


Abstract
Wild-type α1β2γ2 γ-aminobutyric acid (GABA)Areceptors and receptors containing a point-mutated subunit γ2F77Y were expressed by transient transfection in human embryonic kidney 293 cells. Mutant receptors bound the benzodiazepine binding site ligand [3H]flumazenil with similar, subnanomolar affinity as wild-type receptor. Displacement studies with diazepam showed that the affinity for this compound was reduced 250-fold on mutation, indicating that the tyrosine hydroxyl group interferes with diazepam binding. This differential behavior then was used to find the chemical entity presumably interacting with the phenyalanine residue in position 77 of the γ2 subunit of wild-type receptors. Thirty-four substances were analyzed in this respect. Our results suggest that the phenyl substituent of diazepam is located close to γF77. Similarly, we investigated the possible location of α1T206 and γ2M130. Electrophysiological data obtained with the wild-type receptor furthermore suggest a simple overlap between positive allosteric modulators acting at the benzodiazepine binding site with its antagonists.
The GABAA receptor is the most important ion channel conferring fast synaptic inhibition in the mammalian nervous system. Initially, two subunits of the receptor have been purified (Sigelet al., 1983), and its coding DNAs have been cloned (Schofield et al., 1987). Eighteen subunits from mammalian tissue have been cloned: 6α, 3β, 3γ, 1δ, 1ε, 3ρ, and 1π (for reviews, see Macdonald and Olsen, 1994; Rabow et al., 1995). The major adult isoform is most likely α1β2γ2 (McKernan and Whiting, 1996). The receptor channel is modulated by numerous drugs (Sieghart, 1995). Among these, some compounds act at the binding site for benzodiazepines. They act as anxiolytics, sedatives, muscle relaxants, and anticonvulsives and exert a positive allosteric effect on the channel. An antagonist acting at this site also is in clinical use (Hunkeler et al., 1981), whereas negative allosteric modulators such as DMCM are investigational tools.
Amino acid residues H101, Y159, G200, T206, and Y209 on the α1 subunit and F77 and M130 on the γ2 subunit have been identified as putative parts forming the binding pocket for the ligands of the benzodiazepine binding site (Pritchett et al., 1991; Wielandet al., 1992; Buhr et al., 1996, 1997a, 1997b;Amin et al., 1997; Buhr and Sigel, 1997; Wingrove et al., 1997; Schaerer et al., 1998). They are highly homologous to amino acids F64 and I120 on the α1 subunit and Y157, T160, T202, and Y205 on the β2 subunit that take part in the formation of the binding site for the channel agonist GABA (Sigelet al., 1992; Amin and Weiss, 1993; Smith and Olsen, 1994;Westh-Hansen et al., 1997). Thus, the channel agonist and allosteric modulators of the channel seem to bind to pseudosymmetrical structures (Galzi and Changeux, 1994; Sigel and Buhr, 1997).
Many attempts (Borea et al., 1987; Villar et al., 1989; Schove et al., 1994; Zhang et al., 1995) have been made to characterize spatial properties of the benzodiazepine binding pocket. These studies used either in vivo effects or chloride flux experiments in combination with radioligand binding studies on brain membranes of a large number of structurally related compounds. Derived models for the binding pocket are complex and suggest distinct but partially overlapping binding sites for ligands differing in their allosteric effect. Considering the variety of GABAA/benzodiazepine receptors present in brain, it is not surprising that a model that satisfactorily explains all observations is still missing.
It obviously is important to map all the amino acid residues participating in the formation of the benzodiazepine pocket relative to the ligands of this site. Currently, we limit ourselves to α206, γ77, and γ130, which are available in our laboratory, show a high level of expression in HEK 293 cells, and display high affinity to a commercially available radioligand. We investigated binding affinities of a variety of ligands of this binding site in the receptor of the defined recombinant subunit composition α1β2γ2 and point mutants of the three corresponding residues. Based on our observations, we also propose a model of the interaction of positive allosteric modulators and antagonists with the receptor.
Materials and Methods
Substances.
[3H]Flumazenil (87 Ci/mmol) was from DuPont-New England Nuclear (Boston, MA). All nonradioactive ligands of the benzodiazepine binding site were obtained as a kind gift of Hoffmann-La Roche (Basel, Switzerland).
Construction of receptor subunits.
The cDNAs coding for the α1, β2, and γ2S subunits of the rat GABAAreceptor channel have been described elsewhere (Lolait et al., 1989; Malherbe et al., 1990a, 1990b). The mutant γF77Y has a phenylalanine-to-tyrosine substitution at position 77 of the mature peptide and has been described previously (Buhr et al., 1996, 1997a). The same is true for γM130L, which contains a methionine-to-leucine substitution (Buhr and Sigel, 1997). αT206V was prepared using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). In vitro synthesized sequences have been verified by DNA sequencing.
For cell transfection, the cDNAs were subcloned into the polylinker of pBC/CMV. This expression vector allows high level expression of a foreign gene under control of the cytomegalovirus promoter. With standard techniques, the α subunit was cloned into theEcoRI site of the polylinker, and the β and γ subunits were subcloned into the SmaI site.
Transfection of recombinant GABAA receptor in cultured cells.
The cells were maintained in minimal essential medium (GIBCO BRL, Gaithersburg, MD) supplemented with 10% fetal calf serum, 2 mm glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin by standard cell culture techniques. Equal amounts (total of 20 μg of DNA/90-mm dish) of GABA receptor subunits were transfected into HEK 293 cells (CRL 1573; American Type Culture Collection, Rockville, MD) according to the calcium phosphate precipitation method (Chen and Okayama, 1987). After overnight incubation, the cells were washed twice with serum-free medium and refed with complete medium.
Membrane preparation.
Approximately 60 hr after transfection, the cells were harvested by washing with ice-cold phosphate-buffered saline (130 mm NaCl, 16 mm di-sodium hydrogen phosphate, and 4 mmpotassium dihydrogen phosphate) and centrifuged at 150 ×g. Cells were washed with buffer containing 10 mm potassium phosphate, 100 mm KCl, and 0.1 mm K-EDTA, pH 7.4. Cells were homogenized by sonication in the presence of 10 μm phenylmethylsulfonyl fluoride and 1 mm EDTA. Membranes were collected through three centrifugation/resuspension cycles (100,000 × g for 20 min) and then used for ligand binding or stored at −20°.
Binding assays.
Resuspended membranes (0.5 ml) obtained from cells transfected with wild-type receptor, α1β2γ2F77Y, α1T206Vβ2γ2, or α1β2γ2M130L were incubated for 90 min on ice in the presence of [3H]flumazenil (87 Ci/mmol, DuPont-New England Nuclear) and varying concentrations of competing ligands. Membranes (20–50 μg of protein/filter) were collected by rapid filtration on GF/C filters presoaked in 0.3% polyethylenimine. After three washing steps with 4 ml of buffer, the filter-retained radioactivity was determined by liquid scintillation counting. Total binding was measured at 2 nm[3H]flumazenil, nonspecific binding under the same condition but in the presence of 10 μm unlabeled flumazenil. Displacement curves containing numerous points for wild-type and three mutant receptors clearly would have exceeded capacity. Therefore, a simplified procedure was chosen. The affinity of a substance first was approximately estimated in a binding experiment using 1 nm and 1 μm of the displacing ligand. Displacement then was determined in at least in two independent experiments at two or three concentrations of 0.1 nm, 1 nm, 100 nm, or 10 μm depending on the initial estimates. From these data, theKi value was calculated in each case according to the Cheng-Prusoff equation (1973). Two determinations for a Ki value do not allow application of statistical procedures; therefore, the error was estimated by the following procedure. The maximal deviation observed in a total of >200 individual Ki determinations from the corresponding mean value amounted to 28%. In most cases, the individual determination deviated far less. Using error propagation, it can be estimated that under the current conditions, the ratioKi (mutant)/Ki (wild-type) deviates in the worst case by 1.56-fold from the true value and that the ratio is in most cases more accurate. We therefore take any value for the ratio, either >1.6 or <0.62, as a significant change in affinity on mutation. Protein concentration was determined with the BioRad (Hercules, CA) protein assay kit with bovine serum albumin as standard.
Expression and functional characterization.
Xenopus laevis oocytes were prepared, injected, and defolliculated and currents were recorded as described previously (Sigel, 1987; Sigelet al., 1990). Briefly, oocytes were injected with 50 nl of cRNA dissolved in 5 mm K-HEPES, pH 6.8. This solution contained the transcripts coding for the different subunits at a concentration of 10 nm for α1, 10 nm for β2, and 100 nm for γ2. Transcripts were quantified on agarose gels after staining with Radiant Red RNA Stain (BioRad) by comparing staining intensities with varying amounts of molecular weight markers (RNA-Ladder, GIBCO BRL). Electrophysiological experiments were performed by the two-electrode voltage-clamp method at a holding potential of −80 mV. The medium contained 90 mm NaCl, 1 mm KCl, 1 mm MgCl2, 1 mm CaCl2, and 10 mmNa-HEPES, pH 7.4. Allosteric modulation via the benzodiazepine site was measured at a GABA concentration eliciting 3–5% of the maximal GABA current amplitude by the application of GABA alone and the coapplication of GABA with the modulatory compound, usually at a fixed concentration of 100-fold Ki . Allosteric modulation is expressed as the relative current amplitude and was calculated as the modulated current amplitude, divided by the control current amplitude, and the result was multiplied by 100%. GABA was applied for 20 sec, and a washout period of 4 min was allowed to ensure full recovery from desensitization. Positive or negative modulation of GABA currents was expressed as a percentage of the respective control current amplitudes determined in the absence of modulator. To avoid contamination, the perfusion system was cleaned between drug applications by washing with dimethylsulfoxide.
Results
Wild-type α1β2γ2 GABAA receptors and the following point mutants α1β2γ2F77Y,α1T206Vβ2γ2, and α1β2γ2M130L containing GABAA receptors were expressed separately by transient transfection in HEK 293 cells. Cell membranes were harvested and investigated for [3H]flumazenil binding. All four subunit combinations bound this ligand with an affinity in the subnanomolar range (Table 1). The ability to displace [3H]flumazenil binding was determined for 34 substances of different structures (Fig.1) at wild-type and α1β2γ2F77Y receptors, for 15 substances at α1T206Vβ2γ2 receptors, and for 18 substances at α1β2γ2M130L receptors (Table 1).
Ki values at α1β2γ2 receptors
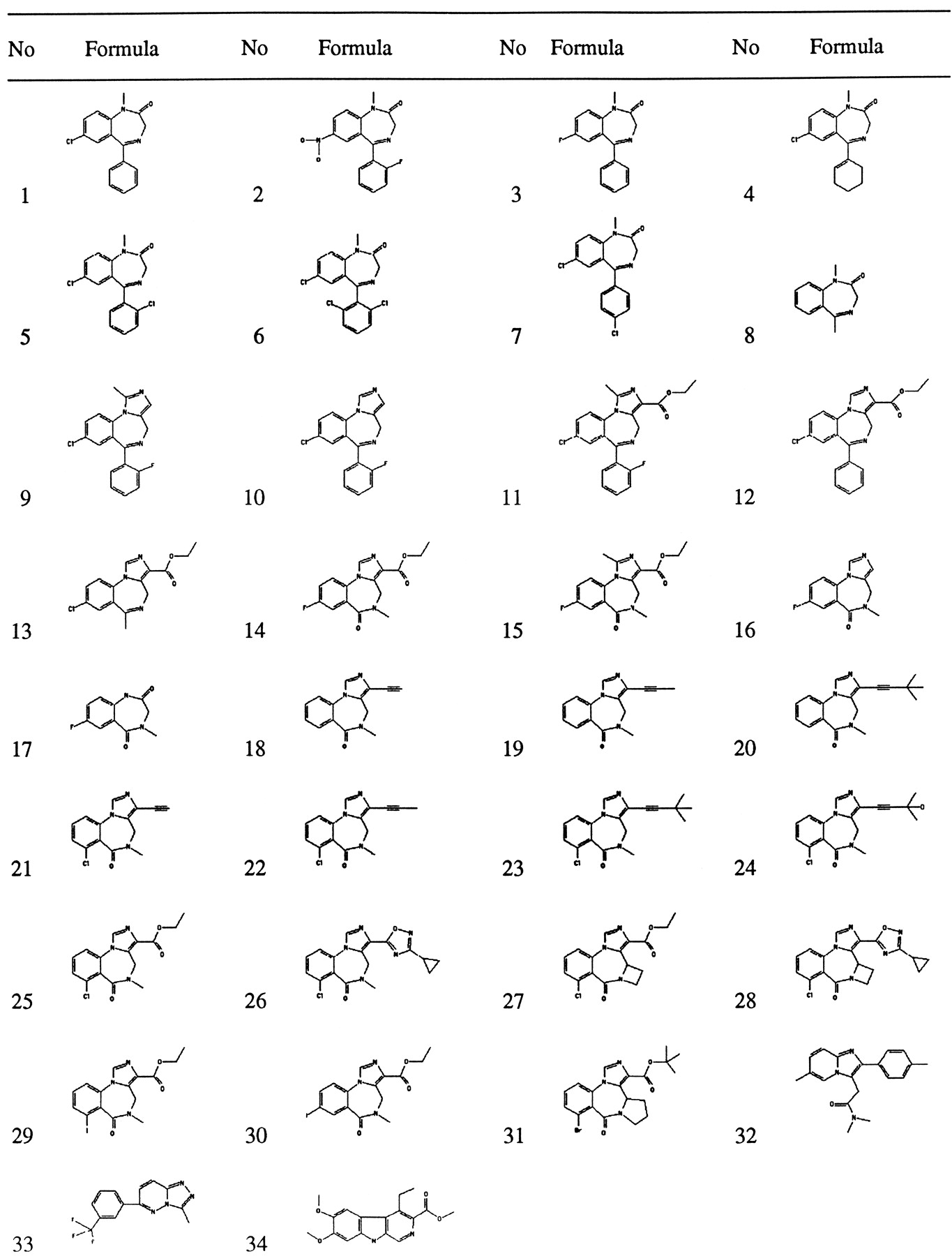
Chemical structures of the compounds used.
Affinity to the wild-type receptor.
Halides in theortho position of the phenyl substituent [compounds2 (flunitrazepam), 5, 6, and9-11; Fig. 1] increased the affinity to the receptor (Table 1). Interestingly, partial saturation of the phenyl substituent (4) of class I compounds (Fig.2) decreased binding affinity but did not abolish binding. Comparisons of compounds 9 (midazolam) with10 and 12 with 11 show that a methyl substituent at the imidazo ring obviously is tolerated in class II compounds. This tolerance is lost when the ligand has class III structure [14 (flumazenil) and 15].Para substitution of the phenyl substituent in class I compounds (7) as removal of the CO2C2H5substituent in class III compounds [14 (flumazenil),16] compromises affinity. The imidazo ring present in class II compounds had no effect on the affinity. Comparisons of 9(midazolam) with 11 and 10 with 12show that a CO2C2H5substituent at the imidazo ring only slightly decreases wild-type affinity.
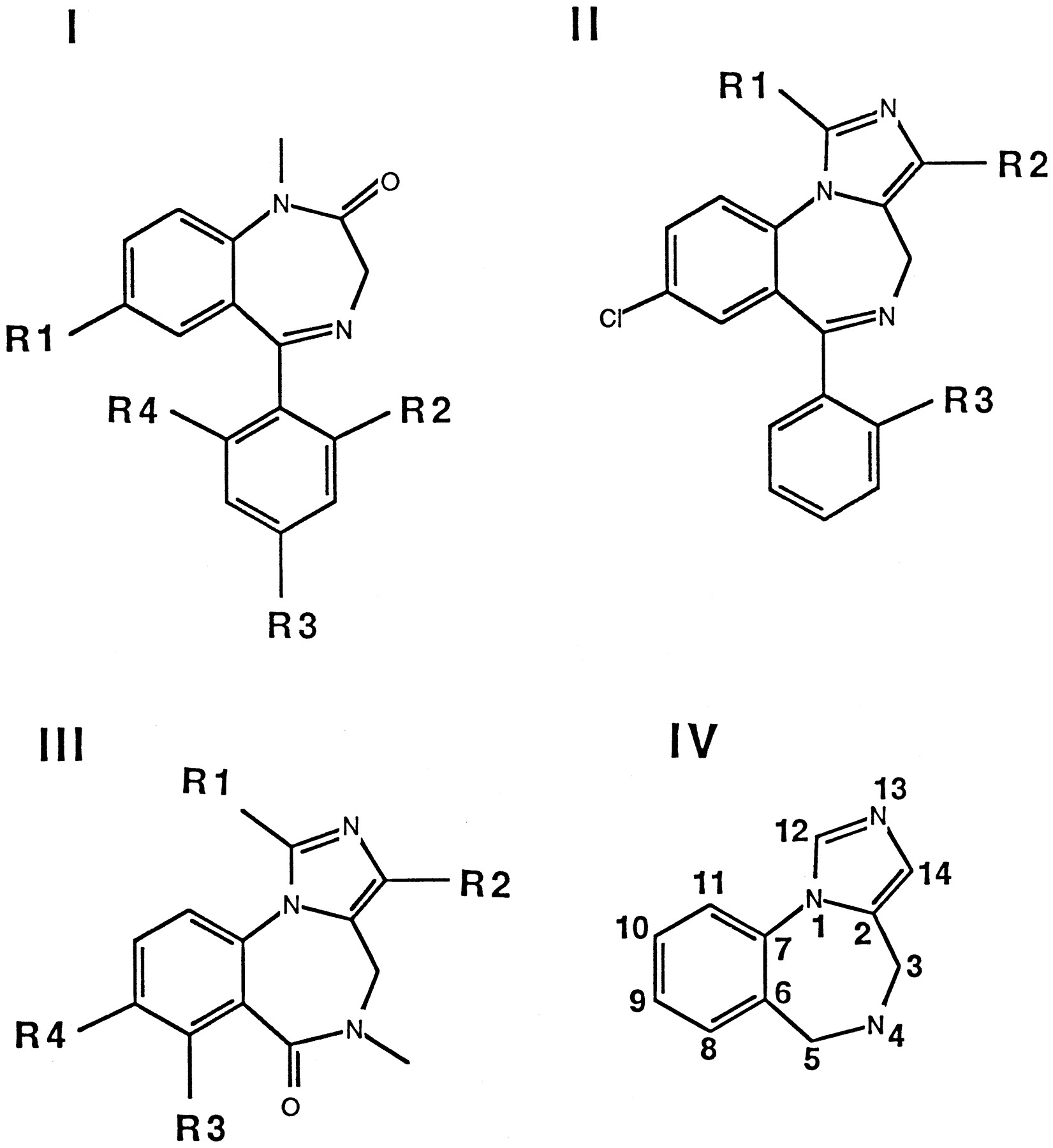
Classification of the compounds used and a model ligand for the benzodiazepine binding site (IV).
Effect of the mutations on the structure of the receptor.
When point mutations are introduced in such a complex protein, its overall structure may be affected. For three reasons, we think we can exclude this. First, functional expression of the channel in the X.laevis oocyte was unaffected by the mutations (Buhr et al., 1997a, 1997b; Buhr and Sigel, 1997). This indicates that subunit recognition, assembly, and transport of the assembled receptor to the surface membrane are unaffected. Second, the apparent affinities of the expressed channels for gating by the agonist GABA were not affected significantly in the two cases investigated: γ2F77Y (Buhret al., 1997a) and γ2M130L (Buhr and Sigel, 1997). In the case of the α1T206V mutation, no functional data are available, but the more drastic mutation α1T206A resulted in channels with only a small reduction in apparent GABA affinity (Buhr et al., 1997b). The fact that the apparent affinity for GABA is little if at all affected by the mutations is surprising in view of the allosteric coupling between this site and the binding site for benzodiazepines. Third, all of the tested mutations resulted in an increase in the binding affinity for some of the tested ligands. This increase was most pronounced for α1T206V, where a 41- and 38-fold increase was observed for compounds 34 (DMCM) and 33 (Cl 218872), respectively (Schaerer et al., 1998). An 8-fold increase was observed in the affinity of 33 (Cl 218872) for the γ2M130L mutation and a ∼5-fold increase was observed for various ligands for the γ2F77Y mutation (Table 1). This gain of function contradicts a generalized disruption. Based on the above considerations, we think it likely that changes in structure introduced by the mutations affect only the binding pocket for ligands of the benzodiazepine binding site and that an analysis based on binding affinities of multiple ligands (Table 1) to wild-type and mutant receptors is justified.
The γ2F77Y mutation strongly affected binding affinities up to 700-fold (Table 1), whereas the α1T206V and γ2M130L mutations had smaller maximal effects of ∼40- and ∼45-fold, respectively. This amounts to a maximal changes in standard free energy of binding of 3.9, 2.2, and 2.2 kcal/mol, caused by the three mutations, respectively.
Effect of the γ2F77Y mutation.
Table 1 shows theKi value for each compound tested that was estimated from the displacement experiments. Compounds1-6 and 9-12, all structures displaying a phenyl residue (Fig. 2, classes I and II), lost affinity to the benzodiazepine binding site on mutation of the wild-type γ77 phenylalanine to tyrosine. All compounds of class III structure (14-16 and 18-31; Fig. 2) (i.e., lacking the phenyl substituent characteristic for class I and II structures) displayed high affinity to the tyrosine mutant of γ77. Despite the fact that compound 12 has a phenyl substituent as diazepam, there is a smaller loss of affinity on mutation than in all other investigated ligands, but this loss is still significant at 6-fold (Table 1). This compound might retain a reasonable affinity to the mutated receptors due to its imidazol/ethyl ester moiety, even though it may have lost some affinity due to disruption of the phenyl moiety/protein interaction.
Comparisons of compounds 9 (midazolam) with 10and 12 with 11 show that a methyl substituent at the imidazo ring obviously is tolerated by both wild-type and mutant receptors in class II compounds. This tolerance is lost in both when the ligand has class III structure [14 (flumazenil) and15]. The negative allosteric modulator 34 (DMCM; Fig. 1), which has an entirely different structure, lost considerable affinity on mutation (Table 1). The positive allosteric modulators32 (zolpidem) and 33 (Cl 218872; Fig. 1), again with a different structure, showed a 3–5-fold increase in affinity on the mutation (Table 1).
Effect of the α1T206V mutation.
This mutation results remarkably in all tested cases in a significantly increased affinity for compounds of class III structure as well as for 34(DMCM; Fig. 1) and 33 (Cl 218872; Fig. 1, Table 1; Schaereret al., 1998). The compounds of class II structure show either a small decrease [9 (midazolam), 10], no change (11), or an increase in affinity. The compounds of class I structure tested here show a decrease in affinity on mutation. Compound 32 (zolpidem, Fig. 1) also loses affinity (Table2).
Classification of the investigated compounds and their allosteric modulation of recombinant α1β2γ2 receptors
Effect of the γ2M130L mutation.
This mutation had quantitatively much smaller but qualitatively similar effects as γ2F77Y. Compounds 34 (DMCM) and 33 (Cl 218872) showed a 6–8-fold increase and compound 32 (zolpidem) showed a very large 45-fold decrease in affinity.
Allosteric action of some selected compounds.
We studied allosteric effects on recombinant α1β2γ2 GABAA receptors functionally expressed inX. laevis oocytes. Some interesting compounds were selected for analysis due to the large amount of work involved.X. laevis oocytes were preferred to HEK 293 cells for this purpose for the same reasons. Different expression systems have been shown to result in the assembly of identical receptors as exemplified for poly(A)+ RNA injected inX. laevis oocytes (Kellenberger et al., 1996) compared with Semliki Forest virus-mediated expression in baby hamster kidney cells (Gorrie et al., 1997).
Allosteric effects (Table 2) were determined as described in Materials and Methods. Compounds 9 (midazolam), 10, and14 (flumazenil) were tested in the same experiments. The relative currents were 376 ± 96% (five determinations), 240 ± 66% (five determinations; different from the current stimulation by midazolam; α < 0.05, Wilcoxon-Mann-Whitney U test), and 105 ± 4% (five experiments), respectively. Compound15 has a relatively low affinity and therefore was tested at 0.1, 1, and 10 μm. In all cases, only a very small effect on the current amplitude was found, and relative currents amounted to 97 ± 3% (five determinations), 95 ± 3% (five determinations), and 95 ± 3% (five determinations), respectively. In addition, the relative current induced by subsaturating concentrations of 1 (diazepam; 50 nm), which amounted to 182 ± 43% (six determinations), was reduced to 108 ± 8% (six determinations) on coapplication of 10 μm compound 15. These properties indicate that this compound acts at the benzodiazepine binding site as an antagonist. Compounds 11 and12 increased relative currents to 324 ± 90% (three determinations) and 179 ± 43% (four determinations). These values were obtained in two different batches of oocytes, and we consider the relative current amplitudes as not necessarily different. Thus, despite the ethyl ester substituent at the imidazo group, these compounds allosterically stimulated the GABA response strongly, whereas compound 13, lacking the phenyl substituent, acted nearly as an antagonist with a relative current amplitude of 112 ± 6% (three determinations).
Structurally similar compounds have divergent allosteric effects.
In discussions, we were alerted to the fact that structurally very similar compounds acting at the binding site for benzodiazepines can have highly variable biological actions when tested in whole animals in vivo (Hunkeler, 1993; data on file at Hoffmann-La Roche, Basel). The three compounds21-23, which share a very similar structure, all bind with high affinity (0.11–1.5 nm) to the benzodiazepine binding site. The action of these on the currents induced by GABA was characterized by electrophysiological techniques at a membrane potential of −80 mV. Concentration-response curves (Fig.3) in the range of 1 nm to 1 μm showed that compound 23 acted as a positive allosteric modulator, compound 22 acted as a very weak positive allosteric modulator, and compound 21 acted as an antagonist. The weak, but significant, effects place compound22 very close to antagonists. Maximal stimulation by compound 23 reached similar values as those by diazepam in the same experiment (not shown). The current stimulation by compound23 was prevented by the simultaneous application of either compound 14 (flumazenil) or compound 21 (Fig.4). Compounds 21 and22 (not shown) both counteracted the enhancement of the currents by diazepam. Results obtained for compound 21 are shown in Fig. 5. Three ligands with a very similar structure thus have differing allosteric actions. Because they all compete with flumazenil binding, their site of action overlaps with the binding site for compound 14 (flumazenil). Based on the structural similarity of these three drug molecules with differing allosteric properties and on their similarity with compound14 (flumazenil), it is tempting to perfectly superimpose these four molecules onto each other.

Concentration-response curves for compounds21-23. X. laevisoocytes expressing α1β2γ2 were voltage-clamped at −80 mV. Control currents elicited by 5 μm GABA were stimulated by the simultaneous application of various concentrations of compounds23 (•), 22 (○), and 21(▪). Compound 23 strongly stimulates control currents elicited by GABA, whereas 22 weakly stimulates and21 only very weakly inhibits them. Mean and standard deviation values of five concentration-response curves carried out on oocytes from two different batches are shown for each substance.

Compound 23 acts as a positive allosteric modulator at the benzodiazepine site. X.laevis oocytes expressing α1β2γ2 were voltage-clamped at −80 mV. Control currents elicited by 5 μm GABA were stimulated by the coapplication of 100 nm compound 23. In independent experiments, 100 nm compound 23 was coapplied with 1 μm 14 (flumazenil) or with 21, respectively. Values are mean and standard deviation of five experiments carried out in two different batches of oocytes. If the coapplication of flumazenil followed the application of 100 nm compound 23 in the same oocyte, inhibition of the stimulation amounted to only ∼50%, possibly due to a slow dissociation of 23 from the receptor.

Compound 21 prevents the current stimulation by diazepam. X. laevisoocytes expressing α1β2γ2 were voltage-clamped at −80 mV. Control currents elicited by 5 μm GABA (trace 1) were stimulated by the simultaneous application of 0.3 μm 1 (diazepam; trace 2). This stimulation was counteracted in the presence of 1 μmcompound 21 (trace 3). Four additional experiments carried out in oocytes from two different batches of oocytes gave very similar results.
Discussion
The affinities of 34 ligands or potential ligands of the benzodiazepine binding site for the recombinant wild-type α1β2γ2 GABAA receptor were determined and compared with that for the α1β2γ2 GABAA receptor carrying the point mutation in the γ subunit F77Y. Thus, the consequences of a minimal chemical change, namely, the addition of a hydroxyl group to an aromatic ring, were studied. Furthermore, the affinities of 15–18 ligands were determined for receptors carrying the point mutations α1T206V and γ2M130L. Thus, after previous work on the variation of the amino acid in these positions, we systematically varied the nature of the ligand. The facts that all three mutations lead to an increase in affinity, at least for some ligands, and that the apparent affinities for the channel agonist GABA for gating of the channel remain unaltered may be taken as indications that the overall structure of the receptor remains unperturbed and that the mutations only lead to local changes in structure.
During our work, we made some observations on wild-type receptors pertinent to the shape of the binding pocket for ligands of the benzodiazepine binding site. These observations are discussed before the mutation work. We assume but do not prove here that alteration in binding affinity reflects an alteration of ligand/receptor interaction that is due to a direct change of the binding pocket and not to allosteric transmission. We discuss our data in the framework of Fig.2, IV, to facilitate naming of areas around the ligand. High affinity may be mediated by the phenyl substituent in position 5 or by a substituent in position 14 of at least three atoms in length. One of the two is disposable, but both may be present in the same molecule.
Three structurally very similar compounds have a different allosteric action.
The actions of the three structurally similar compounds on a recombinant GABA receptor were tested. From the experiments, it was concluded that compound 23 acts as a strong positive allosteric modulator, compound 22 acts as a very weak positive allosteric modulator, and compound 21acts as an antagonist at the benzodiazepine site. The structures of these three compounds are intriguingly similar: only the substituent distal to the triple bond attached to the imidazo ring varies from a hydrogen atom for the antagonist to a methyl group for the weak positive allosteric modulator and to a tertiary butyl group for the positive allosteric modulator. Thus, it seems that in these compounds, a bulky substituent favors positive allosteric modulation.
The binding pocket for ligands of the benzodiazepine binding site.
So far, overlapping, but spatially different, arrangements of positive allosteric modulators and antagonists of this binding site have been envisaged by most authors. However, in the light of these results (Figs. 3-5), it is tempting to suggest a simple superposition of these ligands. Classic benzodiazepines and midazolam may be quite precisely superimposed due to their structural similarity. The same argument holds true for the superposition of compound 9(midazolam) with compound 14 (flumazenil). As a consequence of this superposition of ligands, their interaction with the receptor protein must be similar, at least in the structurally identical parts.
The receptor must exist in three conformations: (A), (I), and (R). (A) is the conformation displaying a preferential affinity for positive allosteric modulators. It is stabilized by this interaction and has an increased affinity for the channel agonist GABA. (I) is the conformation displaying a higher affinity for negative allosteric modulators. It is stabilized by the interaction with these compounds and has an decreased affinity for the channel agonist GABA. (R) is the conformation displaying a preferential affinity for antagonists. It is stabilized by this type of interaction. The affinity of (R) for GABA is the same as for unliganded receptor. The shape of the pocket for ligands of the benzodiazepine site also must be affected by the different conformations of the protein.
Differential tolerance for an additional methyl group.
We have evidence for the fact that the environment of the binding pockets belonging to different allosteric states of the receptor is not identical, at least for positive allosteric modulators and antagonists. This is illustrated by the fact that the tolerance to an additional methyl group at the imidazo ring is very different for positive allosteric modulators and antagonists. A methyl group may be attached to compound 10 to give compound 9 (midazolam) with a 3-fold increase in the binding affinity, whereas there is a 500-fold loss in this parameter on the addition of a methyl group in the same position to convert compound 14 (flumazenil) into compound 15 (Table 1). Both compounds 10 and9 (midazolam) are positive allosteric modulators of recombinant α1β2γ2 GABAA receptors, but compounds 14 (flumazenil) and 15 are antagonists. We interpret these findings with different conformations of the receptor either accepting or excluding an additional methyl group. Steric hindrance may be the cause of the exclusion [i.e., in the resting receptor conformation (R) stabilized by antagonists, the corresponding space may be occupied by the protein], whereas in the receptor conformation stabilized by positive allosteric modulators (A), the corresponding space is available.
Localization of γ2F77 relative to ligands.
By systematic variation of the ligand structure, we derived the following conclusions. For compounds carrying a phenyl substituent in position 5 (Fig. 2, IV), the ratioKi (mutant receptor)/Ki (wild-type receptor) is large (i.e., they lose considerable affinity to the receptor carrying the point mutation γ2F77Y, whereas compounds not carrying this substituent do not) (Table 1). Obviously, the extra hydroxyl group in tyrosine interferes with this phenyl substituent; therefore, it is concluded that γF77 is close to the phenyl substituent in class I and II structures.
As predicted from the binding studies, functional data on this mutation show little stimulation of the mutated receptor by 0.3 μmdiazepam, whereas a massive stimulation is observed with 1 μm compound 32 (zolpidem) (Buhr et al., 1997a). Mutations of phenylalanine in γ77 to leucine or isoleucine have been described (Buhr et al., 1997a). These represent drastic changes, replacing a planar aromatic substituent of the side chain by small, nonplanar ones, in both cases. Although there is a 1.5–7.7-fold decrease in compound 1 (diazepam) and compound 2 (flunitrazepam) affinities, compound14 (flumazenil) affinity is reduced ∼28- and ∼2020-fold by these two mutations (Buhr et al., 1997a). This may indicate that there is a favorable interaction of the aromatic ring of wild-type γF77 with ligands of class III structure. If our proposed superposition of compound 9 (midazolam) with compound14 (flumazenil) holds true, such an interaction would (a) require positioning of γF77 close to the proximal part of the phenyl moiety of class I/II compounds and (b) that the favorable interaction is not disturbed by the additional hydroxyl group in α1β2γF77Y receptors. However, both mutations, to leucine and to isoleucine, also may affect the structure of the binding pocket.
Localization of αT206 relative to ligands.
Compounds12, 13, 14 (flumazenil),21-23, 26, 33 (Cl 218872), and 34 (DMCM) display an increased affinity, whereas compounds 1 (diazepam), 2 (flunitrazepam), and32 (zolpidem) display a decreased affinity to the mutated receptor. This mutated receptor contains a valine instead of a threonine in position 206 of the α subunit. In valine, a methyl group occupies the place of the hydroxyl group in threonine. Thus, a bulkier, unpolar group is introduced in place of the wild-type hydrophilic group. The fact that this leads in some cases to an increase in affinity [class II and III structures 33 (Cl 218872),34 (DMCM)] indicates that the hydroxyl group present in the wild-type receptor might unfavorably interact with an electronegative center present in these compounds. A decrease in the binding affinity on mutation specifically is observed for compounds displaying anN-methyl amide group, as in diazepam. Either there is steric interaction between these groups and the new methyl group, or the removal of the hydroxyl group removes a favorable interaction, for example, in the form of a hydrogen bond. We hypothesize that affinity changes could correlate with the electronic charge of the atom in place of the oxygen atom of the amide group in diazepam. If this atom is rather negatively charged [1 (diazepam), 2(flunitrazepam)], there is a decrease in affinity on mutation, and if it is rather positively charged due to an electron pulling of the corresponding substituent, affinity is increased (13,14, 21-23, 26,34). In 11, electron donor activity of the methyl substituent of the imidazo ring balances this electron-withdrawing effect. However, it should be mentioned that compound 12does not easily fit in this hypothetical arrangement. This failure may be due to the relatively low affinity of this ligand to wild-type receptors.
Localization of γM130 relative to ligands.
The fact that only compounds 32 (zolpidem) 34 (DMCM), and33 (Cl 218872) displayed large changes in affinities indicates that this considerable alteration in the chemical nature of the side chain from methionine to leucine probably specifically affects a space occupied by these and not the other ligands tested.
A view of the benzodiazepine binding pocket.
H101 of the α1 subunit was the first amino acid residue implied to be involved in benzodiazepine binding (Wieland et al., 1992; Duncalfeet al., 1996). This residue recently has been characterized further by mutation to several other residues combined with a characterization of the resulting altered binding properties (Davieset al., 1998). The authors suggest that H101 may interact with aromatic portions of benzodiazepine binding site ligands in an effect called π-π-type stacking, but they refrain from identifying the aromatic portion. They go on to speculate that H101 may be involved in the recognition of positive allosteric modulators and antagonists but not in the recognition of negative allosteric modulators. Very recently, evidence has been presented that the corresponding histidine residue interacts with the phenyl substituent of the compounds designated here as class I and II compounds (McKernan et al., 1998). Based on our results, the GABAAreceptor complexes ligands of the benzodiazepine binding site such that the same phenyl substituent is located in the region that contains residue 77 of γ2 subunits. Wingrove et al. (1997)speculated in a recent report that the γ2M130 may interact in some way with the same moiety. These suggestions are not necessarily mutually exclusive. The phenyl moiety of diazepam is large, and several amino acid residues may simultaneously be located around this region of the molecule. Several additional amino acid residues are involved in the binding of ligands of the benzodiazepine binding site (for a review, see Sigel and Buhr, 1997). Mapping of all amino acid residues will represent the beginning of a three-dimensional understanding of the benzodiazepine binding pocket.
Several studies (Villar et al., 1989; Schove et al., 1994; Zhang et al., 1995) using binding studies in whole forebrain membranes in conjunction with molecular models of the binding pockets for positive allosteric modulators, antagonists, and negative allosteric modulators have suggested the existence of common ligand contact points for these molecules. At present, it seems too early to speculate which amino acid residues correspond to which contact points. Such a correlation may at be misleading due to the presence of various isoforms of the GABAAreceptor in forebrain membranes. Clearly, there is a need to combine the advantages of the two approaches, namely, the use of recombinant receptors combined with the analysis of point mutations on one side and the evaluation of the emerging data using molecular modeling on the other side. It is to be hoped that this combination will be used in future and will lead to a more precise picture of the binding pocket. In view of the present data, results should be analyzed by assuming a simple overlap of at least positive allosteric modulators and antagonists but maybe also of negative allosteric modulators, in combination with subtly different conformations of the binding pocket.
Acknowledgments
We are greatly indebted to Dr. W. Hunkeler (Hoffmann-La Roche, Basel, Switzerland) for helpful discussions and for the generous gift of many substances used in this study. We are grateful to Prof. H. Reuter, in whose institute this work was carried out, for continuous encouragement.
Footnotes
- Received May 4, 1998.
- Accepted September 3, 1998.
-
Send reprint requests to: Dr. Erwin Sigel, Department of Pharmacology, University of Bern, Friedbühlstr. 49, CH-3010 Bern, Switzerland. E-mail: sigel{at}pki.unibe.ch
-
This study was supported by grants 31–37192.93 from the Swiss National Science Foundation and EU Grant BIO4-CT96–0585 (BBW 96.0010).
Abbreviations
- GABA
- γ-aminobutyric acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- DMCM
- methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- HEK
- human embryonic kidney
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}