


Abstract
We investigated the effect of adenovirally mediated overexpression of adenylyl cyclase type 6 (AC6), a major form of AC expressed in mammalian heart, on G protein-coupled receptor regulation of cAMP production in neonatal rat ventricular myocytes. Following gene transfer of AC6, isoproterenol- and forskolin-stimulated increases in cAMP were markedly enhanced, whereas basal levels of cAMP and responses to several other agonists that stimulate cAMP formation, e.g., prostaglandin E2 (PGE2), H2agonist, glucagon, and A2 agonist were not increased. Studies to test whether the selective enhancement in β-adrenergic receptor (AR) response might result from inhibition of AC6 by Gαi and Gβγ indicated that pertussis toxin-sensitive inhibition by the muscarinic cholinergic agonist carbachol was unaltered in myocytes overexpressing AC6. Pertussis toxin treatment failed to reveal an enhancement by AC6 overexpression of basal or PGE2-stimulated cAMP. Immunoblot analysis of membrane fractions indicated that β1-AR and AC6 are expressed in fractions enriched in caveolin-3 and morphologic caveolae. The data suggest that loss of Gi-mediated inhibition is not the mechanism for enhancement of β-AR-stimulated cAMP formation and that key components of β-AR-mediated activation of AC exist in caveolae of cardiac myocytes, providing a means by which β-AR response is selectively enhanced by increasing AC6 expression.
Sympathetic nervous system and hormonal activation of β-ARs is the principal physiologic mechanism for production of the second messenger cAMP in the mammalian heart and, in turn, for the regulation of cardiac rate and force of contraction. Heart failure has been associated with decreased receptor number and desensitized β-AR responses as well as increased expression of both Gi and G protein receptor kinase (as recently reviewed by Post et al., 1999). Accordingly, there has been considerable interest in augmenting β-AR signaling in cardiac tissue.
Analysis of the stoichiometric relationship between the components of the β-AR signaling pathway in cardiac myocytes indicate that receptor, G protein and adenylyl cyclase (AC) are expressed in an approximate molar ratio of 1:200:3 (Post et al., 1995). Expression of AC appears to set the limit on the maximal efficacy of β-AR stimulation. Overexpression of either β-AR subtypes or Gαs in vitro or in transgenic mouse models has led to only modest increases in maximal efficacy (Milano et al., 1994;Gaudin et al., 1995; Drazner et al., 1997; Engelhardt et al., 1999), whereas overexpression of AC6 leads to proportional increases in maximal cAMP response (Gao et al., 1998). It has been further demonstrated that increasing expression of AC6 in the heart improves cardiac performance in normal animals and in animals with a transgenic model of heart failure (Gao et al., 1999; Roth et al., 1999).
Nine mammalian isoforms of AC have been identified, each with distinct regulatory patterns. AC types 5 and 6 are the predominant isoforms expressed in cardiac tissue and share the property of being inhibited by multiple signaling pathways inside the cell, including Gαi and Gβγ, protein kinase A, protein kinase C, and Ca2+ (Sunahara et al., 1996; Hanoune et al., 1997; Bayewitch et al., 1998). β-Adrenergic receptors are the primary positive regulator of AC activity in cardiac cells, acting through the stimulatory G protein, Gs, whereas muscarinic cholinergic receptors are the primary negative regulator acting via Gi.
The goal of this study was to determine whether the regulation of AC activity by Gs- and Gi-coupled receptors present in normal cardiac myocytes is altered by overexpressing AC6. In myocytes overexpressing AC6, we demonstrate a selective enhancement in β-AR-stimulated cAMP formation with a retention of muscarinic cholinergic-mediated inhibition of cAMP production. Moreover, we find that both β-ARs and AC6 are not evenly distributed in the plasma membrane, but instead are specifically expressed in caveolar microdomains in the sarcolemma. Therefore, the colocalization of β-ARs and AC in microdomains of the plasma membrane provides a means for rapid and specific signal transduction in cardiac myocytes and, by inference, other cells as well.
Experimental Procedures
Materials.
AC6 adenovirus was a gift from H. Kirk Hammond (Veterans Affairs Hospital, San Diego). Hemagglutinin-β1-green fluorescent protein (HA-β1-GFP) adenovirus was a gift from Brian Kobilka (Stanford University). Primary antibody for AC5/6 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Primary antibody for HA was obtained from Roche Molecular Biochemicals (Indianapolis, IN). Primary antibody for caveolin-3 was obtained from Transduction Laboratories (Lexington, KY). Radiolabeled chemicals were obtained from NEN Life Sciences (Boston, MA). All other chemicals and reagents were obtained from Sigma Chemical (St. Louis, MO).
Measurement of cAMP Accumulation.
Neonatal cardiac myocytes were prepared and maintained as described previously (Gao et al., 1998). Cells were infected two days after plating with an adenoviral construct containing AC6 for 20 h (2–5 m.o.i./cell). Control cells were treated with the identical adenoviral construct containing the lacZ gene. After infection, cells were washed extensively and allowed to equilibrate for 24 h. Cells were then labeled with [3H]adenine (1.5 μCi/well) for 90 min and washed three times with PBS followed with serum-free and NaHCO3-free Dulbecco's modified Eagle's medium supplemented with 20 mM HEPES, pH 7.4 (DMEH). Cells were equilibrated with DMEH for 30 min then assayed for cAMP accumulation by incubation with drugs of interest and 0.2 mM isobutylmethylxanthine, a cyclic nucleotide phosphodiesterase inhibitor, for 5 min. In experiments where responses to adenosine receptor agonists were measured, an alternative phosphodiesterase inhibitor, Ro20–1724, was used instead of isobutylmethylxanthine. To terminate reactions, assay medium was aspirated and 250 μl of ice-cold trichloroacetic acid (7.5% w/v) was immediately added to each well. Approximately 1000 cpm of [32P]cAMP internal standard was added to each well, and the final volume brought to 1 ml with water. cAMP was then separated from incorporated adenine nucleotides using the double column method described by Salomon et al. (1974) and counted by liquid scintillation. [3H]cAMP was corrected for recovery of internal standard and expressed as a percentage of total incorporated 3H-nucleotides. Data are expressed as fold stimulation over basal.
In other experiments, cAMP content in trichloroacetic acid extracts obtained as described above (omitting incubation with [3H]adenine) was determined by radioimmunoassay as described previously (Gao et al., 1998). Briefly, samples were acetylated according to the manufacturer's protocol (Calbiochem), and 50-μl aliquots were incubated with approximately 12,000 cpm of125I-cAMP and 50 μl of rabbit anti-cAMP antibody (diluted 1:3,000) overnight at 4°C. Fifty microliters of goat anti-rabbit antibody with magnetic bead was added and incubated with mixture for 2 h with constant shaking at 4°C. Separation of bound from free was achieved by filtration, and bound radioactivity was counted and compared with a standard curve. Production of cAMP was normalized to the amount of protein per sample as determined using a dye-binding protein assay (Bio-Rad, Hercules, CA).
Membrane Fractionation.
Neonatal rat cardiac myocytes were fractionated using a detergent-free method adapted from Song et al. (1996). Approximately 30 million cells were washed twice in ice-cold PBS and scraped off the plates in a total of 2 ml of 500 mM sodium carbonate, pH 11. Cells were homogenized with a tissue grinder with three 10-sec bursts and then a sonicator with three 20-sec bursts. The homogenate was brought to 45% sucrose by addition of an equal volume of 90% sucrose in 25 mM 2-[N-morpholino]ethanesulfonic acid (MES), 150 mM NaCl, pH 6.5 (MBS) and loaded in an ultracentrifuge tube. A discontinuous sucrose gradient was layered on top of the sample by placing 4 ml of 35% sucrose prepared in MBS with 250 mM sodium carbonate then 4 ml of 5% sucrose (also in MBS/Na2CO3). The gradient was centrifuged at 39,000 rpm on a SW41Ti rotor (Beckman Instruments) for 18 to 20 h at 4°C. One-milliliter fractions were then collected from the top of the gradient to yield a total of twelve fractions. Adaptin-β, a clathrin-coated pit marker and mannosidase II, a Golgi marker, exclusively localized to the bottom, high sucrose fractions numbered 8 to 12 as determined by immunoblotting (data not shown), consistent with those observed by others who have prepared caveolin-rich fractions from cardiac myocytes (Rybin et al., 1999;Schwencke et al., 1999a).
Immunoblot Analysis.
Individual fractions and whole-cell lysates were concentrated to half-volume and separated by SDS-polyacrylamide gel electrophoresis. Proteins were transferred to a polyvinylidene difluoride membrane by electroblotting. Membranes were blocked in 20 mM PBS with 3% nonfat dry milk and incubated with primary antibody (see Materials) overnight at 4°C. Bound primary antibodies were visualized using the appropriate secondary antibody with conjugated horseradish peroxidase (Santa Cruz Biotechnology) and ECL reagent (Amersham). The amount of protein per fraction was determined using a dye-binding protein assay (Bio-Rad).
Transmission Electron Microscopy.
The caveolin-enriched fraction from neonatal rat cardiac myocytes was collected by taking the light-scattering band (2 ml) from the 35 to 5% sucrose interface of the above fractionation procedure. This fraction was diluted into 20 ml of MBS and spun at 51,000 rpm in a Ti60 rotor (Beckman Instruments) for 1 h. Pellets were fixed in 4% paraformaldehyde, 0.1% gluteraldehyde in 20 mM sodium cacodylate buffer with 50 μM CaCl2 (pH 7.2) and embedded in 1.5% agarose for antibody incubations. Agarose blocks were blocked in PBS with 1% bovine serum albumin and 3% normal goat serum, then incubated with primary antibodies overnight at 4°C. Blocks were washed 5 times with 20 mM PBS with 0.5 mM NaCl and incubated with secondary gold-conjugated antibodies for 2 h at 25°C. Blocks were washed again, then postfixed with 1% gluteraldehyde, followed by OsO4 before being stained in 1% aqueous uranyl acetate. Samples were dehydrated in a series of ascending ethanol concentrations and embedded in Durcupan ACM resin for thin sectioning.
Results and Discussion
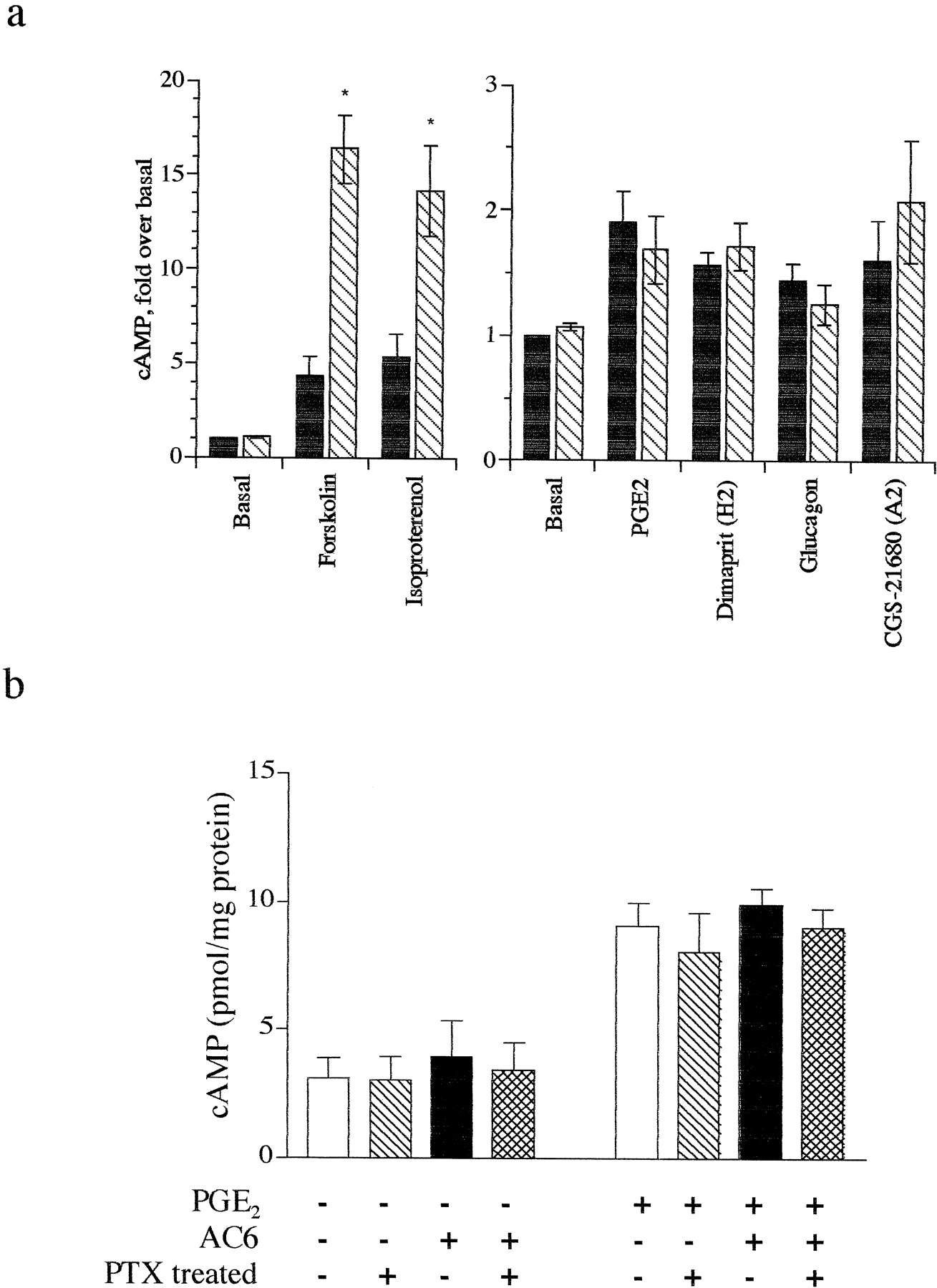
We measured production of cAMP in lacZ (control) and AC6-overexpressing neonatal rat ventricular myocytes, generated by use of an adenoviral construct. Previous data document that this method of gene transfer yields transfer to a high proportion (≥95%) of myocytes and that the adenovirus containing the AC6 cDNA does not alter expression of Gαs, Gαior β-ARs (Gao et al., 1998). Although basal cAMP accumulation did not differ between control cells and cells overexpressing AC6, AC6 overexpression enhanced responses to forskolin (10 μM) and isoproterenol (1 μM) 3- to 4-fold compared with control cells (Fig.1a). To our surprise, responses to other agonists that increase cAMP in cardiac myocytes, including prostaglandin E2 (PGE2) (10 μM), CGS-21680 (10 μM, adenosine A2 agonist), glucagon (10 μM), and dimaprit (10 μM, histamine H2 agonist) were unchanged in cells overexpressing AC6. Inclusion of a low concentration of forskolin (0.1 μM) to “sensitize” cAMP responses of these “weak” agonists failed to uncover an enhancement in response to these agonists in cells overexpressing AC6 (data not shown). cAMP levels in the extracellular medium were proportional to the levels measured intracellularly, and thus do not account for the lack of enhancement of basal or weak agonist responses (data not shown). Therefore, AC6 overexpression in cardiac myocytes selectively enhances cAMP generation by β-ARs but not by other Gαs-coupled receptors.

Overexpression of AC6 selectively enhances β-AR-mediated cAMP response. a, cAMP accumulation was measured inlacZ control (closed bars) or AC6-overexpressing (hatched bars) neonatal rat cardiac myocytes using an [3H]adenine prelabeling method (see Experimental Procedures). Responses shown are to 10 μM forskolin, 1 μM isoproterenol, 10 μM PGE2, 10 μM dimaprit (histamine H2-selective agonist), 10 μM glucagon, and 10 μM CGS-21680 (adenosine A2-selective agonist). Data is expressed as fold over basal percentage conversion, mean ± S.E. of five experiments. *P < .05 by pairedt test as compared with lacZ controls. b, basal and PGE2-stimulated cAMP production in bothlacZ control and AC6-overexpressing myocytes treated with either buffer or 100 ng/ml pertussis toxin for 18 h. cAMP levels were detected by radioimmunoassay (see Experimental Procedures) and data are expressed as mean ± S.E. picomoles of cAMP/mg of total protein of four experiments.
AC6 is one of the isoforms of AC that is subject to inhibition of enzymatic activity by Gi, perhaps by both Gαi and Gβγ subunits (Sunahara et al., 1996; Bayewitch et al., 1998). AC activity may be tonically inhibited by Gαi in cells such that only strong stimulatory signals can overcome this inhibition and recruit enzyme activity. To test whether such tonic inhibition might prevent the ability of a weak agonist to stimulate cAMP formation, we treated cells with pertussis toxin (100 ng/ml, 18 h, an effective protocol to eliminate Gi tone as shown in Fig.2b) following gene transfer of AC6. We found that neither basal nor PGE2-stimulated cAMP accumulation was increased by overexpression of AC6 (Fig. 1b). These data imply that tonic negative regulation of overexpressed AC6 via Gi is not responsible for the inability of weakly coupled receptors to activate AC6.

Inhibition of cAMP accumulation by Gi-coupled muscarinic cholinergic receptors is not dampened by overexpression of AC6. a, cAMP responses in lacZcontrol (open, hatched) or AC6-overexpressing (closed, cross-hatched) neonatal rat cardiac myocytes in the absence (open, filled) or presence (hatched, cross-hatched) of 1 μM carbachol. Responses were measured under basal, 1 μM isoproterenol- or 10 μM forskolin-stimulated conditions. b, cAMP responses in lacZ control or AC6-overexpressing neonatal rat cardiac myocytes stimulated with 1 μM isoproterenol in the absence (open, closed) or presence (hatched, cross-hatched) of 1 μM carbachol. Cells were treated with either buffer (open, hatched) or 100 ng/ml pertussis toxin for 18 h (closed, cross-hatched). Data are expressed as mean ± S.E. picomoles of cAMP/mg of total protein of four experiments. *P < .05 by paired t test as compared with absence of carbachol.
The cholinergic agonist carbachol (1 μM) was added along with stimulatory agents to determine the ability of muscarinic cholinergic receptors to inhibit stimulated AC activity following overexpression of AC6. Carbachol inhibited isoproterenol-stimulated cAMP levels 52 ± 10.3% in control cells and 66 ± 8.3% in cells overexpressing AC6 and forskolin-stimulated cAMP levels by 68 ± 7.1% in control cells and 74 ± 7.3% in cells overexpressing AC6 (Fig. 2a). Carbachol-mediated inhibition was eliminated by pertussis toxin treatment in both control and AC6 overexpressing cells (Fig. 2b). These data indicate that overexpressed AC6 can be inhibited by an endogenous regulator, muscarinic acetylcholine receptors, acting through Gi/o and that the observed enhancement of responses by β-ARs is not due to a loss in Gitone.
One explanation for the ability of AC6 overexpression to enhance β-AR-mediated cAMP accumulation would be the colocalization of AC6 and receptors. We tested the hypothesis that caveolae may be one site for such colocalization. Using a detergent-free method, we detected endogenously expressed AC5/6 protein (as a 139-kDa band) in buoyant membrane fractions (numbered 4 and 5) that are enriched in caveolin-3 (23-kDa band, Fig. 3a). Overexpressed AC6 localized in identical fractions of cardiac myocytes (AdV-AC6, Fig. 3a). These data indicate that AC is enriched in caveolin-containing fractions of cardiac myocytes and that AC6 expressed using adenoviral-mediated gene transfer localizes similarly to natively expressed AC. We also overexpressed a HA-β1-AR-GFP chimeric protein in myocytes using adenovirally mediated gene transfer. Immunodetection of the HA epitope indicated that the β1-AR is also exclusively expressed in caveolin-containing fractions (72-kDa band, Fig. 3a). Total protein concentrations of the individual fractions indicate that the majority of cellular protein also remains in these bottom fractions (Fig. 3b). Fractions 4 and 5 contain primarily membranes displaying distinct caveolar morphology, i.e., 50- to 100-nm vesicular or curved membranes, as determined by electron microscopy (Fig. 3c) (de Weerd and Leeb-Lundberg, 1997; Rybin et al., 1999). Other data (not shown) confirm published findings (Huang et al., 1997) regarding localization of a portion of Gαs and Gαi proteins to cardiac caveolar fractions. Further data by others have shown localization of muscarinic cholinergic receptors to caveolar fractions in cardiac cells (Feron et al., 1997). Taken together, these results indicate that both β-AR and AC6 colocalize and may exist, perhaps together with muscarinic receptor, Gαs and Gαiin prearranged signaling complexes in caveolar microdomains of the cardiac sarcolemma.

β-AR and adenylyl cyclase colocalize to caveolar microdomains in neonatal rat cardiac myocytes. a, immunoblot analysis of fractions (1–12) and whole-cell lysate (wcl) from sucrose gradient centrifugation resolved by SDS-polyacrylamide gel electrophoresis (seeExperimental Procedures) indicates that fractions 4 and 5 are enriched in caveolin-3. Detection of an expressed HA-β1-AR-GFP chimera with an HA antibody indicates that this protein is specifically expressed in caveolin-enriched membrane fractions. Both native AC types 5/6 and overexpressed AC6 (AdV-AC6) were localized to these same fractions as detected with an antibody recognizing both AC5 and AC6. b, total protein content of individual fractions and wcl as determined by dye-binding assay (seeExperimental Procedures). Note that fractions were concentrated for SDS-polyacrylamide gel electrophoresis before determining protein content, whereas wcl was not. c, electron micrograph of the caveolin-enriched fraction from neonatal rat cardiac myocytes shows curved and vesicular membrane structures resembling caveolae. Magnification of 20,000×, bar represents 0.1 μM.
Why is it that responses to agonists more weakly coupled to the activation of AC are not enhanced in cardiac myocytes that overexpress AC6? Several mechanisms could account for this unexpected observation. First, weakly coupled receptors may not have access to the newly expressed AC enzyme. This could result from either a preference of certain receptor types for coupling to particular isoforms of AC (Pucéat et al., 1998) or from selective compartmentation of receptors with specific populations of the effector enzyme. Second, weakly coupled receptors may not elicit enough of a stimulatory signal to overcome the negative regulation that can occur for AC6 (even at high levels of expression of AC). This latter mechanism may be unique to this isoform of AC because it is negatively regulated by protein kinase A, protein kinase C, Gβγ, Ca2+, and Gi (Sunahara et al., 1996; Chen et al., 1997;Hanoune et al., 1997; Lai et al., 1997; Bayewitch et al., 1998). Our data indicate that Gi appears not to be the factor responsible. Last, the observed selective effect may be due to low expression of receptors for weak agonists such that AC expression is not limiting maximal cAMP production. Further studies will be required to test these other alternatives. Nevertheless, the evidence that β-ARs colocalize with AC5/6 in native cardiac myocytes and in cells that overexpress AC6 implies that AC signaling is highly compartmentalized, possibly without all receptor types having access to this compartment.
The idea that the components of G protein-coupled receptor-Gs-AC signaling pathway are randomly distributed in the plasma membrane and interact via a stochastic “collision coupling” appears increasingly unlikely (Neubig, 1994;Chidiac, 1998). Evidence of compartmentation of signaling has been noted for many years, but the cellular and molecular basis for such compartmentation has been elusive (Buxton and Brunton, 1983; Harper et al., 1985; Milligan, 1996; Zhou et al., 1997; Kuschel et al., 1999;Ostrom and Insel, 1999). Caveolae, sphingolipid, and cholesterol-rich invaginations of the plasma membrane, decorated intracellularly with the protein caveolin, concentrate many different molecules involved in signal transduction, including G protein-coupled receptors, G proteins, and effector kinases and enzymes (as recently reviewed in Okamoto et al., 1998; Shaul and Anderson, 1998). Recent studies have suggested that the generation of and response to cAMP signals occurs in caveolae and may be regulated by caveolin (Schwencke et al., 1999a,b). Although these and many other studies have described that signaling molecules localize to caveolae (Okamoto et al., 1998; Shaul and Anderson, 1998), the present data are the first to imply a functional consequence of this compartmentation in native cells.
Acknowledgments
We thank the following individuals for their contributions to this work: Meihua Gao, Jian Zhu, and H. Kirk Hammond for providing adenovirus AC6; Monica Kim, John Adams, and Joan H. Brown for myocyte preparations; Eric Devic and Brian Kobilka for providing adenovirus HA-β1-AR; Ying Ling, Maryanne Martone, and Mark Ellisman for assistance with the electron microscopy; Kathryn Gabot and Brian Torres for excellent technical assistance.
Footnotes
-
Send reprint requests to: Paul A. Insel, M.D., Dept. of Pharmacology, 0636, University of California, San Diego, La Jolla, CA. E-mail: pinsel{at}ucsd.edu
-
↵1 These authors contributed equally to this work.
-
This work was supported by research and training grants from the National Institutes of Health.
- Abbreviations:
- AC
- adenylyl cyclase
- AC5 and AC6
- AC types 5 and 6
- PGE2
- prostaglandin E2
- HA
- hemagglutinin
- AR
- adrenergic receptor
- GFP
- green fluorescent protein
- Received December 16, 1999.
- Accepted February 7, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}