cell development, maturation, survival, growth, and death (Navarro-Tableros et al., 2007a; Hiriart and Aguilar-Bryan, 2008; Yang et al., 2014a; Zhou et al., 2015).
Molecular Structure of Cav Channels
VDCC consists of heteromeric complexes composed of Cavα1, Cavα2, Cavβ-, Cavδ-, and Cavγ-subunits (Fig. 3). The Cavα1 subunit forms the ion-conducting pore, and Cavα2, Cavβ-, Cavα2δ-, and Cavγ-subunits regulate its conductivity and trafficking (Yang et al., 2014a). The Cavα1 subunit is formed by four homologous domains that consist of six transmembrane segments (S1 to S6), a membrane-associated pore loop (P-loop) between each S5 and S6 segments, three intracellular linkers (LI-II, LII-III, LIII-IV), and N and C termini. As mentioned above, S5 and S6 segments and the P-loop form the Ca2+ conducting pore. The S4 segments work as the primary voltage sensor involved in the inactivation and the reactivation of the channel (Catterall, 2000b). The three-dimensional structure of Cav channels has been described, using electron microscopy and X-ray crystallography. The Cavβ subunit is intracellular; the Cavγ subunit is formed by four transmembrane segments, and the Cavα2δ is extracellular and attached to the membrane through a disulfide linkage to the transmembrane delta subunit (Catterall, 2000b).

Representation of the calcium channel subunit structure, signaling that regulates them, and hormone and drug effects on calcium influx through calcium channels. Greek letters depict each calcium channel subunit, and Roman numbers represent each domain in the alpha-1 subunit. PKA, protein kinase A; PKB, protein kinase B; PKC, protein kinase C; PKG, protein kinase G; SNARE, soluble N-ethylmaleimide sensitive factor attachment protein receptor; Gβγ, G protein subunits beta-gamma; CAMKII, calcium/calmodulin-dependent kinase II; GLP-1, glucagon-like peptide-1; NGF, nerve growth factor; IL-1, interleukin 1; TNF-a, tumor necrosis factor alpha; IFNg, interferon gamma. Green arrows/names represent an increasing effect and red lines/names depict an inhibitory effect on calcium influx through Cav channels. Question mark symbols represent an unknown effect on calcium channels. Magenta arrow describes physical interaction.
We can classify VDCC according to different criteria. A functional characterization considers their open probability, in low-voltage activated (T-type channels) and high voltage-activated (L-, N-, P/Q-, and R-type channels). A second structure-based classification considers the type of α1 subunits being expressed and subdivides Cav channels into three families, namely Cav1, Cav2, and Cav3. They are associated with L-, P/Q-, N-, R-, and T- type Cav currents (Hiriart and Aguilar-Bryan, 2008; Yang et al., 2014a; Zamponi et al., 2015). There are also several subtypes of L channels, depending on their alpha subunit, alpha1C (CACNA1C, Cav1.2) and alpha 1D (CACNA1D, Cav1.3). L-type Cav1.2 and Cav1.3 channels are expressed in rats and mice and in rats, mice, and humans, respectively. However, Cav1.3 contributes the most to glucose-stimulated insulin secretion in rats and humans and Cav1.2 in mouse beta cells (Hiriart and Aguilar-Bryan, 2008; Rorsman et al., 2012).
Beta cells from diverse species and cell lines express different combinations of VDCC. For example, human beta-cell expression of CACNA1D mRNA exceeds CACNA1C mRNA by 60 times (Reinbothe et al., 2013). Although the same predominance of Cav1.3 is observed in the rat, in the mouse, Cav1.2 channels are dominant (Hiriart and Aguilar-Bryan, 2008; Rorsman et al., 2012). Cav3.1 and Cav3.2 (T-type) currents are present in INS-1 cells, dog, human, and rat, but not in mouse. Moreover, human beta cells lack Cav2.3 (N-type) currents (Taylor et al., 2005; Hiriart and Aguilar-Bryan, 2008), and N-type Cav2.2 channels have been identified in adult rat beta cells (Navarro-Tableros et al., 2007b). Also, human beta cells express P/Q-type Cav2.1, and mouse beta cells show P/Q-type Cav2.1 and R-type Cav2.3 channels (Yang et al., 2014a).
To date, 10 mammalian α1 genes and four distinct Cavβ genes (Cavβ1, Cavβ2, Cavβ3, and Cavβ4) have been identified in beta cells. Rat and mouse native beta cells express β2 and β3 subunits, although β2 is more abundant than β3 in the rat (Yang et al., 2014a). To date, it has been suggested that Cavβ2 and Cavβ3 subunits modulate GSIS possibly by regulating the interactions between VGCCs and PKC isozymes (Rajagopal et al., 2014).
Pharmacology of Cav Channels in Pancreatic Beta Cells
Many pharmacological agents modulate the activity of Cav channels. For example, L-type Cav1 channels are sensitive to La3+, dihydropyridines, alkyl phenyl amines, and benzothiazepines. Recently, it was observed that the natural polyphenolic flavonoid quercetin (3,3′,4′,5,7-pentahydroxyflavone) enhances L-type Cav1 current, also stimulating insulin secretion in INS-1 cells. Quercetin interacts with L-type Cav1 in a different site from that of the agonist Bay K 8644 (1,4-Dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-3-pyridinecarboxylic acid, methyl ester). The mechanism acts by shifting the voltage-dependent activation toward negative potentials (Bardy et al., 2013). L-type Cav1 channels are the major contributors to the whole cell Cav current and insulin granule exocytosis in human, rat, and mouse beta cells. Despite Cav1.2 being the principal L-type Ca2+ channel in mouse, Cav1.3 is the most important L-type channel in rat and human beta cells. Cav1.3 plays a role in cAMP homeostasis through adenylate cyclase 1 and may participate in insulin secretion in an indirect manner (Kitaguchi et al., 2013). Recent work suggests that Cav1.2 LII-III loop interactions may play a significant role in the glucose-stimulated excitability. They may regulate channel density on the plasma membrane, the Ca2+-induced-Ca2+ release from the endoplasmic reticulum (ER), as well as the activity of other channels such as Ca2+-activated potassium channels (Catterall, 2000b; Wang et al., 2014).
P/Q-type Cav2.1 channels are sensitive to ω-agatoxin IVA, N-type Cav2.2 channels are blocked by ω-conotoxin GVIA but are insensitive to dihydropyridines, alkyl phenyl amines, and benzodiazepines. R-type Cav2.3 channels are resistant to almost all the Cav channel blockers, but it is blocked by the toxin SNX-482. Although SNX-482 has shown a high affinity for Cav2.3 channels, it is capable of blocking other Cav channels Yang and Berggren, 2006; (Catterall, 2011; Yang et al., 2014a).
Calcium entry into the cell through P/Q-type Cav2.1 channels is the second most important entry route in rodent beta cells after glucose stimulation. Nevertheless, P/Q channels contribute in a similar fashion as L-type channels to the whole cell Cav current in human beta cells (Yang and Berggren, 2006; Yang et al., 2014a). N-type Cav2.2 channels are expressed in adult rat beta cells, although 50% of the protein is distributed in the membrane and 50% at the cytoplasm (Navarro-Tableros et al., 2007b).
T-type Cav3.1 and Cav3.2 channels play a significant role in the regulation of insulin secretion. In rat beta cells, they activate around −40 mV and generate a hump at the beginning of the whole cell I(Ca)-V relationship, with rapid inactivation and slow deactivation, and they may play the role of a pacemaker at glucose-stimulating levels (Hiriart and Matteson, 1988; Hiriart and Aguilar-Bryan, 2008). T-type Cav currents have been suggested to be involved in the beta-cell response to cytokines (Wang et al., 1999). Finally, R-type Cav2.3 channels are expressed rodent but not human beta cells (Hiriart and Aguilar-Bryan, 2008). They may play a role in the second phase of insulin secretion by supplying new secretory granules to the release sites (Jing et al., 2005; Yang and Berggren, 2006; Rorsman et al., 2012).
Endogenous Regulation of Cav Channels in the Beta Cell
Several mechanisms and pathways, including protein phosphorylation, Ca2+/calmodulin signaling, G protein-coupled receptors, and phosphorylated inositol may endogenously regulate Cav channels activity and density in the beta-cell membrane (Yang and Berggren, 2006). These regulatory mechanisms interact simultaneously to control insulin secretion in the body (Fig. 3).
Serine/Threonine Protein Kinases, Phosphatases, and Tyrosine Kinase Receptors.
Pancreatic beta cells and insulin secretion are regulated by cAMP and PKA. Cav channels possess several protein phosphorylation sites. Different studies have shown the regulation of Cav channels by PKA. An increased Ca2+ influx through VDCC is observed with cAMP analogs or activators of PKA, and this effect disappears with channel blockers (Henquin and Meissner, 1983, 1984). Moreover, mouse beta cells showed not only an increased exocytotic capability assessed by capacitance measurements after PKA activation (Gillis and Misler, 1993) but an increased amplitude in L-type Cav currents in a dose-dependent manner after cAMP-analog exposure (Kanno et al., 1998; Yang and Berggren, 2006). More recently, it has become clear that PKB may also participate in GSIS. PKB may upregulate L-type calcium channel activity via phosphorylation of the Cavβ2 subunit, which promotes calcium channel trafficking to the membrane (Cui et al., 2012).
Effects on Cav by PKC activation are controversial. PKC depletion from RINm5F cells downregulates L-, P/Q-, and non-N-type Cav currents (Yang et al., 2014a), and acute activation of PKC produces an increase in Cav1 channels. Thus, PKC modulation has been suggested to play a tonic role in maintaining Cav channel function (Yang and Berggren, 2006). Pancreatic beta cells express all PKG isoforms (PKG1α, PKG1β, and PKGII) and all four Ca2+/calmodulin-dependent protein kinase II (CaMKII) isoforms (Easom, 1999). Furthermore, activation of PKG in rat beta cells by cGMP increased Ca2+ influx through Cav channels. Nevertheless, further investigation is needed, because cGMP can produce a direct effect on Cav channels (Yang and Berggren, 2006; Yang et al., 2014a).
Also, regulation of Cav channels by calcium/calmodulin-dependent kinase II (CaMKII) is not clear. Although nonspecific CaMKII inhibitors, namely KN-62 and KN-93, could inhibit Cav channels, it has been suggested that both may act directly on beta-cell Cav channels (Yang and Berggren, 2006). Recently, a reduction in insulin secretion and Ca2+ influx through Cav channels was observed in islet beta cells after CaMKII inhibition (Dadi et al., 2014).
Several types of protein phosphatases are present in beta cells. Experiments using okadaic acid to inhibit protein phosphatases in mouse and rat beta cells have shown a small increase in basal conditions but a significant increase in Cav channel activity with a simultaneous activation of PKA. Thus, it was suggested that Cav channels from mouse beta cells show low phosphorylation level in basal conditions because of a dominant effect of tonically activated phosphatases that surround Cav channels over the protein kinases activity (Ammälä et al., 1994).
Former work in our laboratory showed that TrkA tyrosine kinase receptors regulate rat beta-cell Cav channels. Acute incubation of rat beta cells with nerve growth factor (NGF) induces activation of Cav1 channels probably by tyrosine phosphorylation that produces increased Ba2+ currents (Rosenbaum et al., 2001). Moreover, long-term exposure to NGF potently increases Ba2+ currents due to an increased Cav1 synthesis (Rosenbaum et al., 2002). Insulin receptor signaling might also upregulate Cav channel activity. Indeed, acute incubation of mouse beta cells with an insulin mimetic drug L-783,281 increased intracellular Ca2+ concentration in a dose-dependent manner, and this effect was partially blocked by nifedipine (Roper et al., 2002).
G Protein-Coupled Receptors.
The Cav2.1 and Cav2.2 channels can be modulated by direct membrane-interaction with G proteins. The Gβγ subunits play a fundamental role in the regulation of Cav channels. The Gβγ subunits bind to the intracellular linkers, LI-II of Cav2.1 and Cav2.2 channels, and to the LI-II of Cav1.3 channels. This interaction is characterized by a positive shift in the voltage dependence and a slowing of channel activation. Furthermore, the N and C termini of the Cavα1 subunit are also involved in Gβγ binding (Dolphin, 2003; Yang et al., 2014a). Moreover, it was observed that Cav1.2 channels are negatively regulated by a direct membrane-interaction with G proteins (Ammälä et al., 1992; Ivanina et al., 2000).
It is well accepted that chronically increased concentrations of free fatty acids impair beta-cell function and reduce insulin secretion (Zraika et al., 2002). However, some observations suggest that prolonged exposure to high levels of some unsaturated free fatty acids could protect beta-cell function and increase GSIS (Tuo et al., 2012). It was suggested that the medium- and long-chain free fatty acid receptor GPCR40 could mediate this effect by activation of the Gαq protein, Ca2+ release from the endoplasmic reticulum, and influx via L-type Ca2+ channels (Fujiwara et al., 2005).
Glucagon and Glucagon-like Peptide Receptors.
It has also been speculated that activation of glucagon receptors regulate Cav channels. Experimental evidence for a regulatory role of the G-coupled glucagon receptor over Cav channels comes from experiments in the insulin-secreting HIT cells. These cells showed an increase in intracellular Ca2+ concentration in response to glucagon that was blocked by either chelation of extracellular Ca2+ or application of Cav1 channel blockers (Prentki et al., 1987; Yang et al., 2014a).
Glucagon-like peptides (GLP-1) potentiate nutrient-induced beta-cell insulin secretion and proliferation and decrease their apoptosis (Phillips and Prins, 2011). The GLP-1 effect on mouse beta-cell insulin secretion is in part due to a slower time-dependent inactivation of Cav currents. Similar effects have been observed on Cav1 channels in the rat and human pancreatic beta cells (Yang et al., 2014a). Other works have suggested that PKA activation may modulate the VDCCs pore activity. Moreover, it was observed that PKA inhibitors block the GLP-1-mediated increase of Ca2+ currents (Meloni et al., 2013). Also, Melissa and collaborators suggested that the intracellular II-III loop domain of Cav1.2, and 1.3 may target these channels to specific membrane microdomains, allowing their interaction with proteins involved in GLP-1 signaling (Jacobo et al., 2009).
Somatostatin Receptors.
Activation of somatostatin receptors decreases Cav channel activity via a pertussis toxin-sensitive G protein in HIT and RINm5F cells (Prentki et al., 1987). Recent work determined that somatostatin reduces GSIS in mouse islets partially by modulating L-type Ca2+ channels and suggested that somatostatin may exert this effect through a Gβγ protein-dependent mechanism (Schwetz et al., 2013).
Catecholamine and Acetylcholine Signaling.
Parasympathetic and sympathetic innervation of islets modulates insulin secretion. Sympathetic innervation activates adrenoceptors alpha2 and decreases insulin secretion and parasympathetic innervation releases acetylcholine, which activates muscarinic receptors and stimulates insulin secretion (Hiriart et al., 2014). Stimulation of mouse beta-cell alpha2 adrenoceptors using clonidine inhibits Cav channel activity and insulin secretion. Moreover, this effect is abolished by the selective alpha2 antagonist yohimbine. However, further work is needed to describe the mechanism by which alpha2 adrenoceptors modulate beta-cell Cav channels (Yang and Berggren, 2006). The sympathetic nerve terminals also release galanin. This neuropeptide also inhibits Ca2+ currents and thus insulin secretion in mouse beta cells; this effect could be mediated by a Go2-alpha protein (Tang et al., 2012).
Interestingly, pancreatic beta cells synthesize and store dopamine within their secretory granules. Also dopamine is cosecreted with insulin and exerts an autocrine or paracrine negative feedback loop on beta-cell insulin secretion, acting through D2 and D3 receptors. Furthermore, D3 signaling activates Gβγ proteins, which may negatively regulate both, directly and indirectly, Cav1.2 channels activity (Ivanina et al., 2000; Ustione et al., 2013).
Little information exists on acetylcholine signaling in beta cells. Despite the fact that the parasympathetic neurotransmitter acetylcholine stimulates insulin secretion, it also inhibits the Cav channel activity in mouse beta cells (Gilon et al., 1997). However, studies in HIT cells have observed that muscarinic receptors increase the amplitude of whole cell Cav currents and the Cav1 channel open probability (Yang and Berggren, 2006).
Exocytotic Proteins.
Exocytotic proteins such as syntaxin 1A, SNAP-25, and synaptotagmin also interact with L-type VDCC. Beta cells predominantly express Syn-1A, Syn-2, and Syn-4 in the membrane and Syn-3 in the secretory granules. This interaction tranduces bidirectional signaling that modulates Cav1 channel activity and the exocytotic machinery and could mediate exocytosis of predocked insulin-containing vesicles during the first phase of GSIS (Yang et al., 2014a). Syn-1A deletion in mice blunts the first phase of GSIS (Kang et al., 2002; Xie et al., 2016). Furthermore, syntaxin 3A binds and modulates Cav2.3 channel opening, as well as the fusion of newcomer insulin vesicles during the second phase of GSIS. Syn-3 decreases with the RNAi in INS-1 cells after the second phase of insulin secretion. Moreover, overexpression of Syn-3 also inhibits L-type channels. Furthermore, Syn-3 depletion in INS-1 832/13 cells increases Cav activity. Syn-3 preferably binds to R-type calcium channels and a lesser extent to L-type Ca channels, which show a preference for Syn-1A (Xie et al., 2016).
Cytokines.
Cytokine action in beta cells could be linked to calcium influx through VDCC. Some studies observed that blocking L-type calcium channels prevented IL-1B-induced apoptosis in rodent and human islets, although ERK1/2, p38, and JNK pathways may also be involved (Maedler et al., 2004; Fei et al., 2008; Pratt et al., 2016). Additional data from nonobese diabetic mice showed a beta-cell increased expression of the T-type calcium channel, which results in elevated basal intracellular Ca2+. In addition, the treatment of mouse beta cells with a cytokine cocktail induced expression of the T-type calcium channel (Wang et al., 1996). Moreover, mouse islet exposure to a low-dose cytokine combination, namely, tumor necrosis factor-alpha, interleukin-1 beta, and interferon-gamma, induced beta-cell dysfunction, partly due to an increased calcium influx through L-type calcium channels (Dula et al., 2010). However, cytokine-induced changes in VDCC activity and insulin secretion were not apparent. Early studies observed that interleukin-1 could inhibit glucose-stimulated calcium influx in rat islets (Wolf et al., 1989), but other studies showed the opposite effect (Ramadan et al., 2011). This effect is probably mediated by diacylglycerol production and protein kinase C activation (Welsh et al., 1989; Eizirik et al., 1995). The variability observed in these studies may reflect dosage dependence and the different cocktail combinations used in experiments (Ramadan et al., 2011).
The Role of Cav Channels in the Pathophysiology of MS and T2DM
VDCC alterations, because of mutation, the downregulation of their expression, activity, and/or density at the membrane, could lead to beta cell dysfunction (Yang and Berggren, 2006; Velasco et al., 2012). Changes in calcium influx through VDCC play a vital role in beta-cell dysfunction in metabolic syndrome (MS) and diabetes models. In an MS rat model induced by consumption of a 20% sucrose solution during 24 weeks, changes in whole cell calcium influx were observed, which may be related not only to beta-cell hypersecretion but dysfunction and exhaustion (Velasco et al., 2012). Additional studies have shown that a 48-hour islet exposure to 28 mM glucose increased insulin secretion due to an increased nifedipine-sensitive VDCC influx (Qureshi et al., 2015).
It is well accepted that beta-cell chronic exposure to high long-chain free fatty acids contributes to increasing basal insulin secretion and, at the same time, reduces GSIS during obesity (Zraika et al., 2002). In part, these effects involve a reduced calcium influx through VDCC, associated with an increased activity of the membrane metalloendopeptidase neprilysin (Zraika et al., 2013). Other groups have found that beta-cell long-term exposure to free fatty acids results in selective suppression of exocytosis elicited by brief depolarizations. They suggested that depolarization-induced Ca2+ influx is limited to discrete areas within beta cells due to calcium channel aggregation and that FFAs induce a diffused Ca2+ influx along the membrane surface. This effect could impair insulin exocytosis, because release-competent granules may not be exposed to exocytotic local levels of Ca2+ (Rorsman et al., 2012). Furthermore, two single-nucleotide polymorphisms in the CACNA1D gene (Cav1.3) reduce its mRNA expression, impair insulin secretion, and are associated with type 2 diabetes (Reinbothe et al., 2013). Finally, blocking L-type calcium channels with diazoxide could inhibit hyperglycemia-induced beta-cell apoptosis (Zhou et al., 2015).
Conclusions
Electric activity in beta cells is essential for insulin secretion. A great deal is known about the different channels that participate in depolarization and action potentials. However, there are other ion channels in the plasma membrane of beta cells and in the organelles that are not so well understood, such as chloride and potassium channels, which appear to be involved with membrane repolarization. Studies of channelopathies for KATP have provided important insight and are beginning to emerge for other channels. In the future, it will be important to develop specific drugs for different channels that could be used to directly test functional roles in beta cells and insulin secretion. In addition, it will be important to continue to investigate the epigenetic changes of the channels and explore therapeutic strategies to reverse associated deficits.
Acknowledgments
ASPET thanks Dr. Katie Strong for copyediting of this article.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Velasco, Díaz-García, Larqué, and Hiriart.
Performed figures: Velasco, Díaz-García, and Larqué.
Footnotes
- Received February 17, 2016.
- Accepted July 18, 2016.
↵1 Current affiliation: Department of Neurobiology, Harvard Medical School, Boston, MA.
This work was supported by Dirección General de Asuntos del Personal Académico (DGAPA)-UNAM [Grant IN-213114] to Marcia Hiriart; [Grant IN-211416] to Myrian Velasco; and Carlos Larqué had a CONACyT doctoral fellowship.
Abbreviations
- ABC
- ATP binding-cassette
- AMPK
- AMP-activated protein kinase
- CaMKII
- calmodulin-dependent kinase II
- Cav
- voltage-dependent Ca2+ channel
- CHI
- congenital hyperinsulinism
- ER
- endoplasmic reticulum
- FFA
- free fatty acid
- GLP-1
- glucagon-like peptide
- GLUT1
- Type-1 glucose transporter
- GSIS
- glucose stimulation insulin secretion
- HNF
- hepatocyte nuclear factor
- KATP channels
- ATP-sensitive potassium channels
- Kv
- voltage-dependent potassium channels
- LC-CoA
- long-chain acyl-CoA esters
- MS
- metabolic syndrome
- Nav
- voltage-gated Na+ channels
- NBD
- nucleotide-binding domain
- NDM
- neonatal diabetes mellitus
- NGF
- nerve growth factor
- PIP2
- phosphatidylinositol-4, 5-biphosphate
- SNARE
- soluble N-ethylmaleimide sensitive factor attachment proteins receptor
- SU
- sulfonylurea
- SUR1
- Type 1 sulfonylurea receptor
- Syn-1A
- syntaxin-1A
- T2DM
- type 2 diabetes mellitus
- TMD
- transmembrane domain
- TNDM
- transient neonatal diabetes mellitus
- TRP channel
- transitory receptor potential channel
- TTX
- tetrodotoxin
- VDCC
- voltage dependent calcium channels
- VGSC
- voltage-gated sodium channel
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}