


Abstract
The μ-opioid receptor gene undergoes extensive alternative splicing to generate an array of splice variants. One group of splice variants excludes the first transmembrane (TM) domain and contains six TM domains. These 6TM variants are essential for the action of a novel class of analgesic drugs, including 3-iodobenzoyl-6β-naltrexamide, which is potent against a spectrum of pain models without exhibiting the adverse side effects of traditional opiates. The 6TM variants are also involved in analgesic action through other drug classes, including δ-opioid and κ-opioids and α2-adrenergic drugs. Of the five 6TM variants in mouse, mouse μ-opioid receptor (mMOR)-1G is abundant and conserved from rodent to human. In the present study, we demonstrate a new function of mMOR-1G in enhancing expression of the full-length 7TM μ-opioid receptor, mMOR-1. When coexpressed with mMOR-1 in a Tet-Off inducible CHO cell line, mMOR-1G has no effect on mMOR-1 mRNA expression but greatly increases mMOR-1 protein expression in a dose-dependent manner determined by opioid receptor binding and [35S] guanosine 5′-3-O-(thio)triphosphate binding. Subcellular fractionation analysis using OptiPrep density gradient centrifugation shows an increase of functional mMOR-1 receptor in plasma membrane–enriched fractions. Using a coimmunoprecipitation approach, we further demonstrate that mMOR-1G physically associates with mMOR-1 starting at the endoplasmic reticulum, suggesting a chaperone-like function. These data provide a molecular mechanism for how mMOR-1G regulates expression and function of the full-length 7TM μ-opioid receptor.
SIGNIFICANCE STATEMENT The current study establishes a novel function of mouse μ-opioid receptor (mMOR)-1G, a truncated splice variant with six transmembrane (TM) domains of the mouse μ-opioid receptor gene, in enhancing expression of the full-length 7TM mMOR-1 through a chaperone-like function.
Introduction
The μ-opioid receptor has a special place in the opioid receptor family since it mediates most clinically relevant opioids, such as morphine and fentanyl as well as heroin. The single-copy μ-opioid receptor gene (OPRM1) undergoes extensive alternative pre-mRNA splicing, producing multiple splice variants or isoforms with a pattern that is conserved from rodent to human (Pan, 2005; Pasternak and Pan, 2013). These splice variants can be categorized into three structurally distinct types: 1) full-length seven transmembrane (TM) C-terminal variants, 2) truncated 6TM variants excluding the first TM, and 3) truncated single-TM variants that contain only the first TM (Pasternak and Pan, 2013).
The functional significance of the full-length 7TM C-terminal variants has been demonstrated by differences in regional- and/or cell-specific expression, μ agonist–induced G protein coupling, phosphorylation, internalization, and postendocytic sorting as well as in vivo morphine activity (Koch et al., 1998, 2001; Pan et al., 1999; Abbadie et al., 2000a,b, 2004; Bolan et al., 2000; Caron et al., 2000; Pan et al., 2001; Tanowitz et al., 2008; Xu et al., 2017). The truncated single-TM variants do not bind any opioids. Rather, they function as molecular chaperones that facilitate expression of the 7TM MOR-1 receptor and enhance morphine analgesia (Xu et al., 2013).
Using Oprm1 gene-targeting mouse models, we previously demonstrated the functional importance of the truncated 6TM variants in mediating a novel class of opioids, such as 3-iodobenzoyl-6β-naltrexamide (IBNtxA) (Majumdar et al., 2011b, 2012) IBNtxA is potent against thermal, inflammatory, and neuropathic pain in animal models and does not produce respiratory depression, physical dependence, or reward behavior (Majumdar et al., 2011b; Wieskopf et al., 2014). In an Oprm1 exon 11–knockout (KO) mouse model devoid of all 6TM variants with preservation of 7TM C-terminal variants, IBNtxA analgesia was completely eliminated while morphine analgesia was preserved (Majumdar et al., 2011a,b). Conversely, in a triple-KO mouse model lacking μ exon 1–associated 7TM variants, δ receptors and κ receptors (Schuller et al., 1999), morphine analgesia was completely abolished, but IBNtxA analgesia remained intact (Majumdar et al., 2011a,b; Pasternak and Pan, 2013). Gain-of-function studies further confirmed that 6TM variants are both necessary and sufficient for IBNtxA analgesia (Lu et al., 2015, 2018). In a complete Oprm1 KO mouse lacking both 7TM and 6TM variants, both morphine and IBNtxA analgesia were lost. Restoring expression of any of the individual 6TM variants through lentiviral-mediated delivery in the complete Oprm1 KO mice rescued IBNtxA, but not morphine, analgesia.
The current study establishes a novel function of mouse μ-opioid receptor (mMOR)-1G, a conserved and dominant 6TM variant, in enhancing expression of the full-length 7TM mMOR-1 at the protein level through heterodimerization, providing new insights into the roles of the 6TM variants in μ-opioid pharmacology.
Materials and Methods
Materials.
[3H][D-Ala2,N-MePhe4,Gly(ol)5]enkephalin (DAMGO) (53.4 Ci/mmol) and [35S] guanosine 5′-3-O-(thio)triphosphate (GTPγS) (1250 Ci/mmol) were purchased from PerkinElmer (Boston, MA). Opiates and opioid peptides were a generous gift from the Research Technology Branch of the National Institute on Drug Abuse Drug Supply Program (Rockville, MD). Be(2)C cells were obtained from American Type Culture Collection, whereas Tet-Off CHO cells were obtained from Clontech (Mountain View, CA). The oligodeoxynucleotides were synthesized and purified through Sigma-Aldrich (St. Louis, MO). The anti-FLAG antibody, EZview Red Anti-FLAG M2, EZview Red Anti-HA Affinity Gels, 3x FLAG peptide, and HA peptide were from Sigma-Aldrich. The anti-HA antibody and anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody were from Cell Signaling (Beverly, MA). All other materials were obtained from the sources listed.
Reverse Transcription and Quantitative Polymerase Chain Reaction.
Total RNA was isolated from Tet-Off CHO cells with the RNeasy Kit (Qiagen) and treated with DNase I using Turbo DNA-free Reagents (Invitrogen). Reverse transcription with random primers and Superscript II reverse transcriptase (Invitrogen) were performed as described previously (Xu et al., 2013). SYBR qPCR with the Hot start SYBR Green Master Mix (Affimetrix, Santa Clara, CA) was then carried out in the CFX96 machine (Bio-Rad) using the first-strand cDNAs as the template. qPCR primers were as follows: sense primer (SP)-1, 5′-GCC CTC TAT TCT ATC GTG TGT GTA GTG G-3′, and antisense primer (AP)-1, 5′-CAA TCT ATG GAC CCC TGC CTG TAT TTT G-3′, for mMOR-1 and SP-2, 5′-GAT CTG GGC CGA TGA TGG AAG CTT TC-3′, and AP-1 for mMOR-1G. qPCRs of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (SP-3, 5′-ACC ACA GTC CAT GCC ATC AC-3′, and AP-2, 5′-TCC ACC ACC CTG TTG CTG TA-3′) and mouse 18S ribosome (m18S) (SP-4, 5′-GTA ACC CGT TGA ACC CCA TT-3′, and AP-3, 5′-CCA TCC AAT CGG TAG TAG CG-3′) were used as reference genes to calculate the normalization factor (NF) [NF = (C(t)GAPDH × C(t)m18S)1/2 (Xu et al., 2014)]. The PCR program was 2 minutes at 95°C followed by 45 cycles of a 15-second denaturing step at 95°C, a 30-second annealing step at 65°C (60°C for GAPDH and m18S), and a 30-second (1-minute for GAPDH and m18S) extension at 72°C followed by melting-curve analysis. mMOR-1 and mMOR-1G expression levels were calculated with the Eq. 2−∆(Ct) (ΔC(t) = C(t)mMOR-1 or mMOR-1G − NF) and normalized to samples treated with 100 ng/ml of doxycycline (Doxy).
Cell Culture, Plasmid Constructs, and Stable or Transient Transfection.
The OPRM1-knockdown (KD) Be(2)C cell line was cultured in minimum Eagle’s medium with nonessential amino acids: F12, 10% fetal calf serum, and 0.1 mg/ml hygromycin. This OPRM1-KD cell line was made by stably expressing two short hairpin RNA constructs in the pSilencer 2.1-U6 hygro vector (Ambion) that specifically targeted human MOR-1 exons 1, but not mouse exon 1, in Be(2)C cells (American Type Culture Collection), a human neuroblastoma line that contains endogenous OPRM1 splice variants with hygromycin selection. The constructs were made by annealing and subcloning two pairs of oligos (A: sense oligo, 5′-GAT CCG CGC CAG CAA TTG CAC TGA TTT CAA GAG AAT CAG TGC AAT TGC TGG CGT TTT TTG GAA A-3′, and antisense oligo, 5′-AGC TTT TCC AAA AAA CGC CAG CAA TTG CAC TGA TTC TCT TGA AAT CAG TGC AAT TGC TGG CGC G-3′; B: sense oligo, 5′-GAT CCG CTT GTC CCA CTT AGA TGG CTT CAA GAG AGC CAT CTA AGT GGG ACA AGT TTT TTG GAA A-3′, and antisense oligo, 5′-AGC TTT TCC AAA AAA CTT GTC CCA CTT AGA TGG CTC TCT TGA AGC CAT CTA AGT GGG ACA AGC G-3′) into pSilencer 2.1-U6 hygro with the restriction enzymes BamHI and HindIII. The selected stable cells were then screened with [3H]DAMGO binding, and a cell line with a 99% reduction in binding was used for transient transfection experiments. C-terminal HA-tagged mMOR-1G constructs were made by first performing PCR with primers containing the HA tag sequence and then subcloning the PCR fragments into the pcDNA3.1 vector. C-terminal FLAG-tagged mMOR-1 constructs were made by subcloning mMOR-1 cDNA into p3XFLAG-CMV-14 vectors (Sigma-Aldrich).
Transient transfections in OPRM1-KD Be(2)C cells were carried out in 100-mm plates with Fugene HD (Promega) following the manufacturer’s protocol. Equal amounts of FLAG-tagged mMOR-1 (mMOR-1/FLAG) and HA-tagged mMOR-1G (mMOR-1G/HA) constructs were used in cotransfection. In transfections with a single construct, the amount of mMOR-1/FLAG or mMOR-1G/HA construct used was the same as in cotransfection, and pcDNA3.1 vector DNA without any insertion was included so the total amount of DNA in each transfection was preserved. The cells were harvested 32–36 hours after transfection and used for solubilization and immunoprecipitation or membrane preparation.
Tet-Off CHO cells were cultured in minimum Eagle’s medium α with 2 mM glutamine media, 10% Tet-free fetal calf serum (Clontech), and 0.1 μg/ml G418. mMOR-1 and mMOR-1G cDNAs were subcloned into the pcDNA3.1 (Invitrogen) and pTRE2hyg vector (Clontech) with appropriate restriction enzymes, respectively. To generate the Tet-Off CHO stable cell line, Tet-Off CHO cells were cotransfected with mMOR-1 and mMOR-1G constructs using the LipofectAMINE reagent (Invitrogen) and selected with G418 (0.9 μg/ml) and hygromycin (0.6 mg/ml) for 2 weeks. The stable cells were then treated with doxycycline for 48–72 hours to suppress mMOR-1G expression before they were harvested for opioid receptor binding, [35S]GTPγS binding, and reverse-transcription qPCR. All cells were grown at 37°C in a 5% CO2/95% air humidified atmosphere.
Opioid Receptor Binding Assays.
Tet-Off cells grown under indicated doxycycline concentrations were harvested for membranes as previously described (Pan et al., 1999), and protein concentration was determined with the Lowry method. [3H]DAMGO binding was carried out at 25°C for 60 minutes in 7.4-pH buffer containing 50 mM potassium phosphate and 5 mM magnesium sulfate. The incubation was then rapidly filtered with glass fiber filter paper and washed three times with 3 ml of ice-cold filtering buffer (50 mM Tris-HCl, pH 7.4). Bound radioactivity was extracted in 3 ml of Liquiscint scintillation fluid overnight before being determined by liquid scintillation spectrophotometry. Specific binding was defined as the difference between total binding and nonspecific binding defined by 10 μM levallorphan. Nonlinear regression analyses (GraphPad Prism 7, San Diego, CA) were used to calculate KD and Ki values.
[35S]GTPγS Binding.
Thirty micrograms of membrane protein from cells were incubated in the presence and absence of various concentrations (0.01–30 µM) of different μ-opioids and opioid peptides for 60 minutes at 30°C in the assay buffer (50 mM Tris-HCl (pH 7.7), 3 mM MgCl2, 0.2 mM EGTA, 10 mM NaCl) containing ∼0.05 mM [35S]GTPγS and 60 µM GDP, as described previously (Pan et al., 2005). The incubation was then filtered through glass fiber filter paper and washed three times with ice-cold filtering buffer. Basal binding was assessed in the presence of GDP and absence of drug. EC50 and % stimulation values over basal level were calculated by nonlinear regression analysis (GraphPad Prism 7).
Immunoprecipitation and Western Blot Analyses.
Whole cells from the transient transfection were solubilized in lysis buffer A [PBS (pH 7.4), 8 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid (CHAPS), and a protease inhibitor cocktail of 2 µg/ml each leupeptin, pepstatin, aprotinin, and bestatin and 0.2 mM phenylmethylsulfonyl fluoride], with shaking at 4°C for 5 hours. The mixture was then centrifuged for 15 minutes at 13,000g and 4°C before the supernatant was incubated with either EZview Red Anti-FLAG (M2) or EZview Red Anti-HA beads and shaken overnight at 4°C. The affinity gels were then washed with wash buffer (PBS, pH 7.4; 5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid) and then eluted with 3x FLAG peptide or HA peptide. The eluted sample was mixed with SDS sample buffer containing 0.15 M dithiothreitol and heated at 100°C for 10 minutes. The samples were then run on an Any kilodalton (kD) SDS-PAGE gel (Bio-Rad) before blot transfer onto PVDF membranes. The membranes were shaken in a blocking buffer containing TTBS (10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.05% Tween 20), 4% nonfat dried milk, and 1% bovine serum albumin at 25°C for 1 hour and then incubated overnight with either the anti-HA antibody (1:1000), the anti-FLAG antibody (1:800 dilution), or the anti-GAPDH antibody (1:3000 dilution) in the blocking buffer at 4°C. After four washes with TTBS buffer for 5 minutes each, the membrane was incubated for 1 hour at 25°C in TTBS with either peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG antibody (1:10,000 dilution; Jackson ImmunoResearch). After washing another four times with TTBS buffer, the signals were visualized with ChemiGrow reagents (Proteinsimple, Santa Clara, CA) and imaged and analyzed on ChemiDoc MP Image System (Bio-Rad).
Brefeldin A Treatment.
OPRM1-KD Be(2)C cells were transiently cotransfected with mMOR-1/Flag and mMOR-1G/HA. Eight hours after transfection, the cells were treated with or without 4 µg/ml of brefeldin A (BFA) for another 24 hours. The whole cells were then harvested and used for either immunoprecipitation and Western blot analysis or for membrane preparation used in [3H]DAMGO binding, as described above.
Subcellular Fractionation Analysis.
The Tet-Off cells stably expressing mMOR-1 and mMOR-1G treated with 100 ng/ml of doxycycline for 36 hours or without doxycycline were lifted from 150-mm plates by incubating with 2 mM EDTA for 5 minutes at 37°C. After washing with PBS, the cell pellets were homogenized with a tight-fitting Dounce homogenizer for 25 strokes in buffer A [10 mM Tris-HCl, pH7.4, 1 mM EDTA, 0.25 M sucrose and the protease inhibitor cocktail (see above)] and then passed through a 25-gauge syringe needle. Postnuclear supernatant was collected by centrifugation of the pass through at 1000g for 5 minutes and loaded on the top of the discontinuous OptiPrep (Fisher Scientific) gradient that was manually made by layering 1.4 ml each of 26%, 22%, 18%, 14%, 10%, 6%, and 2% OptiPrep in buffer A from the bottom of ultracentrifuge tube. After centrifugation at 284,000g for 3 hours in a SORVALL wX+ Ultra Series centrifuge, sequential 0.8-ml fractions were harvested from the top of the tube and used for Western blot analysis and [3H]DAMGO binding. The OptiPrep density in each fraction was determined by using absorbance 340-nm measurement following the manufacturer’s instructions (Axis-Shield).
Statistical Analysis.
All statistical analysis was carried out using GraphPad Prism 7. Two-tailed Student’s t test and one-way or two-way ANOVA with post hoc Bonferroni’s multiple comparisons test were performed as described in the figure legend. Data represented the mean ± S.D. of at least three independent experiments. Statistical significance was set at P < 0.05.
Results
6TM Variant mMOR-1G Enhances Expression of the Functional 7TM mMOR-1 Receptor Protein in a Tet-Off CHO Cell Line.
To investigate the influence of 6TM variants on the expression of 7TM mMOR-1, we generated a stable Tet-Off CHO cell line in which 7TM mMOR-1 is constitutively expressed, whereas the expression of the 6TM variant, mMOR-1G, is regulated by doxycycline, a synthetic drug derived from tetracycline. In contrast to a Tet-On system, the Tet-Off system uses Doxy to suppress expression of a gene of interest, which was mMOR-1G in our study. Thus, in the absence of doxycycline, mMOR-1G was maximally expressed, whereas increased doxycycline concentration resulted in reduced expression of mMOR-1G. There are multiple 6TM variants of the mouse Oprm1 gene. We selected mMOR-1G for this study because it was abundant and uniquely conserved from rodent to human. 6TM variants alone do not bind to available radio-labeled opioids, including [3H]DAMGO, [3H]diprenorphine, and [125I]IBNtxA, when expressed in CHO cells (Supplemental Fig. 1) or human embryonic kidney (HEK) 293 cells (unpublished data). Therefore, we used an opioid receptor binding assay with [3H]DAMGO to determine the expression level of 7TM mMOR-1 protein because [3H]DAMGO binding represents the expression of only 7TM mMOR-1 protein and not 6TM variants in this Tet-Off cell line coexpressing mMOR-1 and mMOR-1G. We also generated another Tet-Off cell line expressing only mMOR-1G regulated by doxycycline and lacking expression of 7TM mMOR1. In this mMOR-1G–only Tet-Off cell line, there is no [3H]DAMGO or [125I]IBNtxA binding, with or without doxycycline (Supplemental Fig. 2).
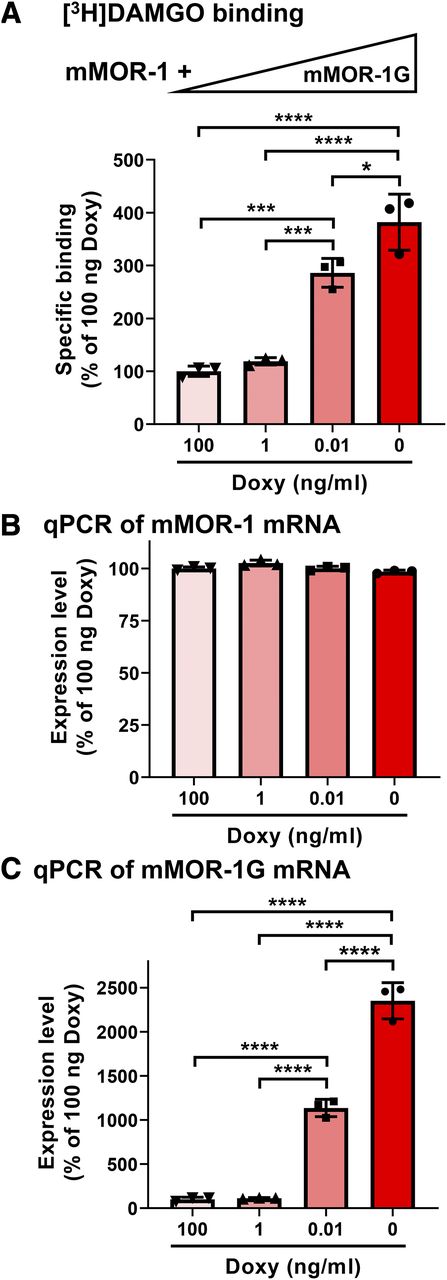
In the Tet-Off cell line with constitutive expression of 7TM mMOR-1 and doxycycline-regulated expression of mMOR-1G, we first observed that expression of mMOR-1G increased [3H]DAMGO binding in a dose-dependent manner (Fig. 1A). When expression of mMOR-1G mRNA was fully suppressed by 100 ng/ml of doxycycline (Fig. 1C), a minimal level of [3H]DAMGO binding was observed, which represents the basal or constitutive expression level of 7TM mMOR-1. However, when mMOR-1G mRNA was increased by lowering doxycycline (Fig. 1C), [3H]DAMGO binding increased in parallel, reaching a maximum when no doxycycline was present and mMOR-1G mRNA was fully expressed. Saturation studies with [3H]DAMGO further demonstrated an increase in functional receptor number (Bmax), whereas receptor affinity (KD) was unaffected (Table 1). However, mMOR-1 mRNA level was not significantly altered by different doses of doxycycline or mMOR-1G expression (Fig. 1B), suggesting that increased [3H]DAMGO binding does not result from changes in mMOR-1 mRNA levels.

Enhanced functional mMOR-1 protein expression by mMOR-1G coexpression in Tet-Off CHO cells. (A) [3H]DAMGO binding (n = 3). [3H]DAMGO binding was performed using membranes isolated from a Tet-Off cell line with constitutive expression of mMOR-1 and Doxy-regulated mMOR-1G in the presence or absence of the indicated concentration of Doxy, as described in Materials and Methods. mMOR-1G expression was controlled by doxycycline, so the highest expression level of mMOR-1G was seen in the absence of doxycycline. Increasing concentrations of doxycycline resulted in progressively reduced levels of mMOR-1G expression. Triangle shows the expression levels of mMOR-1G. (B and C) Expression of mMOR-1 [(B), n = 3] and mMOR-1G [(C), n = 3] mRNAs. Total RNAs isolated from the same Tet-Off cells were used for reverse-transcription qPCR as described in Materials and Methods. GAPDH and m18S were used for normalization. Expression levels were calculated through 2−(ΔC(t)) and normalized using the samples from the Tet-Off cells with doxycycline (100 ng), which was set as 100%. All significant differences were analyzed by one-way ANOVA with post hoc Bonferroni’s multiple comparisons test. *P < 0.05; ***P < 0.001; ****P < 0.0001.
Saturation and competition studies using [3H]DAMGO
Saturation and competition assays were performed using indicated membranes as described in Materials and Methods. In saturation assays, five conc. of [3H]DAMGO (0.125–2 nM) were used. KD values were calculated by nonlinear regression analyses (Prism 7). In competition assays, 1–1.5 nM of [3H]DAMGO was used against three to four conc. of unlabeled morphine and DAMGO (1–25 nM), M6G (5–100 nM), β-endorphin (10–200 nM), and dynorphin A (10–1000 nM). IC50 values were calculated by nonlinear regression analyses (Prism 7). Ki values were calculated by Ki = IC50/(1 + A/KD), in which A is the nM conc. of [3H]DAMGO, and KD value is obtained from saturation study. Results are the mean ± S.D. of three independent experiments. Significant differences between −Doxy and +Doxy groups were analyzed by two-tailed Student’s t test.
We then performed [35S]GTPγS binding assays to assess the effect of 6TM mMOR-1G on modulating μ agonist–induced G protein coupling with mMOR-1 in the Tet-Off CHO cell line coexpressing mMOR-1 and mMOR-1G. When compared with no mMOR-1G expression (+Doxy), induction of mMOR-1G at maximal level (−Doxy) showed no significant effect on drug potency (EC50 value) of morphine, morphine-6β-glucuronide (M6G), and β-endorphin but led to a small and significant decrease of the EC50 value for DAMGO. Meanwhile, the percent stimulation over basal level (% Stim), an indication of drug efficacy by morphine, DAMGO, and M6G, increased significantly when mMOR-1G was maximally expressed (−Doxy) (Table 2), which was consistent with the Bmax value increase seen in saturation studies with [3H]DAMGO. In the Tet-Off CHO cell line expressing only mMOR-1G without expression of 7TM mMOR-1, [35S]GTPγS binding with μ agonists, including DAMGO, morphine, and IBNtxA, showed no G protein stimulation (Supplemental Fig. 3). Thus, the μ agonist–induced G protein stimulation seen in the Tet-Off CHO cell line coexpressing 7TM mMOR-1 and 6TM mMOR-1G is mediated through 7TM mMOR-1. Together, these results suggest that 6TM mMOR-1G does not change mRNA expression of 7TM mMOR-1 but enhances expression of functional 7TM mMOR-1 at the protein level.
[35S]GTPγS binding in Tet-Off cells expressing mMOR-1/mMOR-1G
[35S]GTPγS binding was performed using membranes from −Doxy and +Doxy cells with six conc. of indicated drugs (0–10 µM), as described in Materials and Methods. EC50 and % stimulation values over basal level (% stim) were calculated by nonlinear regression analysis (Prism 7). So, the basal level is, as always, 0%. Results are the mean ± S.D. of three independent experiments. Significant differences between −Doxy and +Doxy groups were analyzed by two-tailed Student’s t test.
To further confirm that mMOR-1G can increase expression of the 7TM mMOR-1 at the protein level, we used an immunoprecipitation (IP) approach in an OPRM1-KD Be(2)C cell line transiently transfected with FLAG-tagged mMOR-1 (mMOR-1/FLAG) or HA-tagged mMOR-1G (mMOR-1G/HA) or both mMOR-1/FLAG and mMOR-1G/HA. When compared with transfection with mMOR-1/FLAG alone, cotransfection of both mMOR-1/FLAG and mMOR-1G/HA significantly increased mMOR-1/FLAG level in both immunoblots (Figs. 2 and 3, A and B) and [3H]DAMGO binding (Fig. 3C), which was consistent with the results seen in the Tet-Off cells with nontagged constructs.

Increased mMOR-1/Flag protein expression by mMOR-1G/HA coexpression through their heterodimerization in OPRM1-KD Be(2)C cells. (A) Co-IP study (n = 3). The transient transfection, solubilization, and immunoprecipitation/immunoblot were performed as described in Materials and Methods. Equal amounts of mMOR-1/Flag and mMOR-1/HA plasmid DNAs were used in transient cotransfection (lane 1). The amount of mMOR-1/Flag or mMOR-1G/HA in single transfection (lanes 2 or 3) was same as the cotransfection, but pcDNA3.1 DNA was included to keep the total amount of DNA constant in all transfections. Top panel: immunoblots (IBs) with the Flag antibody on the elution fractions immunoprecipitated with EZview Red Anti-Flag (M2) beads. The second panel from the top, immunoblots with the HA antibody on the elution fractions immunoprecipitated with EZview Red Anti-HA beads. The third panel from the top, immunoblots with the Flag antibody on the elution fractions immunoprecipitated with EZview Red Anti-HA beads. The fourth panel from the top, immunoblots with the HA antibody on the elution fractions immunoprecipitated with EZview Red Anti-Flag (M2) beads. Bottom panel, immunoblots with GAPDH antibody on lysates used for immunoprecipitation. ab, antibody. (B) Quantification of mMOR-1/Flag in the top panel of (A) (n = 3). The band intensities were analyzed by using Image Laboratory software (Bio-Rad). The expression level of mMOR-1/Flag was calculated by normalizing mMOR-1/Flag level as the mMOR-1/Flag/GAPDH ratio with the bands from transfected with mMOR-1G/HA (lane 3). Significant difference was calculated by using one-way ANOVA with post hoc Bonferroni’s multiple comparisons test. **P < 0.01; ***P < 0.001.
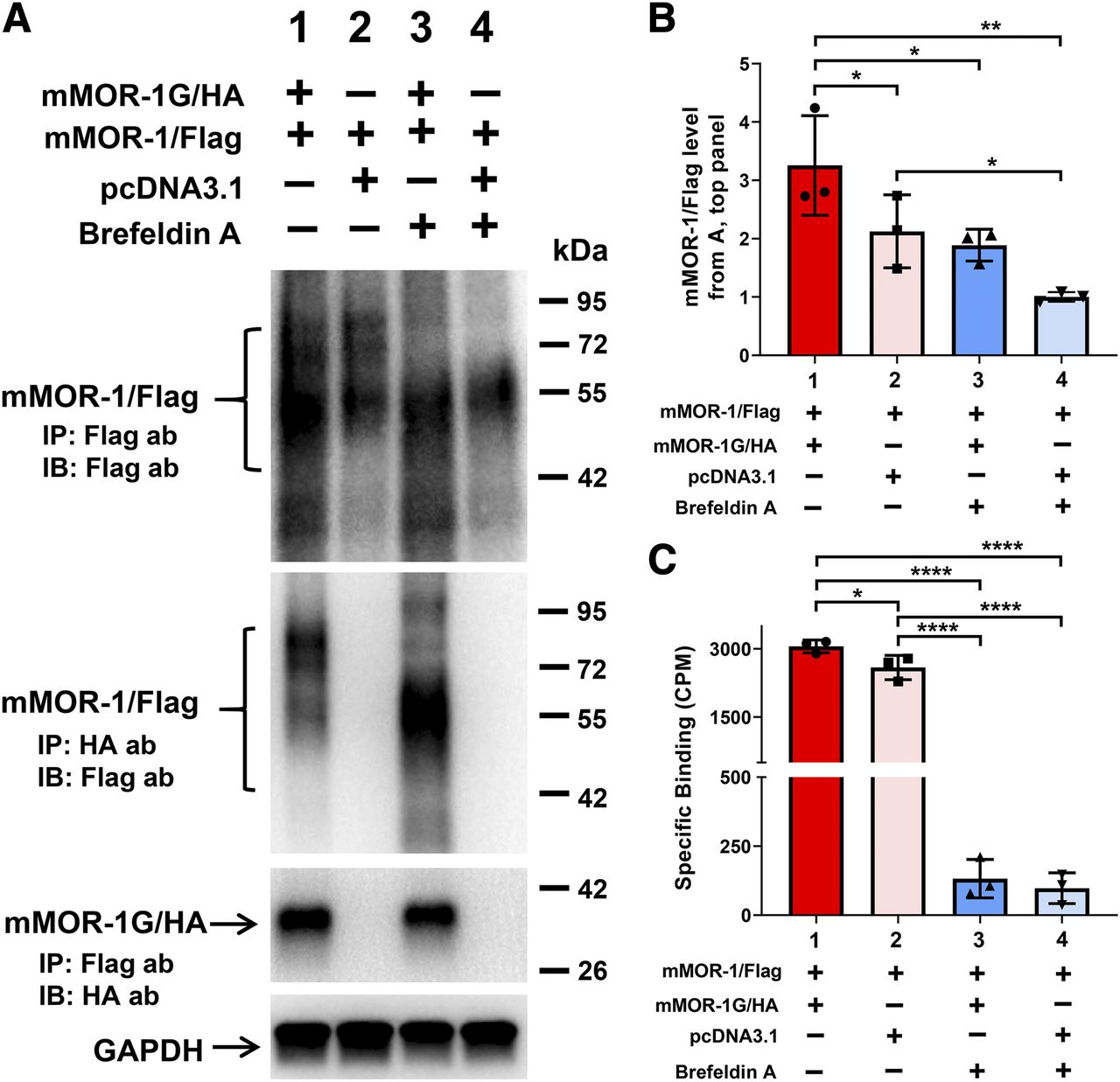
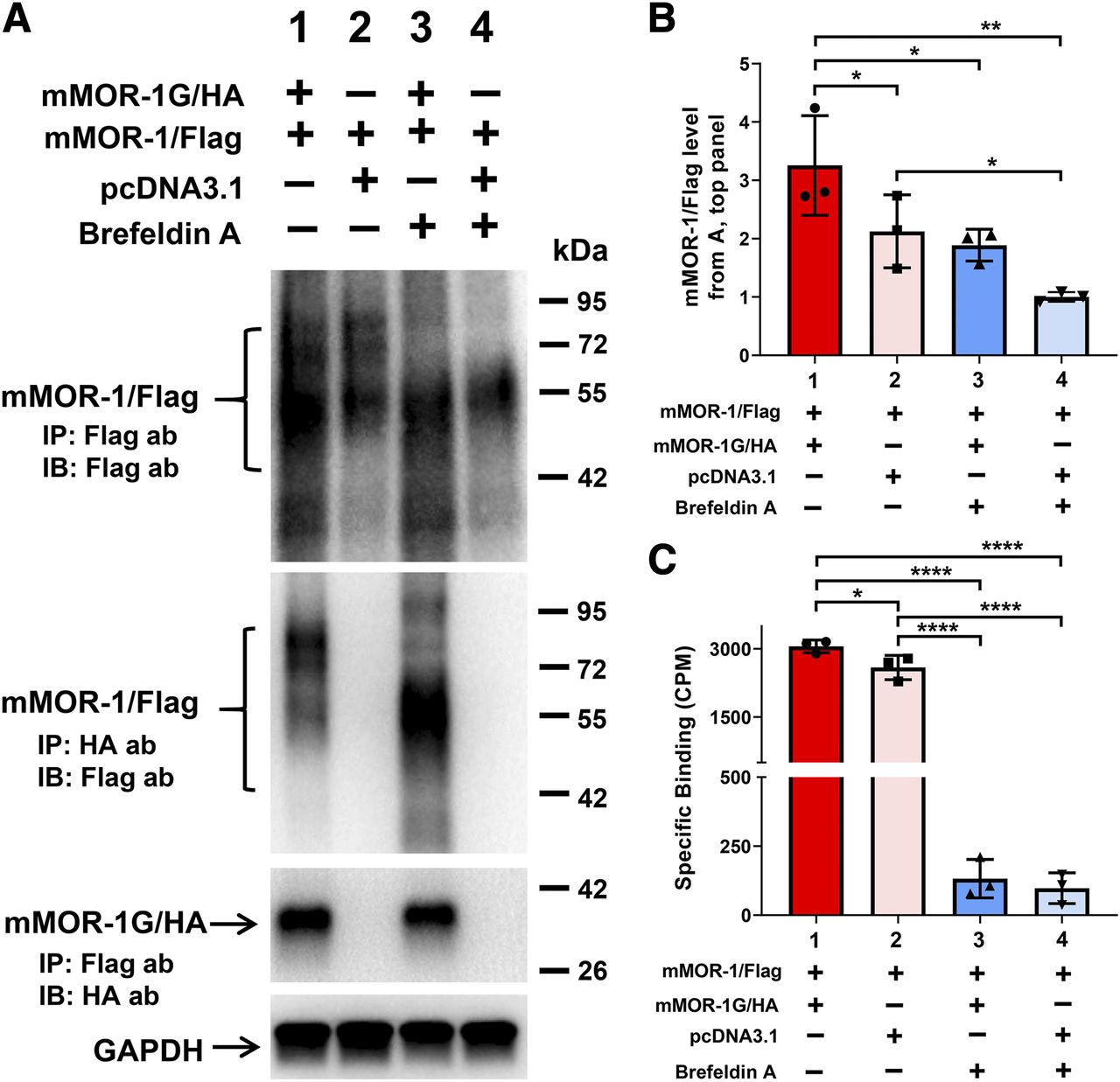
Effect of brefeldin A on expression of mMOR-1/Flag induced by mMOR-1G/HA and heterodimerization of mMOR-1/Flag and mMOR-1G/HA in OPRM1-KD Be(2)C cells. (A) Co-IP study (n = 3). The transient transfection, BFA treatment, solubilization, and immunoprecipitation/immunoblot were performed as described in Materials and Methods. Again, equal amounts of mMOR-1/Flag and mMOR-1G/HA plasmid DNAs were used for cotransfection (lanes 1 and 3). The amount of mMOR-1/Flag in single transfection (lanes 2 and 4) was same as the cotransfection (lanes 1 and 3), but pcDNA3.1 was added to keep the total amount of DNA constant. Eight hours after transfection, the cells were treated with or without 4 µg/ml of BFA for additional 24 hours before harvesting. Top panel, immunoblots (IBs) with the Flag antibody on the elution fractions immunoprecipitated with EZview Red Anti-Flag (M2) beads. The second panel from the top, immunoblots with the Flag antibody on the elution fractions immunoprecipitated with EZview Red Anti-HA beads. The third panel from the top, immunoblots with the HA antibody on the elution fractions immunoprecipitated with EZview Red Anti-Flag (M2) beads. Bottom panel, immunoblots with a GAPDH antibody on lysates used for immunoprecipitation. ab, antibody. (B) Quantification of immunoblots in (A) (n = 3). Band intensities were analyzed using Image Laboratory software (Bio-Rad). Significant difference was calculated by one-way ANOVA with post hoc Bonferroni’s multiple comparisons test. *P < 0.05; **P < 0.01. (C) [3H]DAMGO binding using membranes isolated from cells with indicated constructs and BFA treatment (n = 3). Significant difference was calculated by one-way ANOVA with post hoc Bonferroni’s multiple comparisons test. *P < 0.05; ****P < 0.0001. CPM, counts per minute.
We next performed [3H]DAMGO competition assays with several opioids to investigate whether the coexpression of mMOR-1 with mMOR-1G can modulate μ-binding selectivity and affinity in the Tet-Off CHO cells. When mMOR-1G was not present (+Doxy), μ drugs, such as morphine, DAMGO, fentanyl, and M6G, all competed binding quite potently, whereas κ drug U50,488H and δ drug [D- Pen 2,D- Pen 5]enkephalin (DPDPE) were both ineffective (Table 1). Coexpression of mMOR-1G (−Doxy) did not alter the Ki values of most drugs, demonstrating that coexpressing mMOR-1G does not affect their binding affinity or selectivity for mMOR-1 (Table 1). However, coexpressing mMOR-1G led to a small but significant decrease of the Ki values for M6G and β-endorphin, which raises the interesting question of whether mMOR-1G coexpression has a structural impact on the binding pocket of mMOR-1.
6TM Variant mMOR-1G Increases 7TM mMOR-1 Receptor Levels at the Plasma Membrane in a Tet-Off CHO Cell Line.
We next defined the subcellular distribution of the 7TM mMOR-1 expression in the Tet-Off CHO cells treated with 100 ng/ml of doxycycline (+Doxy, no mMOR-1G expression) or without doxycycline (−Doxy, mMOR-1G coexpression) via subcellular fractionation analysis using OptiPrep density gradient centrifugation, a common method for separating subcellular organelle membranes. The postnuclear supernatants were resolved by centrifugation through OptiPrep gradients (2%–26%) and collected from the top in 14 fractions that showed a near-linear OptiPrep density from 1.00 to 1.25 g/ml (Fig. 4A). Western blot analysis with the organelle marker antibodies revealed that the plasma membrane indicated by N-cadherin bands were enriched in fractions 7 to 8, whereas the endoplasmic reticulum (ER) membrane shown by the calnexin bands was peaked in fractions 10 to 11 (Fig. 4, B and C). Similar results were obtained using Na+/K+-ATPase antibody, another plasma membrane marker, and calreticulin antibody, another ER marker (unpublished data). Although slightly overlapping, the peaks of these two markers were adequately separated.

Increased expression of the functional mMOR-1 by mMOR-1G in the Tet-Off cells at the plasma membrane–enriched fractions. (A) Subcellular fractionation analysis. The Tet-Off cells coexpressing mMOR-1 and mMOR-1G were treated with 100 ng/ml of doxycycline (+Doxy) and without doxycycline (−Doxy), homogenized, and fractionated in OptiPrep density gradient (2%–26%) through ultracentrifugation, as described in Materials and Methods. OptiPrep density of each fraction was determined by measuring absorbance 340-nm following the manufacturer’s instructions (Axis-Shield). One hundred twenty-five microliters per tube of each fraction was used in [3H]DAMGO (1.5 nM) binding as described in Materials and Methods. Specific binding was calculated by subtracting total binding with nonspecific binding in the presence of 10 µM levallorphan. Data represented the mean ± S.D. of three experiments. Significant difference was calculated by two-way ANOVA with post hoc Bonferroni’s multiple comparisons test. Compared with −Doxy, **P < 0.01; ****P < 0.0001. CMP, counts per minute. (B) Western blot analysis. Equal volume of samples from each fraction was treated with SDS sample buffer, separated on Any kilodalton (kD) SDS-PAGE and transferred onto PVDF membrane, as described in Materials and Methods. The PVDF membranes were used in immunoblot with anti–N-cadherine antibody (1:1800; Novus Biologicals), a plasma membrane marker, or anti-calnexin antibody (1:2000; Novus Biologicals), an ER marker, as described in Materials and Methods. (C) Quantification of Western blots in (B) (n = 3). The band intensities were analyzed by using Image Laboratory software (Bio-Rad).
We then determined the expression of the functional 7TM mMOR-1 receptor in each collected fraction using [3H]DAMGO binding. The peak of [3H]DAMGO binding in either +Doxy or −Doxy cells overlapped well with plasma membrane–enriched fractions (Fig. 4A), suggesting that functional and mature mMOR-1 was expressed mainly in the plasma membrane. Furthermore, we demonstrated a significant increase of [3H]DAMGO binding in −Doxy cells (mMOR-1G coexpression) compared with +Doxy cells (no mMOR-1G expression) in plasma membrane–enriched fractions (Fig. 4A). These data illustrate that an increased expression of the functional 7TM mMOR-1 via mMOR-1G coexpression also occurs mainly in the plasma membrane.
Physical Association of 6TM Variant mMOR-1G with 7TM mMOR-1.
Enhanced expression of mMOR-1 protein by coexpression of mMOR-1G in the Tet-Off cell line raised the question of whether 6TM mMOR-1G can physically associate with 7TM mMOR-1. We addressed this question by performing coimmunoprecipitation (Co-IP) assays in the OPRM1-KD Be(2)C cells transiently cotransfected with FLAG-tagged mMOR-1 (mMOR-1/FLAG) and HA-tagged mMOR-1G (mMOR-1G/HA) constructs. We observed that mMOR-1/FLAG was detected with anti-FLAG antibody in the IP elution using anti-HA antibody-conjugated beads from the cotransfections (Fig. 2A, lane 1 in the third panel). The converse Co-IP experiment also detected mMOR-1G/HA with anti-HA antibody in the IP elution using anti-FLAG antibody-conjugated beads from the cotransfection (Fig. 2A, lane 1 in the fourth panel). The specificity of the physical association was confirmed by parallel IP using cells transfected with mMOR-1/FLAG or mMOR-1G/HA alone (Fig. 2A, lanes 2 and 3 in the third and fourth panels, respectively). Together, these results indicate physical association or heterodimerization between mMOR-1G/HA and mMOR-1/FLAG. The mMOR-1/FLAG bands were shown with wide range of molecular weights from 50 to 90 kDa, most likely due to differential glycosylation of the receptor, which was consistent with those reported from literature. Only monomer mMOR-1/FLAG or mMOR-1G/HA with their predicted molecular weights were shown on immunoblots, suggesting that they physically associated or heterodimerized during IP but dissociated in the presence SDS in the SDS-PAGE. Furthermore, cotransfection of mMOR-1/FLAG with mMOR-1G/HA significantly increased immunoprecipitated mMOR-1/FLAG protein compared with transfection of mMOR-1/FLAG alone (Fig. 2A, the top panel). Since the same amount of mMOR-1/FLAG plasmid DNA was used in the transfection, and the same amount of cells was used in IP and immunoblots, these results suggest that mMOR-1G/HA can increase expression of mMOR-1/FLAG at the protein level, which was consistent with the increase of [3H]DAMGO and [35S]GTPγS binding seen in the Tet-Off cell line coexpressing mMOR-1 and mMOR-1G.
Since Co-IP was performed using lysates from whole cells, the subcellular compartment location of dimerization between mMOR-1/FLAG and mMOR-1G/HA was unknown. To determine location of dimerization, we transfected cells treated with or without BFA. BFA blocks protein transport from the ER to the Golgi, resulting an accumulation of proteins in the ER and preventing further protein maturation in the Golgi. If dimerization were to occur in compartments downstream from the ER, we would expect to see no dimerization after BFA treatment. The Co-IP experiment revealed that the dimerization between mMOR-1/FLAG and mMOR-1G/HA was preserved in BFA-treated cells (Fig. 3A, lanes 1 and 3 in the second and third panels), suggesting that this dimerization occurs within the ER. BFA-treated cells showed the same increase in expression level of mMOR-1/FLAG protein by coexpression of mMOR-1G/HA, as indicated by immunoblot (Fig. 3, A and B), implying that dimerization within the ER leads to the increased expression of the full-length 7TM FLAG-mMOR-1 protein at the plasma membrane.
Additionally, BFA treatment led to a loss of mMOR-1/FLAG proteins with higher molecular weights (∼70–90 kDa), as shown on immunoblots (Fig. 3A, lanes 3 and 4 in the first and second panels), suggesting that BFA eliminates further glycosylation or maturation of the mMOR-1/FLAG, most likely in the Golgi. This downstream glycosylation or maturation appears to be crucial in producing a functional receptor. BFA treatment abolished almost all [3H]DAMGO binding in either cotransfection or single mMOR-1/FLAG transfection (Fig. 3C), despite the fact that the mMOR-1/FLAG proteins with the lower molecular weight (∼50–70 kDa) were present (Fig. 3A, lanes 3 and 4 in the first panel).
Discussion
In the present study, we demonstrate a novel role of the Oprm1 6TM variant mMOR-1G in enhancing expression of the 7TM mMOR-1 at the protein level. This conclusion was based on the results from [3H]DAMGO- and [35S]GTPγS binding assays in a Tet-Off CHO cell system as well as immunoprecipitation and Western blot analysis in an OPRM1-KD Be(2)C cell line transiently transfected with tagged mMOR-1 and mMOR-1G constructs. Using a Co-IP approach, we further demonstrate that mMOR-1G physically associates with the 7TM mMOR-1 starting at the ER, contributing to the enhanced expression of 7TM mMOR-1 protein at the plasma membrane. The physical association of mMOR-1G does not modify the binding affinity of mMOR-1 in saturation studies, because the KD values remain unchanged but increase mMOR-1 receptor levels, as shown by an increase in Bmax values in saturation study and an increase of % stimulation in [35S]GTPγS binding assays in the Tet-Off cells coexpressing mMOR-1 and mMOR-1G.
Increased expression of 7TM mMOR-1 protein with coexpression of 6TM mMOR-1G is not correlated with mMOR-1 mRNA levels. Instead, immunoprecipitation and Western blot analysis show an increase of 7TM mMOR-1/FLAG protein levels, which corresponds to the increased Bmax in saturation study and % stimulation in [35S]GTPγS binding studies. Our earlier studies showed that Oprm1 single-TM variants dimerize with 7TM mMOR-1 in the ER, leading to increased 7TM mMOR-1 protein expression via chaperone-like function (Xu et al., 2013). Like the single-TM variants, 6TM mMOR-1G can dimerize with 7TM mMOR-1 and increase its protein expression in the ER, as shown by the BFA treatment studies. Our data suggest that 6TM mMOR-1G increases expression of mMOR-1 through a chaperone-like mechanism similar to what is seen with the single-TM variants. The observation that increased mMOR-1 was mainly distributed at the plasma membrane–enriched fractions by subcellular fractionation analysis further supports the chaperone-like role of the 6TM mMOR-1G. This chaperone-like mechanism is also observed in the actions of several opioid agonists and antagonists (Chaipatikul et al., 2003a,b; Chen et al., 2006; Leskelä et al., 2007; Wannemacher et al., 2007).
The physical association between mMOR-1G with mMOR-1 does not alter the binding affinity for morphine, DAMGO, and fentanyl in competition assays. However, it significantly improves the binding affinity for M6G and β-endorphin by lowering their respective Ki values. Interestingly, the improved binding affinity for M6G and β-endorphin by mMOR-1/mMOR-1G dimerization was not seen in the dimerization of the single-TM variants with mMOR-1 (Xu et al., 2013). This suggests that the association of mMOR-1G with mMOR-1 may slightly alter the agonist conformation of the 7TM mMOR-1, favoring binding to M6G and β-endorphin, whereas association with the single-TM variants does not have similar structural changes. It will be interesting to explore these structural changes of mMOR-1/mMOR-1G dimerization using computer simulation models or crystal structures in the future.
Unlike detergent-resistant heterodimerization between δ-opioid receptor-1 (DOR-1) and κ-opioid receptor-1 (KOR-1) (Jordan and Devi, 1999), the heterodimerization between mMOR-1G and mMOR-1 was not stable in SDS-PAGE since only the monomers were observed on immunoblots. The crystal structure of mMOR-1 revealed that the mMOR-1 homodimer was formed mainly through TM5 and TM6 (Manglik et al., 2012). Although both mMOR-1 and mMOR-1G have the same TM domains from TM2 to TM7, the detergent-sensitive interaction between mMOR-1 and mMOR-1G raises questions of whether TM5 and TM6 are the regions responsible for mMOR-1/mMOR-1G heterodimerization or whether additional regions are involved.
The current study only focused on mMOR-1G, one of five 6TM variants in mouse, raising the question of whether other 6TM variants have a similar chaperone-like function. All five 6TM variants share the same TM2 to TM7 and part of intracellular C-terminal sequence encoded by exon, but have different N termini or C termini generated by alternative 5′ or 3′ splicing, respectively (Pan et al., 2001). We have preliminary data to indicate that two other 6TM variants, mMOR-1M and mMOR-1N, can also physically associate with mMOR-1 (unpublished data), implying that they can enhance 7TM mMOR-1 expression at the protein level through a similar mechanism as mMOR-1G. These data need to be verified in future studies.
N-terminal glycosylation has been shown to be important for expression of functional MOR-1. [3H]funaltrexamine labeling studies (Liu-Chen et al., 1993) and Western blot analyses using N-glycanase (Huang et al., 2008; Huang and Liu-Chen, 2009) demonstrate that 7TM MOR-1 is heavily glycosylated at the N terminus. A single nucleotide polymorphism, A118G, located at a N-glycosylation site of the human MOR-1, was shown to alter β-endorphin binding (Bond et al., 1998; Kroslak et al., 2007) and modulate expression of MOR-1 mRNA and protein (Zhang et al., 2005). An A112G mutation that mimics human single nucleotide polymorphism, A118G, in mouse altered MOR-1 glycosylation and modulated MOR-1 stability at the protein level (Huang et al., 2012). Our BFA study further suggests that MOR-1 glycosylation is essential for maturation and function of the 7TM μ-opioid receptor.
Multiple Oprm1 full-length 7TM variants have been identified, all of which contain identical receptor protein structures and differ only at the tip of the intracellular C terminus. The current study focused on mMOR-1, one of the most dominant and abundant C-terminal variants. It is likely that 6TM variants can dimerize with other full-length 7TM C-terminal variants since they share the same TM domains, if the TM regions, particularly TM5 and TM6 that are responsible for MOR-1 homodimer (Manglik et al., 2012), are the main regions involved in dimerization.
Using in vivo Oprm1 gene-targeting mouse models, we have previously shown that the 6TM variants are essential targets for a novel class of opioids, such as IBNtxA (Majumdar et al., 2011b; Lu et al., 2015, 2018). They are also implicated in analgesic actions of δ and κ drugs as well as nonopioid drugs, such as clonidine, an α2-adrenergic agonist (Marrone et al., 2016). However, when expressed in cell lines alone, the 6TM variants do not bind to any known opioids, raising questions regarding the molecular mechanisms underlying their in vivo functions. One hypothesis is that the in vivo actions of the 6TM variants are mediated through their physical association with a GPCR or non-GPCR protein. [125I]IBNtxA bound to a site in a triple-KO mouse brain, which was similar to that in wild-type mouse brain in the presence of the μ-δ-κ blockers (Majumdar et al., 2011b). IBNtxA analgesia in the triple KO was also normal. In the triple-KO mouse, all opioid receptors, including the 7TM μ-opioid receptors, δ- opioid receptor-1 (DOR-1) and κ-opioid receptor-1 (KOR-1), were disrupted, but expression of the 6TM variants was preserved. We speculate that 6TM variants interact with other receptors or nonreceptor proteins constitute the [125I]IBNtxA binding site and produce IBNtxA analgesia observed in the triple-KO mice. Coexpression of mMOR-1G and ORL-1 receptor in human embryonic kidney 293 cells reconstituted [125I]IBNtxA binding, suggesting heterodimerization between these two receptors (Majumdar et al., 2011b). However, it remains unknown whether the interaction of mMOR-1G with ORL-1 corresponds to IBNtxA analgesia in mice. Future studies should explore the action of IBNtxA in ORL-1 KO or E1/ORL-1 double KO or quad KO (the triple KO plus ORL-1 KO). The 6TM variants can also heterodimerize with β2-adrenoreceptor, leading to synergistic analgesia when the 6TM and β2-adrenoreceptor drugs are coadministered (Samoshkin et al., 2015). The current study provides another example of the physical association between 6TM mMOR-1G and 7TM mMOR-1, at least in vitro. However, their in vivo physical interaction and related functions need further exploration.
Eliminating 6TM variants in an E11-KO mouse led to loss of IBNtxA analgesia but did not affect morphine analgesia (Majumdar et al., 2011b). Knocking out all the 7TM variants in an E1-KO or a triple-KO mouse resulted in loss of morphine analgesia but preserved IBNtxA analgesia (Schuller et al., 1999; Pasternak and Pan, 2013). Thus, the potential association between the 6TM and 7TM variants in vivo cannot contribute to either morphine or IBNtxA analgesia. However, increasing evidence suggests that the 6TM variants are involved in several other actions of morphine. The loss of 6TM variants in the E11-KO mice led to reduction of morphine tolerance, hyperalgesia, and locomotor activity (Marrone et al., 2017). mMOR-1K, another mouse 6TM variant, has been implicated in morphine hyperalgesia (Gris et al., 2010; Oladosu et al., 2015). Our study raises intriguing questions of whether the roles of the 6TM variants in these morphine actions are mediated through the physical association between 6TM and 7TM variants in vivo.
Truncated 6TM variants are not limited to the OPRM1 gene. Approximately 6% of human GPCR genes (excluding olfactory receptor genes) have predicted 6TM splice variants based on genomic data base analysis (unpublished observation). The current study opens avenues of inquiry as to whether other 6TM variants may have a similar function in enhancing 7TM receptor expression.
In summary, the current study illustrates that the 6TM mMOR-1G enhances expression of the functional 7TM mMOR-1 at the protein level. mMOR-1G physically interacts with mMOR-1 starting at the ER, enhancing mMOR-1 expression at the plasma membrane through a chaperone-like function. Our study also raises questions for future exploration of the functional relevance of Oprm1 6TM/7TM interactions in vivo.
Authorship Contributions
Participated in research design: Zhang, Xu, Pan.
Conducted experiments: Zhang, Xu, Pan.
Contributed new reagents or analytic tools: Zhang, Pan.
Performed data analysis: Zhang, Xu, Pan.
Wrote or contributed to the writing of the manuscript: Zhang, Pan.
Footnotes
- Received January 8, 2020.
- Accepted July 6, 2020.
This work was supported by grants from National Institutes of Health National Institute on Drug Abuse [Grants DA006241, DA007242, DA042888, DA040858, and DA046714], grants from the Mayday Foundation and the Peter F. McManus Charitable Trust to Y.-X.P., and a core grant from the National Institutes of Health National Cancer Institute [Grant CA008748] to Memorial Sloan-Kettering Cancer Center.
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- AP
- antisense primer
- BFA
- brefeldin A
- Co-IP
- coimmunoprecipitation
- DAMGO
- [D-Ala2,MePhe4,Gly(ol)5]enkephalin
- Doxy
- doxycycline
- ER
- endoplasmic reticulum
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- GPCR
- G protein–coupled receptor
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- HA
- hemagglutinin
- IBNtxA
- 3-iodobenzoyl-6β-naltrexamide
- IP
- immunoprecipitation
- KD
- knockdown
- KO
- knockout
- m18S
- mouse 18S ribosome
- M6G
- morphine-6β-glucuronide
- mMOR
- mouse μ-opioid receptor
- OPRM1
- μ-opioid receptor gene
- ORL
- opioid receptor-like
- PCR
- polymerase chain reaction
- PVDF
- polyvinylidene difluoride
- qPCR
- quantitative PCR
- SP
- sense primer
- TM
- transmembrane
- TTBS
- Tris buffered saline with Tween 20
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}