


Visual Overview

Abstract
Follitropin, or follicle-stimulating hormone (FSH) receptor (FSHR), is a G protein-coupled receptor belonging to the glycoprotein hormone receptor family that plays an essential role in reproduction. Although its primary location is the gonad, the FSHR has also been reported in extragonadal tissues including bone, placenta, endometrium, liver, and blood vessels from a number of malignant tumors. The recently resolved crystal structure of FSH bound to the entire FSHR ectodomain has been instrumental in more clearly defining the role of this domain in ligand binding and receptor activation. Biochemical, biophysical, and structural data also indicate that the FSHR exists as a higher order structure and that it may heterodimerize with its closely related receptor, the luteinizing hormone receptor; this association may have physiologic implications during ovarian follicle maturation given that both receptors may simultaneously coexist in the same cell. FSHR heterodimerization is unique to the ovary because in the testes, gonadotropin receptors are expressed in separate compartments. FSHR self-association appears to be required for receptor coupling to multiple effectors and adaptors, for the activation of multiple signaling pathways and the transduction of asymmetric signaling, and for negative and positive receptor cooperativity. It also provides a mechanism through which the glycosylation variants of FSH may exert distinct and differential effects at the target cell level. Given its importance in regulating activation of distinct signaling pathways, functional selectivity at the FSHR is briefly discussed, as well as the potential implications of this particular functional feature on the design of new pharmacological therapies in reproduction.
Introduction
The pituitary gonadotropin hormones, luteinizing hormone (LH) and follicle-stimulating hormone (FSH), are synthesized in gonadotrope cells and play a key role in the control of gonadal function and reproduction (Huhtaniemi et al., 1982; Bousfield et al., 1996; Richards et al., 2002). Both gonadotropins, as well as placental chorionic gonadotropin (CG) and thyroid-stimulating hormone (TSH) produced by pituitary thyrotropes, belong to the family of glycoprotein hormones (GPH) (Pierce and Parsons, 1981; Ulloa-Aguirre and Timossi, 1998; Szkudlinski, 2015).
Their cognate receptors (FSHR, LHCGR, and TSHR) belong to the highly conserved subfamily of the G protein-coupled receptor (GPCR) superfamily, the so-called Rhodopsin family, and more specifically, to the δ-group of this large class of GPCRs (Simoni et al., 1997; Kleinau et al., 2013; Troppmann et al., 2013). GPH receptors (GPHR) are characterized by a large NH2-terminal extracellular domain, or ectodomain, where recognition and binding of their corresponding ligands occur. This extracellular domain contains a central structural motif of imperfect leucine-rich repeats (12 in the FSHR), a motif that is shared with a number of other membrane receptors involved in ligand selectivity and specific protein-protein interactions (Fig. 1A) (Bogerd, 2007). The carboxyl-terminal end of the large extracellular domain includes the so-called hinge region, which structurally links the leucine-rich extracellular domain with the serpentine, seven transmembrane domain (7 TMD) of GPHRs and leads to receptor activation and signaling functionality. In all GPHRs, the hinge region is involved in high affinity hormone binding, receptor activation, and the intramolecular signal transduction and/or silencing of basal receptor activity in the absence of a ligand (Mueller et al., 2010).

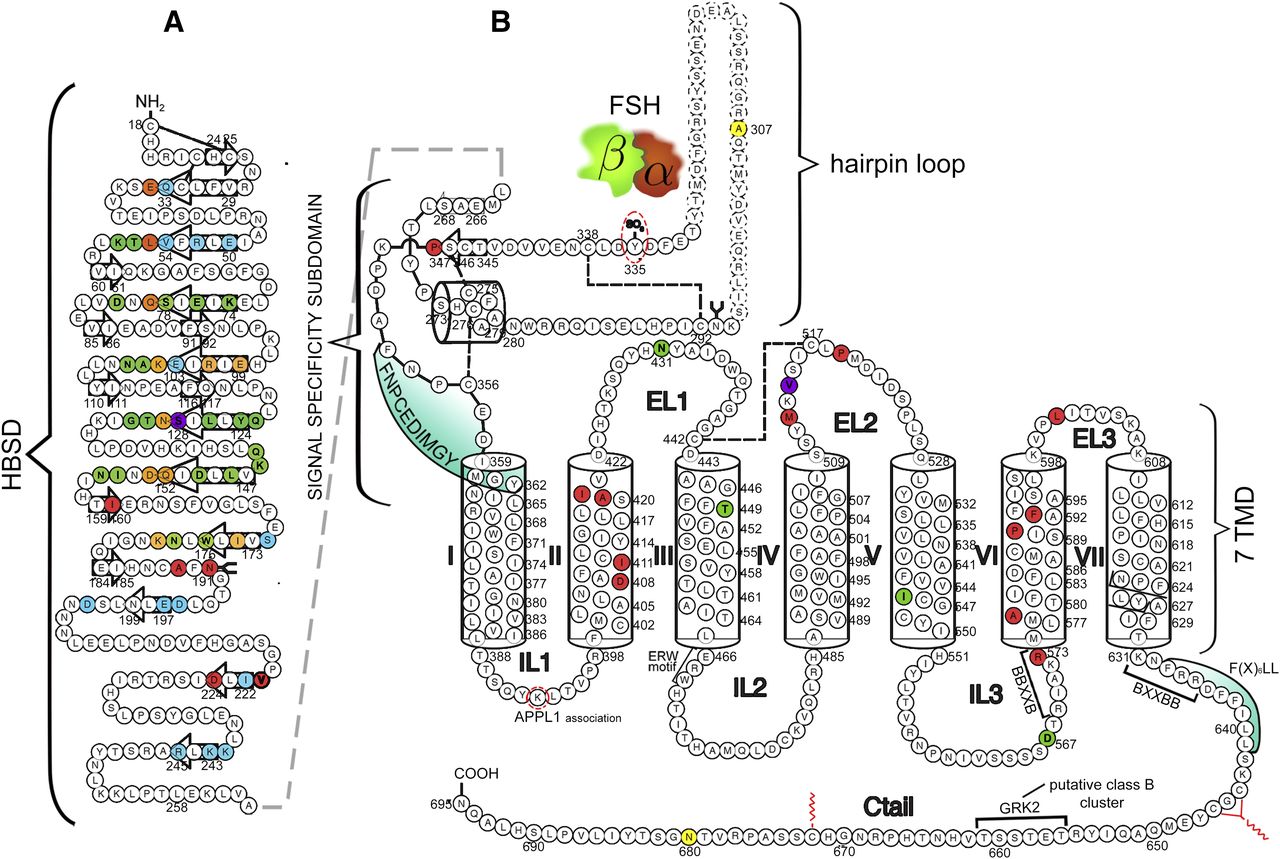
Schematic representation of the human FSHR structure, including the amino acid sequence (circles). Numbering of amino acid residues includes the leader sequence. (A) Hormone binding subdomain (HBSD) of the extracellular domain. The FSHR residues that are buried in the FSH/FSHR interface and located in the high affinity binding site are shown in colored circles. Green shows the FSH α-subunit residues only; blue shows the FSH β-subunit residues only: orange denotes residues from both subunits that interact with the FSHR. Also shown are the locations of naturally occurring receptor mutations in this domain that affect function. The location of the mutation leading to promiscuous ligand binding is shown in magenta (Ser128), whereas the residues whose replacement provokes loss-of-function of the receptor are shown in red; the majority of mutations at these sites lead to trafficking-defective FSHR molecules (reviewed in Ulloa-Aguirre et al., 2014b). β-Strands located in the concave (corresponding to the leucine-rich repeats) or convex surface of the HBSD are indicated by the colorless arrows. Disulfide bonds are shown as broken lines and glycosylation by the arbor-like structure [schematic according to Fan and Hendrickson (2005) structure]. (B) Signal specificity subdomain and seven transmembrane domain (7 TMD) including the extracellular loops (EL), intracellular loops (IL), and carboxyl-terminal domain (Ctail). The regular secondary structure elements of the leucine-rich repeats 11 and 12 are shown as colorless arrows for the β-strands and as a cylinder for the α-helix. α-Helices of the 7 TMD (I to VII) are also shown as cylinders. The Y335 residue, which interacts with a binding pocket located in the interface of the FSH α-and β-subunits (represented by the brown and green freeform drawings, respectively) is surrounded by a broken-line oval. [Schematic according to the structure reported by Jiang et al. (2012).] The location of the naturally occurring loss-of-function mutations reported to date are shown as red-colored circles, whereas the location of gain-of-function mutations are shown in green. Mutations at Asp567, Iso545, and Thr449 lead to promiscuous activation of the receptor by other glycoprotein hormones, whereas the mutation at Asn431 leads to altered desensitization and internalization of the FSHR (Casas-Gonzalez et al., 2012). Mutation at Val514 (magenta circle in the extracellular loop 2) led to increased plasma membrane expression of the receptor and OHSS at low FSH doses (Desai et al., 2015). Also indicated are amino acid residues that are involved in APPL1 interaction (Lys393) with the intracellular loop 1, as well as the most common location of FSHR polymorphisms (N680 and A307) (yellow circles). Additionally, the schematics show the location of some important motifs and sequences involved in receptor function: a) The sequence involved in internal agonist activity (FNPCEDIMGY sequence, shaded in green); b) Motifs involved in receptor trafficking [F(X6)LL and BXXBB, the latter motif found reversed at the NH2-terminal end of the carboxy-terminal tail]; c) Motifs involved in G protein coupling and receptor activation (ERW and NPXXY motifs at the TMD III-intracellular loop 2 junction and within TMD VII, respectively; and another reversed BXXBB motif in intracellular loop 3); d) The Ser/Thr cluster for receptor phosphorylation by GRK 2 (class B cluster at the Ctail); and e) The target residues for palmitoylation (Cys 644, 646, and 672) (Ulloa-Aguirre et al., 2007a; 2014a,b; Desai et al., 2013; Bramble et al., 2016). Disulfide bonds are shown as broken lines.
The FSHR is primarily expressed by specific cells in the female and male gonads (Vassart et al., 2004). In the ovary, the FSHR is expressed in the granulosa cells where FSH-stimulated activation leads to growth and maturation of ovarian follicles and estrogen production. In the testes, the FSHR is expressed in the Sertoli cells of the seminiferous tubules where it supports their growth and maturation and, hence, indirectly promotes spermatogenesis (Dias et al., 2002).
This review summarizes the most important structural and functional aspects of the FSHR that may eventually impact the design and development of new pharmacological approaches in the treatment of reproductive disease.
FSHR Structure
In humans, the FSHR gene is located on chromosome 2p21-p16 (Gromoll et al., 1994). The coding region of this gene consists of 10 exons, ranging in size from 69 to 1234 bp, and 9 introns sized from 108 to 15 kb (Gromoll et al., 1996). The receptor protein is comprised by 695 amino acid residues; the first 17 amino acids encode the signal, or leader sequence, resulting in a mature, cell surface plasma membrane expressed FSHR of 678 amino acid residues with a molecular weight of 75 kDa as predicted from its cDNA (Dias et al., 2002). Nevertheless, N-glycosylation of three of four possible glycosylation sites can yield mature receptor forms with an approximate molecular weight of 80-87 kDa.
Several heterogeneous FSHR splice variants have been detected in various animals species (Gromoll et al., 1993; Khan et al., 1993; Rajapaksha et al., 1996; Yarney et al., 1997a; Tena-Sempere et al., 1999; Babu et al., 2001; Neubauer et al., 2006), including humans (Kelton et al., 1992; Song et al., 2002; Gerasimova et al., 2010; Robinson et al., 2010; Zhou et al., 2013; Karakaya et al., 2014). Because many of the detected alternative transcripts are not translated into a protein or translation yields severely misfolded proteins unable to traffic to the cell surface plasma membrane, the function and regulatory role of FSHR transcript variants has been scarcely studied. Two alternate splice transcripts of the ovine FSHR gene with an altered exon 10 deserve particular attention because of their potential functional consequences (Sairam et al., 1996, 1997; Yarney et al., 1997b; Babu et al., 1999, 2000). The first variant is similar to the full-length wild-type (WT) receptor except that differential splicing leads to divergence in the protein sequence at the carboxyl terminus and the variant is 25 amino acids shorter. This FSHR variant is expressed at the plasma membrane, fails to signal via the cAMP/PKA pathway, and behaves as a dominant negative receptor when coexpressed with the WT receptor in HEK293 cells (Sairam et al., 1996). The other variant is a FSHR consisting of a transcript encoding only exons 1–8, along with a non-FSHR single transmembrane domain, and a carboxyl-terminal extension characteristic of cytokine/growth factor receptors (Sairam et al., 1997). When expressed in granulosa cells, the growth promoting effects of this variant were independent of the cAMP/PKA pathway but were mediated by Ca2+ activation and ERK1/2-dependent pathways (Babu et al., 2000). Further studies demonstrated that this particular variant is also expressed in the mouse ovary and that it is upregulated by gonadotropin stimulation (Babu et al., 2001). More recent studies in ewes (Sullivan et al., 2013) found that the FSH-growth factor-like receptor was more highly expressed than the WT variant in follicles of all sizes, but particularly in medium size follicles obtained during early estrus, suggesting that this receptor isoform may play a role in follicle development in this particular animal species. Additional studies are still necessary to more precisely define the role of these and other FSHR transcript variants in the mediation of the pleiotropic effects of FSH (see below), particularly in humans.
Both the FSHR and its close relative, the LHCGR, exhibit a high degree of amino acid sequence homology. Although the extracellular domain amino acid sequences of the gonadotropin receptors are approximately 46% identical, the 7 TMD sequence portion of the receptors shares nearly 72% homology (Dias et al., 2002; Kleinau and Krause, 2009). Although this relatively high similarity between the 7 TMD of the gonadotropin receptors might suggest similar mechanisms of receptor activation, mutations in this domain leading to constitutive activation of the FSHR are extremely rare (Figs. 1B and 2) (see below), but this is not the case for the LHCGR (Ulloa-Aguirre et al., 2014a). This difference may be due to the relative stability of the FSHR 7 TMD in the inactive state compared with the LHCGR and the TSHR. In fact, constitutively active mutants of the latter receptors are more frequent and share the same location across the serpentine domain, indicating that those two receptors actually share common mechanisms of activation. In contrast, only two of the four FSHR constitutively active mutants identified to date share locations with the LHCGR and the TSHR constitutively active mutants (Ulloa-Aguirre et al., 2014a). Among the three domains of the gonadotropin receptors, the intracellular regions exhibit the lowest amino acid sequence homology (∼27% identity), with the exception of the NH2-ends of the carboxyl termini, which bear cysteine residues for palmitoylation and a primary sequence motif [Phe(x)6Leu-Leu] that regulates anterograde trafficking from the endoplasmic reticulum to the plasma membrane (Duvernay et al., 2004; Uribe et al., 2008). Both of these structural characteristics are common features of the rhodopsin-like GPCR subfamily.

Three-dimensional homology model of the FSH/FSHR complex is shown with mapped positions of the naturally occurring mutations at the FSHR. The 7 TMD constituted by transmembrane helices (H) 1–7 connected by intracellular (IL) and extracellular (EL) loops was modeled based on the determined active structure-conformation of the β2-adrenergic receptor (PMID: 21772288). The (monomeric) extracellular complex between the hinge region, the leucine-rich repeat domain (see Fig. 1A), and FSH were taken as suggested by a structure determined for a fragment (PMID: 22802634). The hinge region structurally links the leucine-rich repeat domain with the 7 TMD. The FSHR (backbone white-7 TMD, light beige-hinge/leucine-rich repeat domain) binds the hormone [FSHβ (brown) and FSHα (yellow), surface representation] at the extracellular side between the leucine-rich repeat domain and the hinge region. The exact orientation between the different components to each other is still unclear. The localization of naturally occurring mutations is indicated by highlighted side chains of wild-type positions (atom spheres without hydrogen). Gain-of-function mutations (green) are characterized by an increase in basal activity or by causing promiscuous ligand binding (magenta spheres or magenta boxes). Few substitutions at the 7 TMD lead to increased basal activity and, simultaneously, to promiscuous hormone sensitivity. Loss-of-function mutations are indicated in red. This spatial arrangement and mapping helps to match functional data with structural information, allowing extraction of important regions for maintenance of properties necessary for signal transduction, including the structural fold or obligate side-chain interactions. Ctail, carboxyl-terminal tail.
Extragonadal Expression of the FSHR
Although for many years the FSHR was thought to be localized exclusively in the gonads, recently it was reported that FSHR exists in extragonadal tissues where it has been purported to have distinct physiologic roles. Extragonadal sites where the FSHR has been localized include bone osteoclasts (Sun et al., 2006, 2010), monocytes (Robinson et al., 2010; Cannon et al., 2011), endothelial cells from the umbilical vein (Stilley et al., 2014b), tumor vasculature and metastases (Radu et al., 2010; Siraj et al., 2013; Planeix et al., 2015), different sites of the female reproductive tract, the developing placenta (Stilley et al., 2014a), and the liver (Song et al., 2016). Data on the expression (mRNA and protein) of either the FSHR WT or the splice variant originally identified in bovine granulosa cells (Rajapaksha et al., 1996) in osteoclasts and monocytes (Robinson et al., 2010) and, consequently, on the role of FSH in bone function are still quite controversial (Seibel et al., 2006; Sun et al., 2006, 2010; Gao et al., 2007; Prior, 2007; Williams, 2007; Allan et al., 2010; Cannon et al., 2011). Meanwhile, immunohistochemical studies using monospecific anti-FSHR antibodies have documented the expression of human FSHR in blood vessels from a number of malignant tumors (Radu et al., 2010; Planeix et al., 2015) and tumor metastases (Siraj et al., 2013). Although the authors posit that FSHR at these locations may contribute to the growth and expansion of tumor tissue by promoting angiogenesis, further studies are still necessary to validate these findings given that such claims can lead to concerns among women undergoing hormone therapy during in vitro fertilization or menopause, conditions where FSH levels may be extremely high. In a series of studies, Stilley et al. (2014a,b) recently identified FSHR in several nonovarian reproductive tissues. Interestingly, feto-placental haploinsufficiency of the Fshr in mice was associated with placental growth defects and fetal loss in both heterozygous and homozygous null Fshr animals (Stilley et al., 2014a). More recently, a role for the FSHR in liver tissue was proposed (Song et al., 2016), and in this report, the authors claimed that the FSHR may be involved in the expression of the low-density-lipoprotein receptor and in the regulation of low-density-lipoprotein cholesterol clearance. However, this study, which focused on menopausal women, did not account for the role of estrogens in this process. In conclusion, further studies are still warranted to elucidate more clearly the expression and physiologic significance of extragonadal FSHRs. It still needs to be determined whether they are structurally similar to those primarily expressed in the gonads or if they are splice variants of the WT receptor that when expressed in the plasma membrane in sufficient quantities may provoke a biologic effect upon exposure to physiologic concentrations of the agonist. Such studies will undoubtedly lead to assertions on the role of FSH in many extragonadal activities.
Ligand Binding and Receptor Activation: Role of the FSHR Ectodomain
The FSHR extracellular domain harbors both the binding site for FSH and the region necessary for ligand-induced receptor activation. Although previous biochemical and in silico studies (Jiang et al., 1995; Schmidt et al., 2001) had predicted the relationship between the GPHRs ectodomain and GPH agonist binding and recognition, it was not until the first structure of the FSH in complex with the extracellular-hormone binding domain of the FSHR (FSHRHB) was established (Fan and Hendrickson, 2005) that this relationship was fully documented. This model, however, did not include the hinge region, which, based on biochemical studies, had been considered a separate structure essential for FSHR activation (Costagliola et al., 2002; Bruysters et al., 2008; Agrawal and Dighe, 2009). As previously predicted, the Fan-Hendrickson structure showed that FSH binds to FSHRHB like a “handclasp,” (Jiang et al., 1995), that most β-strands in the inner surface participate in FSH binding, and that both hormone subunits (noncovalently linked α- and β-subunits, a hallmark of all GPHs) are involved in the specificity of binding to the receptor, with charge and stereochemical dominance dictating specificity. More importantly, the structure revealed that carbohydrates do not participate in the binding interface of the FSH–FSHRHB structure, but rather are sequestered in the periphery of the complex (Fan and Hendrickson, 2005, 2007).
Seven years later, the crystal structure of FSH bound to the entire FSHR ectodomain (FSHRED) was determined (Jiang et al., 2012, 2014a). The structure has more clearly elucidated the role of the FSHR (and other GPHRs as well) extracellular domain in ligand binding and, more importantly, in receptor activation. The FSHR extracellular domain structure identified the hinge region (or signal specificity subdomain) as an integral part of the ectodomain (Fig. 1B). As earlier biochemical studies on the FSHR and TSHR had indicated, this particular region was also confirmed to play an essential role in ligand- and/or mutation-provoked receptor activation (Zhang et al., 2000; Vlaeminck-Guillem et al., 2002; Chen et al., 2003; Vassart et al., 2004; Agrawal and Dighe, 2009). In fact, these and more recent studies (Bruser et al., 2016) strongly suggest that the ectodomain of the GPHRs acts as a tethered inverse agonist that switches to an agonist upon ligand binding (i.e., the unoccupied hinge region inhibits FSHR activation) and activation of an internal agonist unit located in the carboxyl-terminal end of the hinge region (Fig. 1B). The hinge region bears a sulfated Tyr residue (Y335) that interacts with a binding pocket located in the interface of the α- and β-subunits of FSH and formed via conformational changes of the ligand occurring after binding to the hormone-binding subdomain (HBSD) (Fig. 1B). The sulfated Y335 is located right after a rigid hairpin loop that lifts upon Y335 binding to the pocket unlocking the inhibitory effects of the loop on the 7TMD; this process leads to conformational changes in the latter domain and, eventually, to activation of the receptor. This tethered inverse agonist region in the extracellular domain had been previously mapped to the hairpin loop segments 296–331 of the receptor (Fig. 1B) (Agrawal and Dighe, 2009). As part of this process, a fixed short helix formed by residues S273 to A279 rotates and acts as a pivot, further contributing to the conformational change of the FSHR signal specificity subdomain . The importance of this helix movement in FSHR activation is underscored by the finding that the S273I FSHR mutant constitutively activates the receptor (Nakabayashi et al., 2000). In addition, the disulfide bond C275-C346 (Fig. 1B) fastens the last β-strand (leucine-rich repeat 12) to the short helix forming a rigid body. Additionally, the disulfide C276-C356 bridge ties this helix to the last few residues before the first TMD. Because of these constraints, the movement of the hairpin loop that occurs upon ligand binding could directly influence the conformation of the TMD helix 1, thereby promoting rearrangement within the remaining TMDs and ultimately leading to receptor activation. Given the similarity among the structures of GPHs and GPHRs, it is highly possible that all GPHRs share this two-step recognition process (ligand recruitment followed by signal specificity subdomain sulfated tyrosine docking—Y335 in the FSHR, Y331 in the LHCGR, and Y385 in the TSHR). In fact, FSH, LHCG, and TSH receptor mutants lacking this critical sulfated tyrosine residue lose their sensitivity to cognate ligands (Costagliola et al., 2002; Bonomi et al., 2006; Bruysters et al., 2008). Also, FSH mutants with substitutions in amino acid residues located below the sulfated tyrosine binding pocket (αF74E) or at the potential exosite (βL73E) exhibited increased signaling by pushing the hairpin loop up toward the top of the pocket (Jiang et al., 2012).
Biochemical, mutational, and in silico studies suggest that sites in the serpentine domain of the receptor, particularly the exoloops, which are extracellular projections of the TMDs, are also directly involved in receptor-ligand interaction and receptor activation. In fact, as supported by structural studies, exoloops 1 and 3, which are solvent exposed and accessible to gonadotropin hormone, may represent potential secondary gonadotropin binding sites, specifically at its α-subunit tips in the bound state (Fan and Hendrickson, 2005; Dupakuntla and Mahale, 2010). Moreover, according to mutagenic and functional studies, and the 7 TMD theoretical model (Jiang et al., 2014a), it seems that the exoloops interact with the hairpin loop of the hinge region to trigger FSHR activation; lifting of the sulfated Y335 to the FSHα/β binding pocket frees the hinge-tethered exoloops, releasing the inhibitory influence of the ectodomain on receptor activation (Jiang et al., 2014a). Accordingly, mutations in residues located at the FSHR exoloops 1 to 3, in addition to altering the receptor’s intracellular traffic [e.g., the N431I and L501F FSHR mutants (Casas-Gonzalez et al., 2012; Banerjee et al., 2015)], may also lead to attenuation of either agonist binding (Ji and Ji, 1995), FSH-stimulated signal transduction (Touraine et al., 1999; Sohn et al., 2002; Dupakuntla et al., 2012; Banerjee et al., 2015), or foster constitutive activation of the receptor (Casas-Gonzalez et al., 2012). The relationship between the FSHR exoloops and the hinge region is further supported by studies of the TSH receptor in which particular residues [e.g., Y563 and K565 at the exoloop 2 (Y511 and K513 in the FSHR)] are crucial for ligand-stimulated receptor activation (Kleinau et al., 2007).
Another interesting nuance is the FSHR (and TSHR) promiscuity for ligand specificity that the receptors exhibit upon particular structural modifications induced by mutations (Figs. 1 and 2) in the extracellular domain and the 7 TMD, a phenomenon with important consequences in the clinical realm. GPH-GPHR pairs have evolved so that a limited number of residues at the “seat-belt” domain of the ligand and the leucine-rich repeats of the receptor at the HBSD (Fig. 1A) take part in electrostatic interactions at the receptor-hormone interface to define binding and specificity. Because of the structural similarities among the GPHs and the GPHRs, it is conceivable that a given GPHR may be “cross-activated” by a ligand other than its cognate ligand, albeit with low binding affinity and without triggering detectable basal receptor activation. Therefore, it is not surprising that substitutions of key residues that directly or indirectly participate in the interaction of the receptor with its cognate ligand may dampen structurally related ligand discrimination and result in the interaction of the altered receptor with other than its own cognate GPH. For example, the S128Y mutation at the FSHR (Figs. 1B and 2) may lead to pregnancy-associated ovarian hyperstimulation syndrome (OHSS) [a potentially life-threatening condition (Kumar et al., 2011)] due to increased affinity and responsiveness of the mutated FSHR to CG, which circulates at very high levels during the first trimester of pregnancy (De Leener et al., 2008). In this particular mutation, the S→Y replacement allows the FSHR to form a hydrogen bond with the αR95 residue of CG, which leads to receptor activation.
A different scenario is observed in the case of mutations leading to constitutive activation of the FSHR and concomitant promiscuous binding of CG and/or TSH. Most mutations leading to constitutive activation of the FSHR are located in the 7 TMD, and considerably fewer naturally occurring activating mutations have been identified in the FSHR compared with the highly related LHCGR (Figs. 1B and 2) (Ulloa-Aguirre et al., 2014a); this difference suggests that the FSHR is generally more resistant than the LHCGR to mutation-induced constitutive activity (Zhang et al., 2007), albeit that activating FSHR mutations are generally difficult to detect in the clinical setting, particularly in otherwise healthy men, as they may not be associated to an evident phenotype (Casas-Gonzalez et al., 2012). In fact, when mutations leading to ligand-independent activation of LHCGR were introduced into the FSHR, the level of detected constitutive activity was consistently lower (Kudo et al., 1996; Zhang et al., 2007). Meanwhile, the same mutations exhibited stronger FSHR promiscuous activation by CG and TSH (Zhang et al., 2007). Strikingly, similar promiscuous activation has been detected in three of six reported naturally occurring constitutively active mutants of the FSHR [reviewed in (Ulloa-Aguirre et al., 2014a); Fig. 2], indicating a close link between constitutive activation of the FSHR and ligand promiscuity, a phenomenon not observed in the other GPHRs (Montanelli et al., 2004b; Vassart et al., 2004). These FSHR constitutively active mutants leading to spontaneous OHSS [D567N at the junction of TMD 6 and intracellular loop 3; T449A at TMD 3; and I545T at TMD 5) (Fig. 2)] are particularly interesting because the mutations provoke conformational changes in the receptor structure that, aside from triggering modest constitutive activity, “relax” the binding specificity of the receptor and allow the altered receptor to bind and become activated by high concentrations of CG (Smits et al., 2003; Vasseur et al., 2003; Montanelli et al., 2004a; De Leener et al., 2006) or TSH (Smits et al., 2003; Montanelli et al., 2004b; De Leener et al., 2006) without altering receptor affinity to GPH. As discussed above, in the unliganded state, the signal specificity subdomain of the FSHR ectodomain exerts an inhibitory influence on the transmembrane domain, keeping the receptor in relative quiescence; upon agonist binding or when mutations are introduced at Ser273 (at the hinge region) (Nakabayashi et al., 2000), the tethered extracellular loops of the TMDs are freed from this inhibitory influence, which allows the TMDs to attain an active conformation (Jiang et al., 2012). In this scenario, the OHSS pathogenesis associated with these constitutively active mutants has been explained by low affinity promiscuous interaction of CG or TSH with the ectodomain of a mutant FSHR in the setting of a partially “unlocked” FSHR 7 TMD and high agonist concentrations, which leads to excessive follicular recruitment (De Leener et al., 2006). The ability of the FSHR constitutively active mutant D567N to become activated by CG and TSH has also been confirmed in animal models (Allan et al., 2009). Thus, partial activation of the 7 TMD facilitates receptor activation by relaxing the inhibitory constraints present in this domain and by making the FSHR more prone to full activation by high concentrations of other gonadotropins that share an identical α-subunit with FSH, which also directly participates in receptor binding (see above) (Montanelli et al., 2004b). Undoubtedly, knowing the entire FSHR structure in both the inactive and active states will further clarify the molecular mechanisms mediating promiscuous binding and activation of the FSHR in the presence of gain-of-function mutations.
It appears that the relative resistance of the FSHR to ligand-independent activation (and, consequently, to ligand promiscuity) reflects an evolutionary strategy in primates to prevent FSHR activation and the development of disease (e.g., OHSS) resulting from very high levels of circulating CG or TSH during pregnancy or primary hypothyroidism, respectively (Vassart and Costagliola, 2011).
Finally, the last concept concerning agonist binding and receptor activation pertains to FSHR interaction with and response to FSH variants with differential glycosylation. All glycoprotein hormones exhibit up to four N-linked oligosaccharides, and in human FSH, these oligosaccharides contribute to nearly 30% of the glycoprotein’s mass and are located in both the common α-subunit (α52 and α78) and the hormone specific β-subunit (β7 and β24) (Bousfield et al., 2015). Glycans, critical for FSH function, play important roles in subunit assembly, intracellular trafficking, heterodimer secretion, circulatory half-life, receptor binding, and signal transduction at the target cell level (Ulloa-Aguirre et al., 2003). Furthermore, glycans in glycoprotein hormones are not homogeneous structures, but rather highly heterogeneous structures in terms of both oligosaccharide composition and glycan location within the protein core of the heterodimer (Ulloa-Aguirre et al., 2003; Bousfield et al., 2014b). Differential glycosylation of FSHβ appears to be closely related to physiologically distinct functions, because hypoglycosylated variants are more abundant in women of reproductive age, exhibit greater receptor-binding affinity, and occupy more FSH binding sites than fully glycosylated FSH, which predominates in men and in postmenopausal women (Butnev et al., 2015). Several studies have also indicated that the amount of glycans, the particular carbohydrate composition, and the structure of the attached oligosaccharides influence both binding and response to the FSH stimulus in vitro and in vivo (Creus et al., 2001; Barrios-De-Tomasi et al., 2002). An intriguing question is how FSH glycosylation influences FSHR binding and activation and leads to differential signal transduction, given that according to the 3-dimensional structure of the FSHR ectodomain, FSH glycans play no role in the binding interface of the FSH/FSHR complex, but rather appear to be sequestered in the periphery of the complex (Dias, 2005; Fan and Hendrickson, 2005; Jiang et al., 2012, 2014a). In this regard, the structures of FSH and the FSHR ectodomain in complex with FSH (Dias and Van Roey, 2001; Fox et al., 2001; Dias, 2005; Fan and Hendrickson, 2005) predict contact between the GlcNac1 residue at position Asn52 of FSHα [which is indispensable for signaling (Matzuk and Boime, 1989; Matzuk et al., 1989; Flack et al., 1994)] and Tyr58 in FSHβ. It is therefore possible that structural variations in the α52 glycan may influence the stability and/or conformation of the FSH ligand, thereby compromising the ability of the FSH/FSHR complex to affect signal transduction. Recently, extended molecular dynamics simulation studies on complexes of the FSHR HBSD (Fan and Hendrickson, 2005) and different FSH glycoforms were conducted (Meher et al., 2015). This study revealed that FSHR in complex with hypo(di-)glycosylated FSH exhibited greater conformational flexibility and a more favorable kinetic profile than tetraglycosylated FSH. Hypoglycosylated FSH also exhibited stronger binding free energy due to formation of closer and more persistent salt-bridges of both the α- and β-subunits of the variant with the FSHR. Thus, differences in FSH glycosylation appear to influence the stability of FSHR binding, with a more stable FSHR interaction of the hypoglycosylated FSH glycoform relative to the fully (tetra-) glycosylated; these differences in FSH glycoform binding suggest possible mechanisms for the variant biologic effects of fully and partially glycosylated FSH in vivo and in vitro (Walton et al., 2001; Bousfield et al., 2014a; Jiang et al., 2015). Furthermore, the recent model of the FSHR as a functional trimer (see below) (Jiang et al., 2014b) supports in vitro data showing that binding of bulky, fully glycosylated FSH to the FSHR is delayed and occurs at a lower rate compared with that of a more compact, hypoglycosylated variant bearing a high percentage of oligomannose glycans (Bousfield et al., 2014a). In this model, tetraglycosylated FSH with extended glycans would require more time to fit the α52 glycan into the central cavity of the receptor trimer than the hypoglycosylated FSH variant with a more compact glycan structure at this position (Jiang et al., 2014b). This model also may explain the different receptor binding activities and in vitro biologic potency of distinct FSH glycoforms with variations in sialic acid content, in the internal structure of the carbohydrate chains, and in glycan complexity (Ulloa-Aguirre et al., 1995; Creus et al., 2001).
FSHR Self-Association
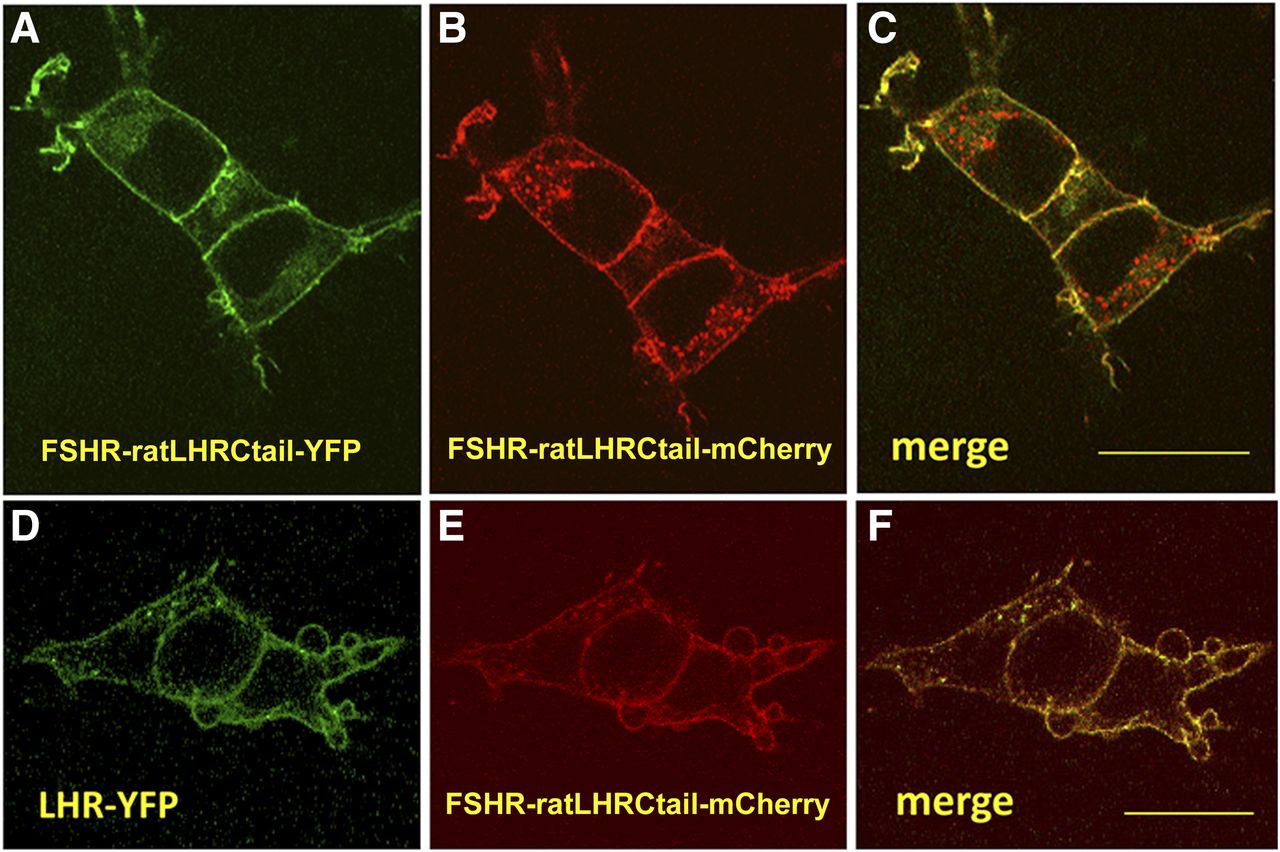
A number of biochemical, pharmacological, and biophysical studies support the concept of receptor self-association or oligomerization as a fundamental process enabling GPCR activity (Milligan, 2007). Mechanisms of action of G protein-coupled receptors confirm this important issue in different ways. Although some receptors are monomeric at the PM and then associate upon agonist binding and receptor activation, others form dimers or oligomers during synthesis at the endoplasmic reticulum or during processing in the Golgi apparatus. As with many other GPCRs, GPHRs also associate to form homodimers/oligomers and even heterodimers, in which two closely related receptors simultaneously expressed in the same cell [e.g., the LHCGR and FSHR in ovarian mature granulosa cells (Richards et al., 2002)] become associated. A number of studies have shown that the LHCGR and the TSHR do indeed self-associate (Horvat et al., 1999; Latif et al., 2001; Tao et al., 2004; Lei et al., 2007; Kleinau et al., 2013). Although the structure of the FSH-FSHRHB (Fan and Hendrickson, 2005) revealed that the FSHR ectodomain may form weakly associated dimers, with each molecule bearing one FSH molecule, later studies based on combined biochemical and biophysical approaches directly demonstrated that the FSHR self-associates early during receptor biosynthesis and that it can be identified as dimers or oligomers at the plasma membrane (Thomas et al., 2007). Furthermore, this study demonstrated that the carboxyl terminus of the receptor is clipped before trafficking to the plasma membrane (Thomas et al., 2007), a finding that partially explained the paucity of studies on FSHR association because of the difficulties in constructing either functional epitope-tagged FSHRs or FSHR fluorescent fusion proteins at this particular domain. More recently, Mazurkiewicz et al. (2015) overcame this problem by constructing functional chimeras, in which the carboxyl terminus domain of the human WT FSHR was replaced with that of the LHR [which in contrast to the FSHR is not clipped during its biosynthesis (Thomas et al., 2007)] fused with fluorescent proteins (FP). Fluorescence correlation spectroscopy and photon-counting histogram analysis showed that unlike the FSHR-FP, the FSHR-LHR-carboxyl terminus-FP chimera localized to the plasma membrane of transfected live HEK293 cells as a freely diffusing homodimer (Fig. 3, A–C). Using an intensity-based quantitative FRET assay (Elangovan et al., 2003; Wallrabe et al., 2007), Mazurkiewicz et al. (2015) also found that the FSHR-LHR-carboxyl terminus-FP chimera formed homodimers on the cell surface plasma membrane of transfected cells and that upon cotransfection with a WT LHR-fluorescent protein construct, both chimeras formed heterodimers (Fig. 3, D–F). Formation of intracellular FSHR/FSHR and FSHR/LHR complexes may explain the dominant negative effects of FSHR loss-of-function mutants on the plasma membrane expression of either WT FSHR or LHR (Zarinan et al., 2010), which, as shown in other GPCRs (e.g., the gonadotropin-releasing hormone receptor, the TSHR and the V2-vasopressin receptor) might eventually lead to disease (Zhu and Wess, 1998; Ulloa-Aguirre et al., 2004; Calebiro et al., 2005; Leanos-Miranda et al., 2005). Formation of FSHR/FSHR complexes also explains intermolecular complementarity between mutant FSHR dimers, in which function of a hormone binding-deficient FSHR protomer stably expressed in HEK293 cells may be rescued by a signal deficient counterpart after agonist exposure (Ji et al., 2004). Dimerization also explains negative cooperativity between FSHR receptor protomers [i.e., upon ligand binding one protomer blocks or decreases the affinity of the second protomer for its ligand or for intracellular signaling partners leading to a right shift of the dose-response curve (Urizar et al., 2005)], a form of negative dominance at the plasma membrane level. Although this effect may vary according to the cell context, interestingly, in the case of the LHR receptor, complementarity has been documented to also occur in vivo (Rivero-Muller et al., 2010), and this observation may herald important physiologic implications for the gonadotropin receptors. For example, temporal association and alternating negative cooperativity between the FSH and the LH receptors in the ovarian granulosa cells might protect the follicle from premature luteinization or, conversely, from ovarian hyperstimulation, depending on the expression level of each receptor (Yamashita et al., 2005; Feng et al., 2013). Heterozygous mutations that inactivate receptors may remain silent by virtue of WT-receptor rescue effect or may conversely be expressed as adverse clinical manifestations by virtue of the inactive receptor (i.e., with a trafficking defect), preventing the WT receptor from trafficking to the cell surface. The occurrence of homo- and heterodimerization as a mechanism for negative cooperativity between GPHRs (and other GPCRs) represents a unique opportunity for therapeutic interventions. FSHR/LHR heterodimerization is unique to the ovary given that cells expressing these receptors in the testes are located in separate compartments.

Confocal microscopy of HEK293 cells expressing fluorescent FSHR-ratLHRCtail/FSHR-ratLHRCtail homodimers and FSHR-ratLHRC tail/LHR heterodimers. (A–C) Representative images of HEK293 cells expressing FSHR-ratLHRCtail-yellow fluorescent protein (YFP) and FSHR-ratLHRCtail-mCherry fluorescent protein. (D–F) Representative images of HEK293 cells expressing LHR-YFP and FSHR-ratLHRCtail-mCherry. The images of the merged channels (C and F) demonstrate that FSHR-fluorescent protein chimeras traffic to the plasma membrane (C) and that LHR-YFP traffics to the plasma membrane along with the FSHR-ratLHRCtail-mCherry chimera (D). Quantitative FRET analyses demonstrated that FSHR can form homodimers and FSHR can form a heterodimer with LHR. FRET was detected between these receptor pairs on the plasma membrane with an energy transfer efficiency (E%) of 12.8 ± 1.7 for FSHR homodimers and 14.4 ± 0.8 for the FSHR/LHR heterodimers. In the FRET analysis, the images were loaded into an ImageJ plugin. See the text for additional details. Ctail, carboxyl-terminal tail. Bar = 20 µm.
The mechanism and extent of FSHR self-association is not known, but it seems reasonable to assume that contacts via the α-helices of the TMD play an important role. The more recent crystal structure of the ligand-bound ectodomain of the FSHR, which includes the signal specificity subdomain, demonstrated an additional mode of FSH association with the entire FSHR ectodomain and a quaternary structure bearing three individual ectodomains (i.e., a trimeric receptor structure). This structure predicts hosting of one fully glycosylated FSH molecule or three deglycosylated molecules (Jiang et al., 2014b). In the former scenario, the FSHR may become activated, whereas in the latter, the structure remains inactive, supporting early observations on the lack of effect of deglycosylated FSH in triggering signal transduction at the FSHR (Sairam and Bhargavi, 1985; Sairam, 1989). The trimeric model also explains the reported differences in receptor binding activity between fully glycosylated and hypoglycosylated FSH isoforms, with the former exhibiting delayed and lower binding activities (Bousfield et al., 2014b; Meher et al., 2015). It also suggests a mechanism for FSHR association with multiple proteins and multiple signaling pathways (see below). In contrast with the previously reported structure (Fan and Hendrickson, 2005), Jiang et al. (2014b) structure additionally supports intermolecular associations between the serpentine domains of the protomers, which when associated can only accommodate one Gs protein heterotrimer. In fact, mutational and interfering peptide fragment studies previously suggested that the association between FSHR monomers might occur through the TM domains and/or the carboxyl terminus (Zarinan et al., 2010), as previously demonstrated in the β2-adrenergic receptor (Hebert et al., 1996) and the dopamine D2 receptor homodimerization (Ng et al., 1996; Lee et al., 2003), and the µ- and ∂-opioid receptor hetero-oligomerization (Fan et al., 2005).
Signal Transduction at the FSHR: Multiple Interacting Partners, Signaling Networks, and Functional Selectivity
It is currently accepted that after FSHR activation, a number of intracellular signaling pathways become activated either in parallel or sequentially. The canonical Gsα/cAMP/PKA signaling pathway, which subsequently activates cAMP response element-binding protein, thereby modulating gene transcription, has been recognized for more than two decades as a key effector mechanism of gonadotropin biologic action (Means et al., 1974; Dattatreyamurty et al., 1987; reviewed in Ulloa-Aguirre et al., 2007b). However, in recent years, it has become evident that the FSHR is connected by conformational selectivity to a nonlinear and complex signaling network mediated either by other G protein subtypes, including the Gi, Gq/11, and Gh proteins (Quintana et al., 1994; Arey et al., 1997; Zeleznik et al., 2003; Lin et al., 2006); other types of receptors such as the epidermal growth factor receptor and the insulin-like growth factor 1 receptor (Wayne et al., 2007); and/or particular receptor-associated proteins including β-arrestins and adaptor protein containing the pleckstrin homology domain, the phosphotyrosine binding domain, and the leucine zipper motif (APPL) (Dias et al., 2005, 2010; Kara et al., 2006; Marion et al., 2006; Nechamen et al., 2007; Thomas et al., 2011). These associations promote activation of a number of signaling pathways, including those mediated by distinct kinases such as PKC, PI3K, PKB/Akt, and ERK1/2 (Quintana et al., 1994; Gonzalez-Robayna et al., 2000; Richards et al., 2002; Zeleznik et al., 2003; Kara et al., 2006; Wayne et al., 2007). This complex network of pathways fine tunes the regulation of the FSH stimulus and related ligands (see below), in which the activation/inhibition of its multiple components vary depending on the target cell (e.g., granulosa versus Sertoli cells versus immortalized cell lines), the particular developmental stage of the host cells (e.g., cells from prepubertal versus adult animal donors or in cells from ovarian follicles at different maturation stages), and the receptor and ligand concentration (Crepieux et al., 2001; Richards et al., 2002; Donadeu and Ascoli, 2005; Wayne et al., 2007; Musnier et al., 2009; Casarini et al., 2012; Breen et al., 2013).
The FSHR-mediated pathways triggered by cAMP accumulation independently of PKA activation, such as those activated by the exchange protein directly activated by cAMP, are of particular relevance (Wayne et al., 2007). In addition to cAMP response element-binding protein, cAMP-activated PKA activates p38 mitogen-activated protein kinases, ERK 1/2, p70S6 kinase (p70S6K), and PI3K via insulin receptor substrate 1, which leads to PKB/Akt transactivation (Hunzicker-Dunn et al., 2012; Baumgarten et al., 2014; Law and Hunzicker-Dunn, 2016); exchange protein directly activated by cAMP promotes p38 MAPK and PKB/Akt phosphorylation, and upregulation of the EGFR (Gonzalez-Robayna et al., 2000; Wayne et al., 2007; Choi et al., 2009). As mentioned above, in addition to G proteins, the FSHR also associates with a number of other interacting proteins including the APPL1 and 14-3-3τ adapters and β-arrestin 1 and 2 scaffolds (Cohen et al., 2004; Nechamen et al., 2004; Kara et al., 2006; Nechamen et al., 2007; Dias et al., 2010). Although linking of the FSHR with APPL1 occurs at the receptor intracellular loop 1, particularly at Lys393 (Thomas et al., 2011) (Fig. 1B), association with 14-3-3τ maps to the intracellular loop 2, which overlaps with the canonical G protein binding sites (Cohen et al., 2004; Dias et al., 2010). Follicle-stimulating hormone stimulation rapidly induces FOXO1a phosphorylation in HEK293 cells, leading to repression of apoptosis via either serum and glucocorticoid-induced kinase (Sgk) or APPL1 interaction with the upstream PI3K/Akt pathway (Gonzalez-Robayna et al., 2000; Cunningham et al., 2003; Nechamen et al., 2004). In addition, APPL1 also participates in the FSH-stimulated inositol 1,4,5 triphosphate pathway and is thus implicated in intracellular calcium signaling in granulosa cells (Thomas et al., 2011). The coexistence of APPL1 and 14-3-3τ associated with the FSHR suggests a scenario in which FSH causes phosphorylated FOXO1a to be sequestered by 14-3-3τ with APPL1 to facilitate this antiapoptotic process (Dias et al., 2010).
The FSHR activates ERK1/2 phosphorylation through two distinct pathways, one mediated by Gsα and the other by β-arrestins (Crepieux et al., 2001; Cottom et al., 2003; Kara et al., 2006), which promote desensitization and internalization of the phosphorylated receptor at high levels of receptor occupancy (Ayoub et al., 2015). In addition, these downstream effectors are also involved in the regulation of intracellular signaling via activation of distinct signaling cascades (Reiter and Lefkowitz, 2006). When stimulated by the Gsα-PKA pathway, ERK1/2 phosphorylation occurs via PKA-promoted dissociation of ERK from a 100-kDa phosphotyrosine phosphatase that inhibits ERK activation and its translocation to the nucleus (Cottom et al., 2003). When stimulated by this PKA-mediated pathway, ERK activation occurs early and is transient, peaking 5–10 minutes after FSH exposure, whereas in the Gsα-independent pathway mediated by β-arrestins, it is slower, sustained, and mainly occurs during the ensuing 10–30 minutes after agonist exposure (Kara et al., 2006). As with other GPCRs, the activated FSHR interacts with G protein receptor kinases (GRK), specifically with GRKs 2, 5, and 6 (Troispoux et al., 1999; Kara et al., 2006), which phosphorylate the receptor while simultaneously promoting β-arrestin recruitment (Lazari et al., 1999; Kara et al., 2006). In this regard, a cluster of five serines and threonines (Fig. 1B) at the carboxyl terminus of the FSHR has been shown to account for the bulk of agonist-induced FSHR phosphorylation by GRK2, although GRK5 and 6 also contribute to the same process, but to a lower extent. However, whereas GRK2-phosphorylated FSHR predominates in the β-arrestin-mediated desensitization/internalization process, GRK5- and 6-promoted receptor phosphorylation is required for β-arrestin-dependent, Gsα-independent signaling (Kara et al., 2006; Reiter and Lefkowitz, 2006).
It is currently known that differential or preferential activation of distinct GPCR-mediated signaling cascades may occur through functional selectivity, also referred to as biased agonism, resulting from stabilization of distinct receptor conformations (Venkatakrishnan et al., 2013; Luttrell et al., 2015) by particular ligands or receptor mutations (Wei et al., 2003; Shenoy et al., 2006; Reiter et al., 2012; Luttrell et al., 2015).
In the case of the FSHR, functional selectivity toward Gi-mediated signaling has been documented for some naturally occurring glycosylated variants of human FSH (Timossi et al., 1998; Timossi et al., 2000), in recombinant FSH expressed in insect cells (Arey et al., 1997; Arey and Lopez, 2011), as well as for selective thiazolidinone allosteric compounds with FSH agonist or antagonist properties (Arey et al., 2008). Bias toward β-arrestin-mediated ERK1/2 MAPK signaling has also been observed after exposure to a modified agonist [truncated equine LHβ (Δ121-149) combined with Asn56-deglycosylated equine LHβ] (Wehbi et al., 2010) in HEK293 cells expressing the FSHR; this molecule is particularly interesting because it preferentially activates the β-arrestin-ERK1/2 phosphorylation module, but simultaneously acts as an antagonist of FSH-stimulated cAMP-PKA signaling when tested at equimolar concentrations (Butnev et al., 2002; Wehbi et al., 2010). Recently, Dias et al. (2011) identified a negative allosteric modulator (ADX61623) that increased the affinity of FSH for the FSHR but inactivated FSH-stimulated cAMP and progesterone production, while having no effect on FSH-mediated estrogen production. Interestingly, another negative allosteric modulator [ADX68693 (Dias et al., 2014)] exhibited similar effects on progesterone and estradiol production in vitro and when administered orally to immature female rats, did not block FSH-induced follicle development, which demonstrates the feasibility of applying the concept of biased agonism to in vivo conditions for therapeutic purposes in the reproduction field. At the receptor level, biased agonism has been documented when FSHR plasma membrane expression is severely impaired as a result of a loss-of-function mutation (A189V) (Aittomaki et al., 1996) (Fig. 2); in HEK293 cells, β-arrestins recruited to the agonist-bound FSHR assembled a MAPK module, whereas G protein-dependent signaling remained impaired (Tranchant et al., 2011). Also, expression of the receptor bearing the missense M512I FSHR mutation in HEK293T cells led to an impaired FSH response in terms of cAMP accumulation and PI3K activation but not ERK1/2 phosphorylation (Uchida et al., 2013). A corollary to these findings is that our traditional manner of analyzing the functional impact of structural alterations in ligands and/or receptors should be redesigned to include measurement of multiple readouts by sensitive assays. This may eventually allow further detailed insight into the complex network of signals triggered by FSHR activation and allow us to unambiguously identify the biased effects resulting from such molecular modifications.
Concluding Remarks
The first experimental evidence that the gonads exhibit binding sites specific for FSH was reported in the early 1970s (Means and Vaitukaitis, 1972). Since then, significant progress in the understanding of the structure and function of the FSH/FSHR system has been achieved. Nevertheless, there are still important issues that remain to be resolved. These include the elucidation of the entire structure of the 7 TMD, which undoubtedly will allow a more precise understanding of the mechanisms of receptor activation, oligomerization, and cooperativity, as well as the distinct conformations adopted by the receptor upon association with different ligands, allosteric modulators, and effector proteins. Such knowledge may have important therapeutic implications in the design of potentially useful molecules for the treatment of infertility and in contraception. In this vein, the recently described sequence at the carboxyl-terminal end of the hinge region of all GPHRs that acts as an internal agonist unit after structural changes in the extracellular domain (Fig. 1B) is worth mentioning (Bruser et al., 2016). The possibility of synthesizing related ligands that may be used to modulate ligand-stimulated FSHR activation, particularly in conditions in which receptor mutations lead to promiscuous activation by TSH and/or CG, along with those associated to putative extragonadal effects of FSH, such as menopausal osteoporosis, hyperlipidemia, and tumoral angiogenesis (Radu et al., 2010; Robinson et al., 2010; Sun et al., 2010; Cannon et al., 2011; Siraj et al., 2013; Planeix et al., 2015; Song et al., 2016), could also be considered.
It is also important to continue assembling the complex signaling network and modules that are activated after FSH binding (Gloaguen et al., 2011). This activity provides a suitable testbed for intentional perturbation that may reveal the intracellular mechanisms that control preferential activation of distinct pathways and the crosstalk between different signaling cascades triggered upon FSHR activation. This information can then be further leveraged to understand how the modulation of activation of these pathways governs transcriptional, translational, and posttranslational processes triggered by FSH. Undoubtedly, the application of “omic” technologies, including genomic, transcriptomic, and proteomic techniques, will enormously facilitate this challenging task. In this regard, it seems that the past is just the prologue to what remains to be unveiled.
Acknowledgments
The authors thank George R. Bousfield, Wichita State University, Wichita, KS, and Gunnar Kleinau, Charité-Universitätsmedizin Berlin, Berlin, Germany, for kindly providing Figs. 1 and 2, respectively, and to Joseph E. Mazurkiewicz from Albany Medical College, Albany, NY, for providing Fig. 3. Ilpo Huhtaniemi, Imperial College London, London, England; James A. Dias, University of Albany, Albany, NY; and Gunnar Kleinau, Charité-Universitätsmedizin Berlin, Berlin, Germany, kindly critically read the manuscript. Ari Kleinberg, from the Research Support Network, UNAM, modified Fig. 1 with permission from George R. Bousfield.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Ulloa-Aguirre, Zariñán.
Footnotes
- Received March 21, 2016.
- Accepted June 28, 2016.
Research in the authors' laboratory is supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT), Mexico [grant 240619], and the Coordinación de la Investigación Científica, Universidad Nacional autónoma de México (UNAM).
Abbreviations
- APPL
- adaptor protein containing pleckstrin homology domain, phosphotyrosine binding domain, and leucine zipper motif
- FSH
- follicle-stimulating hormone
- GPH
- glycoprotein hormone
- GPHR
- GPH receptor
- GRK
- G protein-receptor kinase
- HBSD
- hormone-binding subdomain
- LH
- luteinizing hormone
- LHR or LHCGR
- luteinizing hormone-chorionic gonadotropin receptor
- MAPK
- mitogen-activated protein kinase
- OHSS
- ovarian hyperstimulation syndrome
- PI3K
- phosphoinositide-3 kinase
- PKA
- protein kinase A
- PKB
- protein kinase B
- TMD
- transmembrane domain
- TSHR
- thyroid-stimulating hormone receptor
- WT
- wild type
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Visual Overview
- Abstract
- Introduction
- FSHR Structure
- Extragonadal Expression of the FSHR
- Ligand Binding and Receptor Activation: Role of the FSHR Ectodomain
- FSHR Self-Association
- Signal Transduction at the FSHR: Multiple Interacting Partners, Signaling Networks, and Functional Selectivity
- Concluding Remarks
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters