


Abstract
The ability of chemically distinct ligands to produce different effects on the same G protein–coupled receptor (GPCR) has interesting therapeutic implications, but, if excessively propagated downstream, would introduce biologic noise compromising cognate ligand detection. We asked whether cells have the ability to limit the degree to which chemical diversity imposed at the ligand–GPCR interface is propagated to the downstream signal. We carried out an unbiased analysis of the integrated cellular response elicited by two chemically and pharmacodynamically diverse β-adrenoceptor agonists, isoproterenol and salmeterol. We show that both ligands generate an identical integrated response, and that this stereotyped output requires endocytosis. We further demonstrate that the endosomal β2-adrenergic receptor signal confers uniformity on the downstream response because it is highly sensitive and saturable. Based on these findings, we propose that GPCR signaling from endosomes functions as a biologic noise filter to enhance reliability of cognate ligand detection.
Introduction
G protein–coupled receptors (GPCRs) comprise the largest family of signaling receptors and a very important class of therapeutic targets. Initially, GPCRs were thought to transduce external cues into cellular responses in a linear fashion, with ligand binding to the receptor promoting a binary transition from inactive off to active on states coupled to downstream effector control through G proteins (Kenakin, 1997). Over the past couple of decades, however, experimental evidence has accumulated, indicating the existence of remarkable diversity in GPCR activation states and the effects of chemically distinct ligands. Now it is widely recognized that there are multiple sources of functional diversity at GPCRs, involving not only equilibrium affinity and intrinsic efficacy of the drug, but also kinetics of the drug–GPCR interaction and an extended theoretical formulation of intrinsic efficacy that is now called functional selectivity or agonist bias (Kenakin, 2011).
Recognition of the expanded diversity of ligand action at a target GPCR has exciting implications for drug development but raises a significant biologic problem. In principle, any chemical difference between ligands could differentially bias the receptor's conformational landscape. Indeed, with the development of more sophisticated assays, essentially all drugs, even natural ligands, appear to differ in some way in receptor-based effects (Thompson et al., 2015). However, from an evolutionary point of view, receptor-dependent signaling systems need to mediate reliable transfer of physiologically salient information, and thus fluctuations at the ligand–GPCR interface could be considered a source of biologic noise. These considerations imply a physiologic imperative for receptors to generate a stereotyped rather than divergent integrated cellular response to binding of a cognate ligand. Although functional diversity downstream of the drug–GPCR interface has been widely observed and investigated, the converse possibility has received less consideration: Do cells have the ability to generate a stereotyped, rather than variable, downstream response to chemically distinct drugs? If so, how is diversity inherent to the chemistry of drug–GPCR interactions moderated to produce a uniform response?
We addressed these questions by focusing on the β2-adrenoceptor (β2-AR), a prototypical GPCR with rich pharmacology that mediates cardiovascular regulation by naturally produced catecholamines. We began by assessing the effects of a panel of synthetic and naturally occurring β2-AR ligands on receptor–G protein coupling and internalization, and saw extensive differences in these early signaling events that reflected the different properties of each ligand at the ligand–receptor interface. Next, we focused on isoproterenol (Iso) and salmeterol (Sal), two clinically relevant drugs that act through β2-ARs. In addition to differing in chemical structure, these drugs also differ in binding affinity and intrinsic efficacy: Iso is classified as a full agonist and Sal as a higher affinity partial agonist (January et al., 1998; Nino et al., 2009). Furthermore, they exhibit distinct receptor-binding kinetics, with Sal having a much slower dissociation rate than Iso (Nials et al., 1993; Sykes and Charlton, 2012). Moreover, Sal drives β2-AR internalization less strongly than Iso (Moore et al., 2007) and is reported to be functionally selective, as indicated by a moderate β-arrestin bias (Rajagopal et al., 2011). Accordingly, Iso and Sal differ in multiple pharmacological properties at the ligand–receptor interface.
We report in this study a phosphoproteomic and transcriptional profiling approach to comprehensively compare the integrated cellular response elicited by these drugs in cells expressing native β2-ARs at endogenous levels. We do not observe qualitative differences in the phosphoresponses elicited by the two drugs, but the partial agonist Sal induces a quantitatively less robust response than the full agonist Iso. Remarkably, the two drugs are indistinguishable, both qualitatively and quantitatively, in the transcriptional response that they elicit. This is surprising because full transcriptional signaling mediated by β2-AR activation requires receptor internalization and signaling from endosomes (Tsvetanova and von Zastrow, 2014), and Sal is generally recognized to promote β2-AR internalization less strongly than Iso. We show in this work that the endocytosis-dependent transcriptional signal is very sensitive and saturates at a low level of receptor internalization. We also show that the endosome signal saturates at a level considerably below the cell’s transcriptional capacity. Furthermore, we demonstrate that the stereotyped signaling response is accomplished by increasing the number of receptor-containing endosomes in a ligand dose-dependent manner but keeping the receptor concentration per endosome constant. These results provide, to our knowledge, the first unbiased catalog of drug action on integrated signaling of a GPCR expressed at endogenous levels, and the first demonstration that cells can produce a stereotyped, rather than variable, downstream response to chemically diverse GPCR ligands. They also reveal a previously unanticipated role of endosome signaling in conferring uniformity on the response. We propose that endosomal GPCR activation operates as part of a cellular strategy to reduce downstream transduction of chemical noise that is introduced by variability at the ligand–GPCR interface, thereby enhancing reliability of cognate ligand detection.
Materials and Methods
Adrenergic Ligands.
(-)-Iso hydrochloride, (-)-Epinephrine (Epi), (-)-Norepinephrine (Norepi), Terbutaline (Terb) hemisulfate salt, and Dopamine (Dopa) hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). Sal xinafoate was purchased from Tocris Bioscience (Pittsburgh, PA). Saturating doses of each drug were applied as follows: 1 μM Epi, 10 μM Norepi, 10 μM Terb, 10 μM Dopa, 1 μM Iso, 50 nM Sal. (-)-Iso Hydrochloride, (-)-Epi, (-)-Dopa hydrochloride, and (-)-Norepi were dissolved in water/100 mM ascorbic acid; Terb hemisulfate salt was dissolved in water; Sal xinofoate was dissolved in dimethylsulfoxide (DMSO).
Cell Culture.
Human embryonic kidney (HEK)293 cells endogenously expressing β2-AR were obtained from American Type Culture Collection (Manassas, VA) and grown in a CO2- and temperature-controlled incubator. Stably transfected HEK293 cells expressing FLAG-tagged β2-AR were described previously (Temkin et al., 2011). HEK293 cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Grand Island, NY) with 10% fetal bovine serum (FBS; University of California, San Francisco Cell Culture Facility, San Francisco, CA). For stable isotope labeling with amino acids in cell culture (SILAC) experiments, HEK293 cells were grown in DMEM deficient in L-arginine and L-lysine and supplemented with L-lysine and L-arginine, or doubly labeled 13C-labeled lysine and 13C,15N-labeled arginine to a final concentration of 0.46 mM each with 10% FBS (Thermo Scientific, Grand Island, NY). Cells were maintained in specific isotope conditions for a minimum of six doublings, with frequent medium changes. Primary cultures of human airway smooth muscle cells were established from bronchial explants of lung transplant donors. Bronchi were dissected out of the lung and placed on a dish containing Hanks’ balanced salt solution (Corning, Corning, NY) supplemented with penicillin/streptomycin (University of California, San Francisco Cell Culture Facility). Bronchial segments were dissected into 5- to 8-mm squares and placed in a six-well plate. After adherence, DMEM (Corning) with 20% fetal calf serum (Gibco) was added to cover the explants. Explanted bronchi were subsequently removed when there was a local confluence of the outgrowth of smooth muscle cells. The purity of airway smooth muscle cells was confirmed by anti–α-smooth muscle actin staining (Sigma-Aldrich). Dyngo-4a (Abcam, Cambridge, MA) was dissolved in DMSO to 30 mM, stored protected from light, and added to cells to 30 μM final concentration in serum-free DMEM.
cAMP Measurements Using a Luminescence-Based cAMP Biosensor.
Plasmid pGLO-20F (Promega, Madison, WI) encoding a circularly-permuted firefly luciferase cAMP reporter was transfected into HEK293 cells and assayed, as described previously (Tsvetanova and von Zastrow, 2014). For every experiment, reference wells were treated with 5 μM forskolin (Sigma-Aldrich) and all experimental cAMP measurements were normalized and displayed as percentage of the maximum luminescence value measured in the presence of forskolin.
Sample Preparation for Mass Spectrometry.
Cells were grown to ∼90% confluence in 15-cm–round cell culture dishes containing 20 mL appropriate culture medium. Cells were grown in lysine- and arginine-depleted medium supplemented with regular lysine (Lys 0) and arginine (Arg 0) (referred to as light), or [13C] lysine (Lys 6) and [13C, 15N] arginine (Arg 10) (referred to as heavy). Two dishes (one light, one heavy) were used for each agonist condition [low Iso (10 nM), high Iso (1 μM), or Sal (50 nM)]. Six dishes (three light, three heavy) were used for untreated cells. Prior to drug treatment, cells were washed once in phosphate-buffered saline and twice in serum-free DMEM, and then grown in 20 ml serum-free DMEM for 16 hours. The next day, cells were treated with drug or vehicle (DMSO) for 20 minutes, then detached in prewarmed phosphate-buffered saline with 0.04% EDTA containing drug or vehicle for 10 minutes in an incubator, and collected by centrifugation at 4°C. Each drug condition had a total of two biologic replicates. One biologic replicate was carried out per medium type as follows: 1) drug-treated cells were grown in heavy medium and untreated cells were grown in light medium, and 2) drug-treated cells were grown in light medium and untreated cells were grown in heavy medium. The type of SILAC medium used to label the untreated and agonist-treated cells was swapped for each condition tested to minimize impact of SILAC-based labeling artifacts. Cells were lysed in 5 M urea, 0.2% N-dodecyl-maltoside, and phosphatase inhibitors (Sigma-Aldrich phosphatase inhibitors 2 and 3). Individual lysates (∼2 mg total protein) were sonicated at 12% amplitude using a Fisher sonicator (Fisher Scientific, Pittsburgh, PA) for total of 20 seconds until lysates were clear (10 seconds on, 10 seconds off, 10 seconds on). Nanodrop was used to estimate approximate protein concentrations prior to mixing untreated versus agonist-treated samples at a final 1:1 ratio. Each mixed sample was reduced using 10 mM TCEP (tris(2-carboxyethyl)phosphine) at room temperature for 30 minutes, followed by alkylation with 18 mM iodoacetamide for 30 minutes and quenching with 18 mM dithiothrietol. Urea concentration was adjusted to 2 M before digestion with modified trypsin (1:20 enzyme:substrate ratio; Promega) to each mixed sample and incubating overnight at 37°C on a rotator. Peptides were desalted with SepPak C18 solid-phase extraction (Waters, Milford, MA), according to the manufacturer’s specifications, and lyophilized to dryness. Samples were dried and resuspended in 0.1% formic acid (FA) in preparation for mass-spectrometric analysis.
Phosphopeptide Enrichment.
Phosphopeptides were purified from approximately 1 mg each sample. The use of IMAC/C18 columns is described in the literature (Kokubu et al., 2005; Ficarro et al., 2009; Mertins et al., 2013). Fe3+-IMAC resin was created by taking Ni-NTA Superflow agarose resin (Qiagen, Germantown, MD) and stripping out Ni by incubating with 500 mM EDTA, pH 8 (1:1 v/v), for 5 minutes, and repeating. Fe3+ was added by incubating stripped beads with 10 mM iron chloride (1:1 v/v) for 5 minutes and repeating. After washing beads with 0.5% FA twice and water twice (1:1 v/v), 10 µl Fe3+-NTA resin was placed on top of wetted micro spin C18 columns (NEST group, Southborough, MA) for each sample. Micro spin columns were placed on a 20 port VacMaster vacuum manifold device for this procedure (Biotage, Charlotte, NC). Prior to phosphopeptide enrichment, 1 mg dried peptides described in the previous section were resuspended in 200 µl 80% MeCN and 0.2% trifluoroacetic acid. Each sample was added to the aliquoted Fe3+-NTA beads, mixed, and incubated for 5 minutes. The beads were washed with 200 µl 80% MeCN and 0.1% trifluoroacetic acid four times. The beads were then washed twice with 200 µl 0.5% FA. The samples were eluted from the Fe3+-NTA beads by washing twice with 200 µl 500 mM Na2HPO4, pH 7. The beads and C18 resin were then washed twice with 200 µl 0.5% FA. The enriched peptides were eluted from the C18 resin with 150 µl 50% MeCN and 0.2% FA. The eluted peptides were dried by speed-vac and resuspended in 50 µl 0.1% FA prior to liquid chromatography-mass spectrometry analysis.
Mass Spectrometry and Data Analysis.
Purified phosphopeptides were analyzed in technical duplicate on a Thermo Scientific LTQ Orbitrap Elite mass-spectrometry system equipped with a Proxeon Easy nLC 1000 ultra-high pressure liquid chromatography and autosampler system. Samples were injected onto a C18 column (25 cm × 75 μm I.D. packed with ReproSil Pur C18 AQ 1.9-μm particles) and subjected to a 4-hour gradient from 0.1% FA to 30% acetonitrile/0.1% FA. The mass spectrometer collected data in a data-dependent fashion, collecting one full scan in the Orbitrap at 120,000 resolution, followed by 20 collision-induced dissociation tandem mass spectrometry (MS/MS) scans in the dual linear ion trap for the 20 most intense peaks from the full scan. Dynamic exclusion was enabled for 30 seconds with a repeat count of 1. Charge state screening was employed to reject analysis of singly charged species or species for which a charge could not be assigned.
Raw mass spectrometry data were analyzed using the MaxQuant software package (version 1.3.0.5) (Tyanova et al., 2015). Data were matched to SwissProt reviewed entries for Homo sapiens in the UniProt protein database. MaxQuant was configured to generate and search against a reverse sequence database for false discovery rate calculations. Variable modifications were allowed for methionine oxidation; protein N terminus acetylation; and serine, threonine, and tyrosine phosphorylation. A fixed modification was indicated for cysteine carbamidomethylation. Full trypsin specificity was required. The first search was performed with a mass accuracy of ±20 ppm, and the main search was performed with a mass accuracy of ±6 ppm. A maximum of five modifications was allowed per peptide. A maximum of two missed cleavages was allowed. The maximum charge allowed was 7+. Individual peptide mass tolerances were allowed. For MS/MS matching, a mass tolerance of 0.5 Da was allowed, and the top six peaks per 100 Da were analyzed. MS/MS matching was allowed for higher charge states, water, and ammonia loss events. The data were filtered to obtain a peptide, protein, and site-level false discovery rate of 0.01. The minimum peptide length was seven amino acids. Results were matched between runs with a time window of 2 minutes for technical duplicates.
The data were condensed by a Perl script that takes the maximum intensity of any unique peptide and charge state between the two technical replicates. We calculated fold change between the raw phosphopeptide abundances using the best practices in the DESeq239 manual. For example, technical replicate results were averaged to yield one value per peptide. The filtered data were log transformed (log2) and median centered. To identify high-confidence β2-AR–regulated phosphosites, we considered peptides that had statistically significant log2 values (based on z-scores with P < 0.05) in each of the two media swaps for each experimental condition (high Iso, low Iso, and Sal). The phosphosites are summarized in Supplemental Tables 2 and 3.
We performed randomized simulations to assess the statistical significance of the observed differences in phosphorylation of targets across the three drug conditions. SILAC medium swap experiments were treated as a total of two biologic replicates per drug condition. For each condition, we performed a linear regression analysis of the data from the replicates. We then used the parameters of the regression fit to computationally generate 10,000 independent simulated measurements for each condition (assuming a Gaussian distribution with a mean and S.D. estimated from the residuals of the regression). Next, we calculated the average difference between the observed and simulated values of phosphorylation levels across all targets, and we tabulated the values of this summary statistic across all simulations to estimate a null distribution for this condition. We then evaluated whether the observed average differences in phosphorylation levels from other drug conditions were statistically significant by computing their empirical p values according to the null distribution. This analysis was repeated for each condition independently, and results are summarized in Supplemental Table 4.
Enrichment of amino acid motifs was determined with MotifX software (Chou and Schwartz, 2011) and plotted using WebLogo (Crooks et al., 2004), and associated kinases were assigned with the NetPhorest algorithm (Miller et al., 2008).
Western Blotting.
Cells were grown in 6-cm dishes in serum-free medium overnight. Drugs were added for 30 minutes, and cells were lysed on ice in ice-cold lysis buffer [50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitors 2 and 3 (Sigma-Aldrich), and 1 mM phenylmethylsulfonyl fluoride], spun down to collect the soluble fraction, and concentrated with Amicon Ultra centrifugal filter units with ultracel-10 membrane (Millipore, Billerica, MA). Lysates were boiled at 70°C in LDS sample buffer (Invitrogen, Waltham, MA) and loaded on NuPage 4–12% Bis-Tris gels (Invitrogen). Gels were transferred onto nitrocellulose membranes, blocked in Tris-buffered saline (TBS)/0.05% Tween 20/5% milk, and incubated with 1:500 anti–β-catenin Ser552 (Bioss, Woburn, MA) or 1:1000 anti–ATP-citrate lyase Ser455 (Cell Signaling Technology, Danvers, MA) in TBS/0.05% Tween 20/5% bovine serum albumin overnight at 4°C on a shaker. Membranes were washed, incubated with secondary antibodies conjugated to horseradish peroxidase in TBS/0.05% Tween 20/5% milk for 1 hour at room temperature, washed, and visualized on film. Membranes were subsequently stripped in Restore PLUS Western blot stripping buffer (Thermo Scientific) and reprobed with 1:1000 anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Millipore) in TBS/0.05% Tween 20/5% milk. Bands were quantified using ImageJ.
DNA Microarray Sample Preparation and Data Processing.
Human exonic evidence based oligonucleotide microarrays printed on epoxysilane-coated glass (Schott Nexterion E; Schott, Tempe, AZ) were purchased from the Stanford Functional Genomics Facility (Stanford, CA) and processed according to standard protocols (Tsvetanova et al., 2010). HEK293 cells were grown in six-well plates at ∼90–100% confluency in DMEM supplemented with 10% FBS. Drugs (1 μM Iso, 10 nM Iso, or 50 nM Sal) or vehicle (DMSO for no drug) were added for 2 hours. Total RNA was isolated, amplified, labeled with either Cy5 (drug) or Cy3 (no drug) dyes (GE Healthcare Life Sciences, Pittsburgh, PA), and hybridized to microarrays at 65°C using the MAUI hybridization system (BioMicro, Salt Lake City, UT) for 12–16 hours, as described previously (Tsvetanova and von Zastrow, 2014). Microarrays were washed according to standard protocols and scanned using AxonScanner 4000B (Molecular Devices, Sunnyvale, CA), in which photomultiplier voltages were manually adjusted for every slide scanned to maximize signal without saturation. All data were log-transformed (log2) and median-centered for comparison across conditions. β2-AR target genes were determined for 10 nM Iso and 50 nM Sal independently, as previously described (Tsvetanova and von Zastrow, 2014). Genes were classified as targets for a given condition, if their expression was induced ≥1.5-fold by drug treatment in each of three replicates for a given drug condition, and if their averaged expression showed at least a twofold increase relative to untreated samples, that is, log2 (drug and no drug ≥1). All targets were combined for a total of 84 genes (Supplemental Table 5). All DNA microarray datasets were deposited on Gene Expression Omnibus under accession number GSE87461.
Quantitative Real-Time Polymerase Chain Reaction.
Total RNA was extracted from samples with RNeasy Mini Kit (Qiagen). Reverse transcription was carried out with SuperScript III RT (Invitrogen) and a mix of oligo(dT) and random nonamer primers following standard protocols. The resulting cDNA was used as input for quantitative polymerase chain reaction (PCR) with StepOnePlus (Applied Biosystems, Waltham, MA) and SYBR Select MasterMix (Invitrogen). Statistical significance was established with unpaired t test. All levels were normalized to the levels of a housekeeping gene (ACTA or GAPDH). The following primer pairs were used: PCK1 forward, 5′-CTGCCCAAGATCTTCCATGT-3′ and reverse, 5′-CAGCACCCTGGAGTTCTCTC-3′; ACTA forward, 5′-CTGAGCGTGGCTACTCCTTC-3′ and reverse, 5′-GCCATCTCGTTCTCGAAGTC-3′; GAPDH forward, 5′-CAATGACCCCTTCATTGACC-3′ and reverse, 5′-GACAAGCTTCCCGTTCTCAG-3′; CHC17 forward, 5′-ACTTAGCCGGTGCTGAAGAA-3′ and reverse, 5′-AACCGACGGATAGTGTCTGG-3′; PDE4D forward, 5′-GGACACTTTGGAGGACAATCGTG-3′ and reverse, 5′-CCTTTTCCGTGTCTGACTCACC-3′; DUSP1 forward, 5′-CAACCACAAGGCAGACATCAGC-3′ and reverse, 5′-GTAAGCAAGGCAGATGGTGGCT-3′; CGA forward, 5′-TCCATTCCGCTCCTGATGTGCA-3′ and reverse, 5′-CGTCTTCTTGGACCTTAGTGGAG-3′; and CHC17 forward, 5′-ACTTAGCCGGTGCTGAAGAA-3′ and reverse, 5′-AACCGACGGATAGTGTCTGG-3′.
Flow Cytometry.
For β2-AR internalization assays, we used stably transfected FLAG-β2-AR cells. Cells were treated with indicated doses of ligand for 20 minutes, and cells were lifted and labeled with Alexa647-M1 antibody (1:1000). Flow cytometry of 10,000 cells per sample was carried out using a FACS-Calibur instrument (BD Biosciences, San Jose, CA). Percentage of internalized receptors = 100 − (number of surface receptors after 20 minutes of Iso)/(initial number of surface receptors) × 100.
Endosome Quantitation.
For quantitation of number of receptor-containing endosomes and number of receptors per endosome, we used HEK293 cells transiently expressing FLAG-tagged β2-AR. Data were averaged from >20 cells per condition from two independent transfections. Respective dose of agonist was added for 20 minutes, and then cells were fixed by incubation in 4% formaldehyde diluted in Brinkley Buffer 1980 (80 mM 1,4-piperazinediethanesulfonic acid, pH 6.8, 1 mM MgCl2, 1 mM EGTA, and 1 mM CaCl2) for 15 minutes, permeabilized, and blocked in 0.1% Triton X-100 and 2.5% milk diluted in TBS with Tween 20 buffer for 15 minutes at room temperature. β2-ARs were labeled with mouse anti-FLAG M1 (1:1000; Sigma-Aldrich) and anti-mouse Alexa Fluor 594 (1:1000; Invitrogen). Fixed cells were imaged by epifluorescence microscopy using a Nikon inverted microscope, 60× NA 1.4 objective (Nikon, Melville, NY), mercury arc lamp illumination, and standard dichroic filter sets (Chroma, Bellows Falls, VT). Number and average intensity of endosomes (corresponding to number of endosomes with receptor and number of receptors per endosome, respectively) were analyzed on a cell-by-cell basis using the ICY plug-in in ImageJ that automatically counts the number of vesicles and their fluorescence intensity. Regions of interest were drawn manually around each cell; the undecimated wavelet transform detector was used to detect bright spots over dark background; the size of spots to detect was set to scale 2 and 3 px diameter; the size-filtering option was selected; and size thresholds were set to minimum size of 10 and maximum size of 3000. The number of detected objects (corresponding to endosomes) and the mean intensity of each object (corresponding to number of receptors per endosome) were averaged on a per cell basis.
Results
Pronounced Differences in Upstream Cellular Effects of β2-AR Ligands.
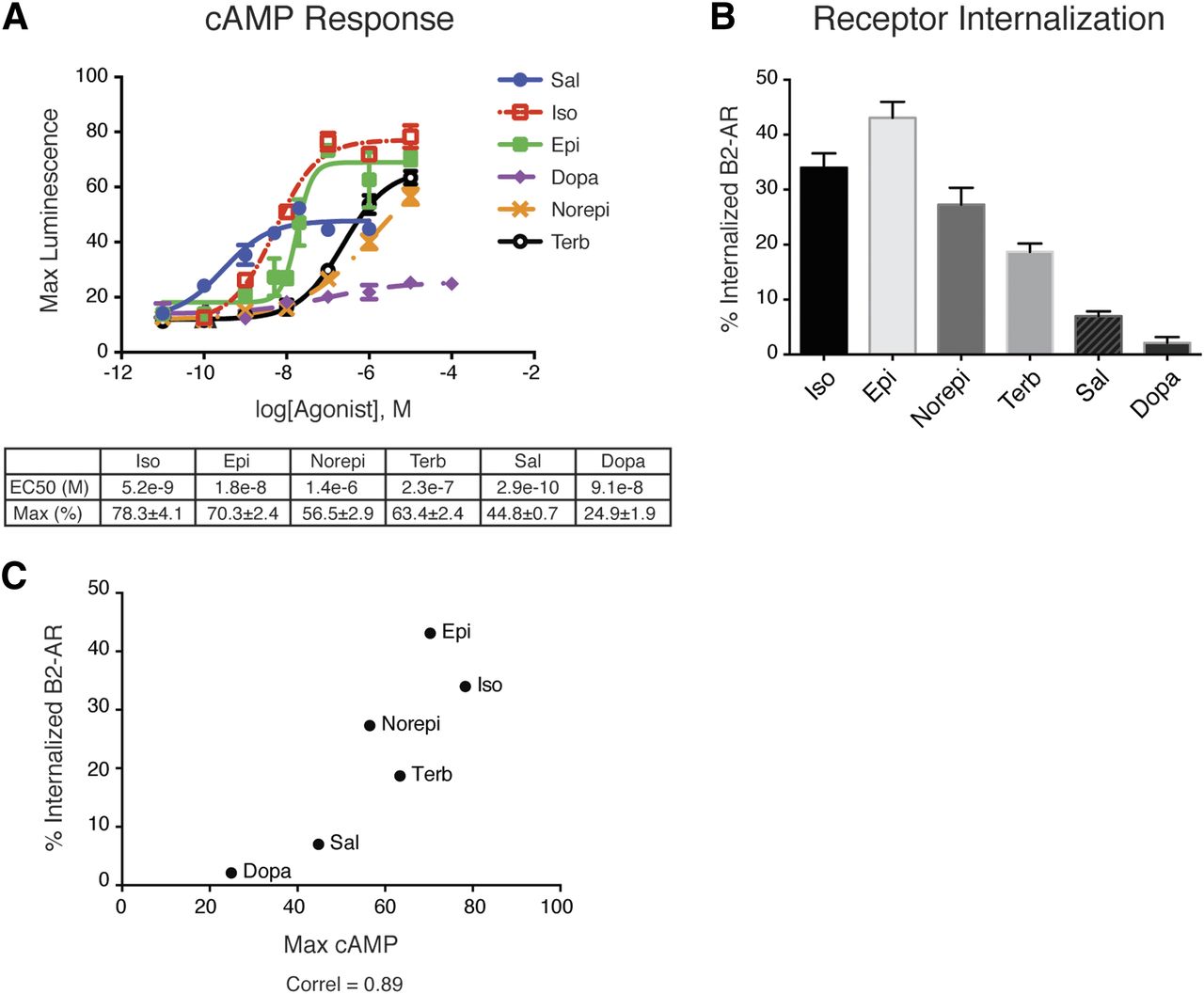
We subdivided β2-AR responses into early (0–20 minutes after ligand binding), intermediate (30 minutes after ligand binding), and late (2 hours after ligand binding). We monitored cAMP production and receptor endocytosis as hallmarks of early responses, and protein phosphorylation and gene transcription as hallmarks of intermediate and late responses, respectively (Fig. 1). We began by assessing the early signaling effects of a panel of six synthetic and naturally occurring β2-AR ligands: Epi, Norepi, Dopa, Iso, Sal, and Terb (Table 1). Using HEK293 cells expressing endogenous β2-ARs at low levels to avoid potential complications of receptor overexpression, we evaluated G protein/adenylyl cyclase activation by each ligand in intact cells with a luminescence-based cAMP biosensor (Irannejad et al., 2013; Tsvetanova and von Zastrow, 2014). We observed the following order of ligand efficacy: Iso > Epi > Terb > Norepi > Sal > Dopa, and of ligand potency: Sal > Iso > Epi > Dopa > Terb > Norepi for G protein signaling (Fig. 2A), which is consistent with previous studies for these compounds (January et al., 1998; Del Carmine et al., 2002; Baker, 2005; Moore et al., 2007). Next, we measured β2-AR internalization as the loss of cell surface receptors after agonist exposure using flow cytometry in cell expressing a flag-tagged receptor. Similar to G protein activation, endocytosis reflected the pharmacodynamic differences of the ligands with the full agonists Iso and Epi yielding comparable steady-state number of internalized receptors (35–40%); the partial agonists Norepi, Terb, and Sal leading to less internalization (7–27%); and the weak partial agonist Dopa yielding no detectable internalization (Fig. 2B). In fact, G protein signaling strongly correlated with receptor endocytosis (Pearson coefficient = 0.89; Fig. 2C), suggesting extensive differences in early signaling events that reflect the different properties of each ligand at the ligand–receptor interface.

Cellular responses from activated adrenoceptors. Responses to activated β2-ARs are subdivided into early (cAMP accumulation and receptor endocytosis), intermediate (protein phosphorylation), and late (gene transcription).
Compounds selected for the study

Pronounced differences in early signaling events from the β2-AR. (A) Dose-response curves for a panel of endogenous (Epi, Norepi, Dopa) and synthetic (Iso, Sal, Terb) β2-AR ligands. cAMP accumulation was measured using the luciferase-based biosensor pGLO-20F (Promega), and data were normalized to forskolin-treated controls. Data are average of n = 3–5 experiments ± S.E.M. EC50 curve fitting was performed using Prism6 software. Ligand EC50 (M) and maximum responses (percentage of forskolin response) are summarized under the graph. (B) β2-AR internalization quantified by flow cytometry in cells overexpressing flag-tagged β2-AR. The 1 μM Iso, 1 μM Epi, 10 μM Norepi, 10 μM Terb, 50 nM Sal, and 10 μM Dopa were added for 20 minutes. Data are average from n = 6–23 ± S.E.M. (C) Correlation between cAMP signaling (A) and receptor endocytosis (B).
We chose two of the ligands, Iso and Sal, for comprehensive analysis of intermediate and late signaling events in HEK293 cells expressing endogenous β2-ARs. These two clinically relevant adrenoceptor ligands differ vastly in chemical structure, efficacy and potency, and bias at the receptor level (Fig. 2; Table 1) (January et al., 1998; Nino et al., 2009; Rajagopal et al., 2011). Thus, Iso and Sal differ in multiple pharmacological properties that would suggest that the two drugs would produce diverse signaling responses at the β2-AR. To determine whether this is the case, we next examined global phosphoproteomic changes after ligand application by mass spectrometry.
Global Analysis of β2-AR Phosphoresponses to Iso and Sal Reveals Drug-Dependent Quantitative Differences in Intermediate Signaling Responses.
To evaluate the intermediate signaling responses to ligands with different pharmacological properties, we chose the following three β2-AR activation conditions: 1) saturating Iso (1 μM, high Iso), 2) saturating Sal (50 nM), and 3) subsaturating Iso (10 nM, low Iso). Low Iso and saturating Sal produced comparable net cAMP amounts (Supplemental Fig. 1; compare blue and red curves), whereas high Iso produced ∼1.5 times more cAMP (Supplemental Fig. 1; black curve). We reasoned that comparisons across these three conditions would allow us to distinguish agonist-specific differences from variations in β2-AR signaling that are due solely to differences in total cAMP production.
To globally identify and quantify proteins that are phosphorylated in a β2-AR–dependent manner, we used stable isotope labeling with amino acids in cell culture (SILAC) and mass spectrometry (Ong et al., 2003). HEK293 cells were exposed to either Iso or Sal (drug) or treated with vehicle (no drug) for 30 minutes. Then proteins were extracted and digested, and samples were enriched for phosphorylated peptides by iron (III)-nitrilotriacetic acid immobilized metal ion affinity chromatography and subjected to liquid chromatography–mass spectrometry (LC-MS and LC-MS/MS) (Supplemental Fig. 2A). To rule out off-target and nonspecific effects, we swapped the SILAC media for each drug condition. We identified ∼4000 high-confidence phosphopeptides for each experiment set (Supplemental Table 1; see Materials and Methods for data analysis). From these data, we next generated a list of 54 high-confidence β2-AR–regulated phosphosites corresponding to 37 different proteins (Supplemental Tables 2 and 3). We considered as targets only peptides that had statistically significant log2 (drug/no drug) values (based on z-score P < 0.05) in each of the medium swaps for each experimental condition (high Iso, low Iso, and Sal; see Materials and Methods for details). These proteins carry out a diverse range of biologic functions, such as synapse organization, angiogenesis, cell communication, chromatic assembly, and developmental maturation, and localize to different subcellular compartments (Supplemental Fig. 2B).
Three lines of evidence strongly suggest that the 54 phosphorylated sites we discovered are indeed induced in response to β2-AR/cAMP signaling. First, 9 of the 54 phosphosites we identified are previously known cAMP-stimulated sites (Berwick et al., 2002; Gunaratne et al., 2010; Lundby et al., 2013; Yip et al., 2014). Second, we found enrichment of the amino acid motif R/K-X-pS (p < 1.0 × 10−8 by Fisher’s exact test; Supplemental Fig. 2C) in agreement with findings reported by Lundby et al. (2013) for phosphosites in β1-AR target proteins in cardiac myocytes (Lundby et al., 2013). Finally, 75% (29 of 38) of the phosphosites we discovered are substrates for basophilic serine/threonine kinases (Supplemental Table 3), which are activated by second-messenger (cAMP, calcium, phospholipid) release, consistent with the current view of β2-AR signaling via cAMP generation and calcium mobilization.
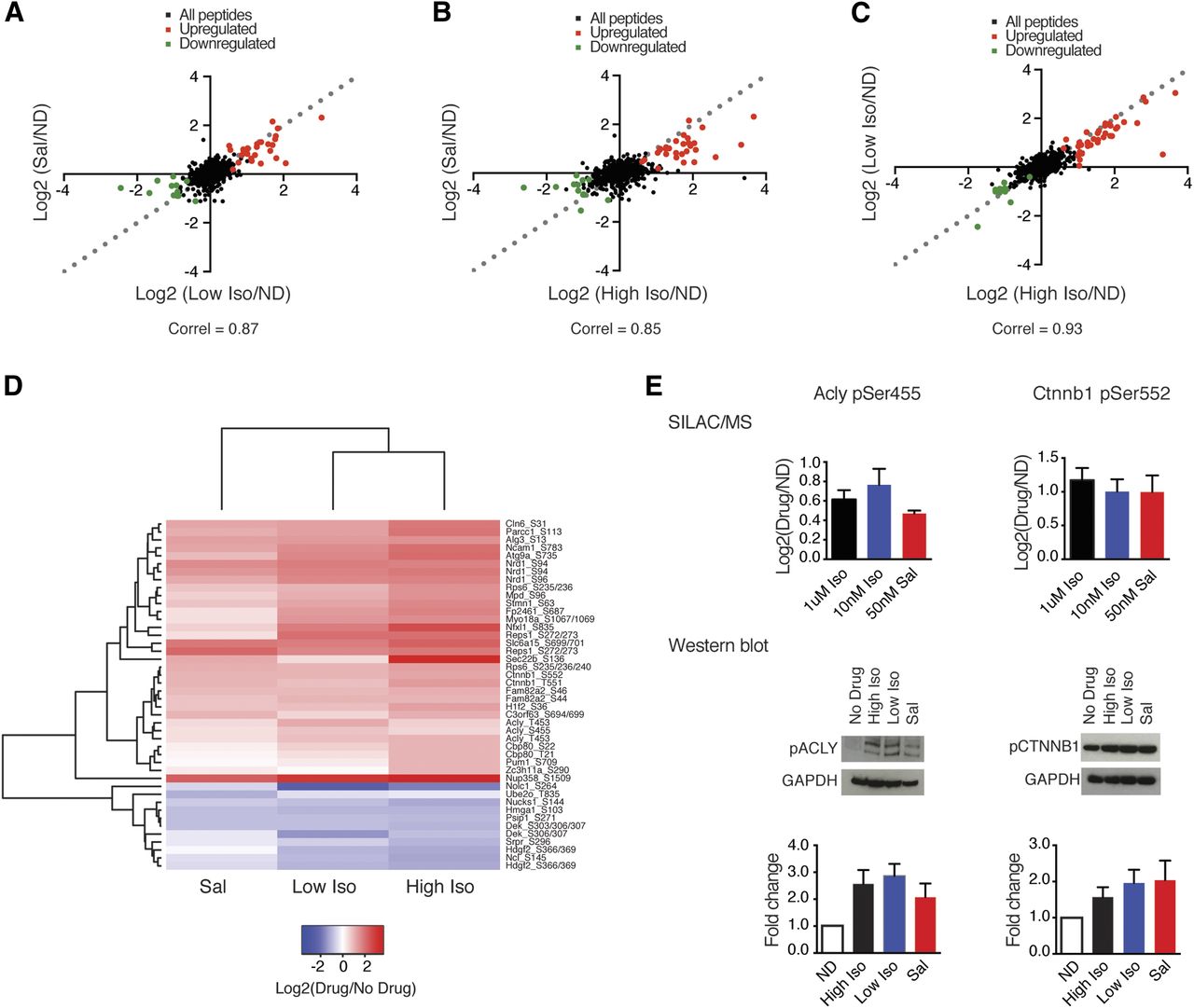
To determine how differences at the ligand–receptor interface contribute to the phosphoprotein response, we next asked whether the proteomic changes induced by Iso are different than the ones elicited by Sal. First, we noticed that the phosphoresponses to the two drugs were qualitatively similar, that is, the same sites were phosphorylated in each case (Supplemental Table 2). However, when we compared the abundance of β2-AR target phosphosites identified across conditions, we saw quantitative differences between Sal and Iso. As a general trend, Iso treatment yielded more robust phosphorylation of target sites (Fig. 3, A–C). We determined whether the observed quantitative differences between Iso and Sal were statistically significant by modeling the variability in the measurements for each condition based on the parameters obtained from its regression analysis (see Materials and Methods for details). This analysis indicated that the phosphoresponse induced by Sal was significantly different from that induced by both low and high Iso (p = 3.6 × 10−3 and p < 1.0 × 10−4, respectively), whereas the two Iso conditions elicited comparable intermediate responses (Supplemental Table 4). As a complementary approach, we carried out unsupervised hierarchical clustering analysis of the data, which revealed higher similarity between the signaling profiles of the two Iso conditions compared with Sal (Fig. 3D). As low and high doses of Iso yield different amounts of net cAMP (Supplemental Fig. 1), but identical phosphorylation responses (Pearson correlation = 0.93; Fig. 3, C and D), quantitative differences in phosphorylation between Iso and Sal must be due to the different pharmacodynamic properties of the two ligands at the β2-AR.

Mass-spectrometric analysis of global phosphoproteomic responses to Iso and Sal. Values from SILAC media swap experiments were log2-transformed and averaged. (A–C) Scatter plots comparing phosphopeptide abundance for high Iso, low Iso, and Sal. Significantly upregulated sites are shown in red, and downregulated sites are shown in green. Pearson’s correlation values are shown. Dashed lines have a slope of 1 (y = x) for comparison. (D) Average linkage hierarchical clustering was performed using Euclidian distance as a similarity metric on the set of β2-AR–regulated phosphosites. (E) SILAC-LC-MS/MS data and Western blot analysis of phosphoserines in Acly (n = 4–6 ± S.E.M.) and Ctnnb1 (n = 3 ± S.E.M.). Protein levels for the no drug (ND) condition were adjusted to 1, and phosphoprotein changes were normalized to expression levels of GAPDH. Original uncropped Western blots are shows in Supplemental Fig. 4.
We independently confirmed the validity of our mass-spectrometry results for one agonist-selective phosphosite (pSer455 in the enzyme ATP citrate lyase) and one site upregulated similarly across all three conditions (pSer552 in β catenin, the key downstream component of the Wnt signaling pathway) by immunoblotting with phospho-specific antibodies (Fig. 3E; Supplemental Figs. 3 and 4). Therefore, our mass-spectrometry analysis reveals that differences in ligand properties at the receptor are partially propagated to the intermediate signaling response, with Iso and Sal eliciting qualitatively identical phosphorylation events, but the weaker agonist Sal inducing a less robust response than the full agonist Iso.
Sal and Iso Induce Indistinguishable Late Signaling Responses.
We next assessed longer-term adrenoceptor signaling responses by measuring the global effects of each drug on gene expression using DNA microarrays. We recently described a comprehensive interrogation of transcriptional changes upon treatment of HEK293 cells with different concentrations of Iso, and showed that both high (1 μM) and low (10 nM) doses of Iso yield identical gene expression changes (Tsvetanova and von Zastrow, 2014). In this study, we took a similar approach and examined changes in gene expression elicited by low Iso and Sal. Analysis of the datasets revealed 83 high-confidence β2-AR target genes (see Materials and Methods for data analysis; Supplemental Table 5). As seen previously (Tsvetanova and von Zastrow, 2014), the target set showed significant enrichment for known and predicted cAMP-responsive genes (32 of 83, P < 1.0 × 10−12 by Fisher’s exact test), consistent with transcription from activated β2-ARs being controlled predominantly via Gs/cAMP-dependent signal transduction.
Unlike the drug-specific differences in intermediate response, we found that the transcriptional responses to Iso and Sal were qualitatively and quantitatively indistinguishable (Pearson correlation = 0.96; Fig. 4A). We confirmed the expression of four target genes by quantitative PCR: CGA, which encodes the α polypeptide of the human chorionic gonadotropin hormone; PCK1, the gene for phosphoenolpyruvate carboxykinase that regulates glyconeogenesis; PDE4D, encoding phosphodiesterase 4D; and DUSP1, encoding a dual phosphatase for mitogen-activated protein kinase extracellular signal-regulated kinase 2. In full agreement with the microarray results, quantitative reverse-transcription PCR analysis verified that Iso and Sal elicit identical transcription (Fig. 4B).

Sal and Iso elicit identical transcriptional responses by DNA microarray analysis. (A) Summary of DNA microarray data comparing transcriptional responses to low (10 nM) Iso and 50 nM Sal. Data are average from n = 3. Dotted line has a slope of 1 (y = x). In red, β2-AR target genes. (B) The expression of genes indicated by arrows in (A) was verified by quantitative PCR. Data are average from n = 4 ± S.E.M. (C) Time course of PCK1 induction measured by quantitative PCR for low Iso and Sal. Data are average from n = 3 ± S.E.M. ND, no drug.
Sal has a long duration of action at the β2-AR that persists extensive washout of antagonist (Ball et al., 1991; Green et al., 1996). Therefore, it seemed plausible that Iso and Sal could differ in the duration of gene expression upregulation. To address this question, we carried out a time course of receptor activation and monitored transcription of one of the β2-AR targets, PCK1, by quantitative reverse-transcription PCR. We found no difference in the transcriptional profiles of Iso and Sal between 1 and 4 hours post-treatment (Fig. 4C). Thus, we concluded that the effects of the two chemically distinct ligands on transcriptional reprogramming—the late cellular response to β2-AR signaling—are indistinguishable.
The Uniform Late Response Is Sensitive, Saturable, and Controlled by the Number of β2-AR–Containing Endosomes.
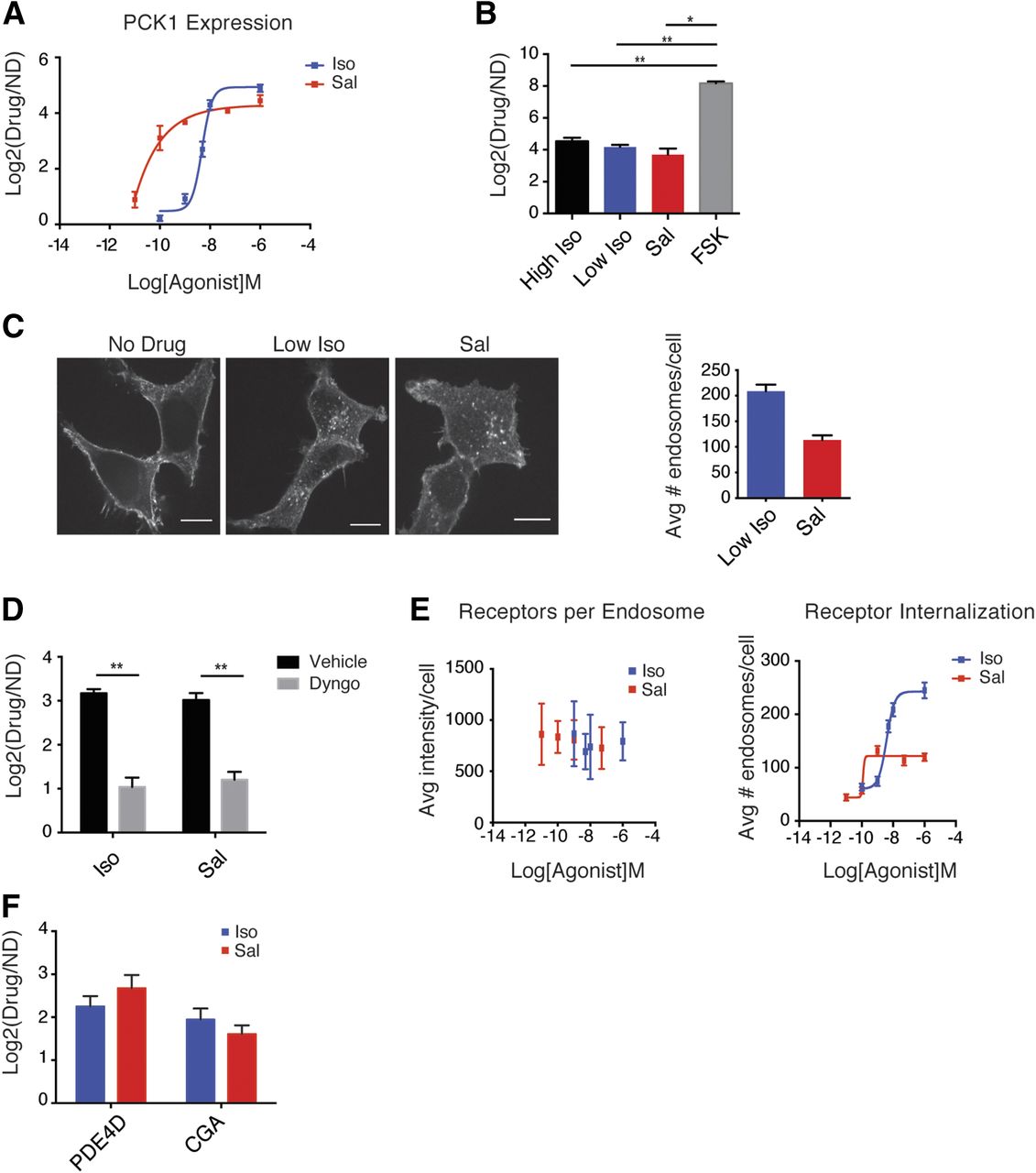
We were intrigued that all three conditions (high, low Iso, and Sal) examined in this and our previous studies elicited quantitatively and qualitatively identical transcriptional responses. Concentration–response analysis using expression of the robust β2-AR target gene PCK1 as readout for receptor-dependent transcriptional response indicated that transcriptional induction produced by both ligands was highly sensitive and saturable (Fig. 5A). In principle, saturation of the response could occur at multiple steps in the signaling pathway. We first asked whether saturation happens downstream, at the level of the cAMP-dependent transcriptional machinery. To do so, we compared the maximal response produced by the β2-AR agonists to that produced by direct activation of adenylyl cyclase with forskolin (Seamon and Daly, 1981), when applied at a relatively high (5 μM) concentration that generates ∼1.3-fold and twofold more cytoplasmic cAMP than high Iso and Sal, respectively (Supplemental Fig. 1). PCK1 induction by forskolin was >16-fold higher than that produced by either Iso or Sal (Fig. 5B), indicating that the downstream transcriptional machinery is not saturated.

Mechanisms underlying the stereotyped adrenoceptor-dependent transcriptional response. (A) Dose–response curves for PCK1 induction by Iso and Sal show sensitive and saturable transcriptional responses. Data are average from n = 3–7 ± S.E.M. (B) The β2-AR–dependent transcriptional response does not reflect saturation of the cellular transcriptional machinery. Treatment with forskolin (5 μM) upregulates PCK1 expression more significantly than Iso or Sal. Data are average from n = 4 ± S.E.M. (C) Receptor internalization was visualized by spinning disk confocal microscopy in cells expressing a flag-tagged β2-AR. Drugs were added for 20 minutes, and cells were fixed, permeabilized, and stained with Alexa-conjugated flag antibody. Scale bar = 10 μm. Number of receptor-containing endosomes was analyzed using ICY and ImageJ, as described in Materials and Methods. (D) Pharmacological blockade of endocytosis with 30 μM Dyngo inhibits receptor-dependent transcriptional upregulation of PCK1. Gene expression levels are measured by quantitative PCR. Data are average from n = 4 ± S.E.M. (E) Quantitation of receptor concentration per endosomes (left) and receptor-containing endosomes (right) was carried out in fixed cells transiently overexpressing flag-tagged β2-AR after treatment with respective doses of ligand for 20 minutes using ICY and ImageJ. Data are average from 27–35 cells ± S.D. from two independent transfections. (F) Expression of PDE4D and CGA in human airway smooth muscle cells treated with 10 nM Iso or 50 nM Sal for 90 minutes. Gene levels were measured by quantitative PCR. Data are average from n = 3 ± S.E.M. **P < 0.005; *P < 0.05 by two-tailed, unpaired Student’s t test.
Transcriptional induction by Iso requires endocytosis and is preferentially induced by cAMP generated from endosomes (Tsvetanova and von Zastrow, 2014). Therefore, we next considered the possibility that saturation of the response might occur at the level of the endosome signal itself. Surprisingly, despite Sal driving β2-AR endocytosis relatively weakly compared with Iso (Figs. 2B and 5C), the transcriptional response induced by Sal, like that induced by Iso, required endocytosis. To demonstrate this, we first inhibited clathrin-dependent endocytosis genetically by small interfering RNA-mediated depletion of clathrin heavy chain (encoded by CHC17). Small interfering RNA knockdown depleted >90% of the CHC17 mRNA and inhibited β2-AR internalization by ∼40–50% as quantified by flow cytometry (Supplemental Fig. 5, A and B). We observed that diminished receptor endocytosis resulted in a significant decrease in gene induction of PCK1 for both Iso and Sal (P < 0.05 by Student’s t test; Supplemental Fig. 5C). To corroborate the clathrin knockdown results, we used a complementary pharmacological approach to acutely inhibit clathrin/dynamin-dependent endocytosis with the drug Dyngo (Harper et al., 2011). Pretreatment of cells with Dyngo was more effective than CHC17 knockdown in blocking Iso- and Sal-stimulated β2-AR endocytosis: we observed complete inhibition as quantified by fluorescence flow cytometry (Supplemental Fig. 5D). Consistent with previous reports (Tsvetanova and von Zastrow, 2014), inhibition of receptor internalization almost completely blocked transcriptional response to Iso (>4.5-fold, P < 5.0 × 10−3 by Student’s t test; Fig. 5D). More interestingly, we observed comparable level of inhibition of Sal-dependent transcription (Fig. 5D). These results show that transcriptional response to both Iso and Sal requires receptor endocytosis.
We therefore looked more closely at the effects of Iso and Sal on β2-AR accumulation in endosomes. Although Sal stimulated β2-AR internalization less strongly than Iso, the fluorescence intensity of receptor-containing endosomes—proxy for relative receptor concentration in these membranes—appeared similar (Fig. 5C). We verified this quantitatively over a range of agonist concentrations. The mean receptor fluorescence per endosome was indistinguishable between agonists, and over a wide range of agonist concentration (Fig. 5E, left panel). Although receptor concentration per endosome was uniform, differences in the degree of net internalization produced by Iso and Sal were manifest at the level of the number of receptor-containing endosomes accumulated in the cytoplasm (Fig. 5E, right panel). We also noted that the transcriptional response produced by both Iso and Sal saturated at concentrations that generate a relatively small number of receptor-containing endosomes in the cytoplasm (Fig. 5, A and E). These properties suggest that the inherent features of β2-AR internalization underlie the sensitivity and uniformity of the transcriptional response produced by distinct agonists.
Discussion
Understanding signaling specificity and diversity through GPCRs constitutes a fundamental challenge in pharmacology. It is increasingly clear that there are multiple ways by which chemically diverse ligands can produce different effects at a target GPCR. The therapeutic promise of such diversity is supported by recent progress in exploiting differences at the ligand–receptor interface to develop drugs with differing in vivo activity profiles (Raehal and Bohn, 2014; Urs et al., 2014; Santos et al., 2015).
In some cases, chemical differences imposed at the ligand–GPCR interface are propagated downstream to produce diversity in the cell or tissue response. For example, classic full agonist and biased partial agonist ligands produce markedly different integrated cellular responses through angiotensin II and parathyroid hormone receptors, as indicated by striking differences in the cell's phosphoproteomic or transcriptional responses (Christensen et al., 2010, 2011; Gesty-Palmer et al., 2013; Maudsley et al., 2015; Santos et al., 2015). Evolutionarily, however, there is selective pressure for cells to generate a stereotyped output in response to the presence of a cognate ligand. From this perspective, chemical variability and fluctuations occurring at the ligand–GPCR interface could be considered a source of biologic noise that may degrade the reliability of cognate ligand detection. This raises the question of whether, in some cases, instead of propagating the variability inherent to the ligand–GPCR interface, cells generate a stereotyped integrated downstream response. To our knowledge, there are currently no examples supporting the latter model.
In this study, we looked at the early, intermediate, and late responses to two chemically and pharmacodynamically distinct drugs, Iso and Sal, acting through the endogenous complement of cellular β2-ARs. We showed that there are extensive differences in early cellular signaling events after treatment with Iso and Sal. Similarly, we report distinct early responses for a panel of chemically distinct adrenoceptor ligands that reflect the different properties of each ligand at the ligand–receptor interface (Fig. 2). Next, we applied a phosphoproteomic and transcriptional profiling strategy to comprehensively examine the integrated response elicited by Iso and Sal. Even though our profiling strategy achieved comparably broad coverage as analyses used previously to detect prominent ligand-specific differences in responses elicited through the angiotensin receptor (Christensen et al., 2010, 2011; Santos et al., 2015), we show in this work that the downstream cellular effects of Sal and Iso mediated through endogenous β2-ARs are remarkably similar. This was true at the level of the cellular phosphoresponse with no evidence of drug-specific clusters and only modest quantitative differences in relative fold-abundance changes determined by SILAC analysis (Fig. 3). Even more remarkably, genome-wide interrogation of the transcriptional response revealed indistinguishable effects of Sal and Iso (Fig. 4). To our knowledge, the present results provide the first direct demonstration that cells can generate a stereotyped rather than divergent integrated downstream response to chemically diverse drugs acting at the same GPCR.
It would be interesting to determine whether such uniformity is a general property of β2-AR signaling or whether it is dependent on the cellular context. Although the β2-AR is ubiquitously expressed and controls a range of physiologic processes, Iso and Sal are classically administered to target its effects in the lung (van der Westhuizen et al., 2014). Quantitative PCR analysis of the transcriptional responses induced by the two ligands in human airway smooth muscle cells reveals comparable upregulation of the expression of two adrenoceptor target genes, PDE4D and CGA (Fig. 5F). Whereas these data support the generality of our findings, a more comprehensive investigation of the downstream signaling responses in this cell type is necessary to obtain conclusive answers.
Diversity in signaling can be propagated downstream through differential receptor coupling to G proteins relative to arrestins (Beaulieu et al., 2007; Reiter et al., 2012; Raehal and Bohn, 2014), or among G protein or arrestin isoforms (Sauliere et al., 2012; Santos et al., 2015). How cells limit or reduce variability in downstream signaling is an important, emergent question (Lemmon et al., 2016). We investigated this question by focusing on the downstream transcriptional response that was indistinguishable between the drugs tested in this study. Given its generally weak endocytic activity relative to Iso (Figs. 2B and 5C), and because β2-AR signaling from endosomes is required for the full transcriptional response to Iso (Tsvetanova and von Zastrow, 2014), we initially expected that Sal would differ greatly from Iso in its transcriptional effects or, perhaps, fail to induce the response altogether. To the contrary, we found that Sal can generate the full transcriptional response (Fig. 4A) and, same as Iso, the response elicited by Sal requires endocytosis (Fig. 5D; Supplemental Fig. 4C). In addition, we found that the magnitude of the endocytosis-dependent response saturates at a very low number of internalized receptors (Fig. 5, A and E) and at a level considerably below the capacity of the downstream signaling machinery that ultimately generates the transcriptional response (Fig. 5B). This indicates that the endocytosis-dependent transcriptional response is not only highly sensitive, but it is also saturable, effectively limiting the downstream response to a uniform level. Together these characteristics define the property of ultrasensitivity (Altszyler et al., 2014), a widely deployed strategy to reduce variability or noise in biologic systems (Fig. 6).

Speculative model for how endosome signaling can reduce the propagation of chemical variability at the ligand–receptor interface by acting as an ultrasensitivity module.
Although much remains to be learned about specific biochemical mechanism, we propose that two properties of β2-AR endocytosis identified in the present study underlie its ability to confer ultrasensitivity. First, we show that cells accumulate different numbers of receptor-containing endosomes in response to chemically diverse agonists, but that the receptor concentration in each individual endosome is remarkably uniform (Fig. 5E). Second, we show that the cellular transcriptional response, which requires β2-AR endocytosis, occurs after accumulation of a relatively small number of receptor-containing endosomes and at the same number irrespective of the agonist (Fig. 5, A and E). These two properties are sufficient, in principle, to produce an ultrasensitive response, because the first would confer sensitivity and the second would account for saturation. Testing this hypothesis will require elucidating the biochemical basis of each property. We speculate that uniformity in receptor number per endosome might arise simply from the mechanics of endocytosis, as each clathrin-coated pit has a fixed cargo capacity (Liu et al., 2010). Saturation of the downstream response could be explained by a limiting component necessary to transduce the endosome signal to the nucleus.
In conclusion, the present study provides the first demonstration that chemically diverse ligands can generate a stereotyped output that is dependent on receptor endocytosis. A function of the endocytic network in receptor-mediated signal processing is an emergent concept that may also contribute to robustness and regulation of growth factor responses (Villasenor et al., 2015). In this study, we propose a discrete function of the endocytic network as part of a noise-reduction strategy that confers uniformity on downstream signaling elicited by GPCR activation.
Acknowledgments
We thank Dr. Dean Sheppard for valuable discussion and generously providing human airway smooth muscle cells, and Dr. Roshanak Irannejad and Dr. James Fraser for valuable discussion.
Authorship Contributions
Participated in research design: Tsvetanova, Trester-Zedlitz, Krogan, von Zastrow.
Conducted experiments: Tsvetanova, Trester-Zedlitz, Newton.
Contributed new reagents or analytic tools: Riordan, Sundaram, Johnson.
Wrote or contributed to the writing of the manuscript: Tsvetanova, Trester-Zedlitz, von Zastrow.
Footnotes
- Received August 3, 2016.
- Accepted November 18, 2016.
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA010711 and DA012864 to M.v.Z.], National Institutes of Health National Institute of Mental Health [Grant MH109633 to N.G.T.], National Institutes of Health National Institute of General Medical Sciences [Grants GM081879, GM082250, and GM107671 to N.J.K.], and National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL124049 to A.B.S.]. N.G.T. is also supported by the American Heart Association.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- β2-AR
- β2-adrenergic receptor
- DMEM
- Dulbecco’s modified Eagle’s medium
- DMSO
- dimethylsulfoxide
- Dopa
- dopamine
- Epi
- epinephrine
- FA
- formic acid
- FBS
- fetal bovine serum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GPCR
- G protein–coupled receptor
- HEK
- human embryonic kidney
- Iso
- isoproterenol
- MS/MS
- tandem mass spectrometry
- Norepi
- norepinephrine
- PCR
- polymerase chain reaction
- Sal
- salmeterol
- SILAC
- stable isotope labeling with amino acids in cell culture
- TBS
- Tris-buffered saline
- Terb
- terbutaline
- Copyright © 2017 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}