


Abstract
The benzothiazepine diltiazem blocks ionic current through L-type Ca2+ channels, as do the dihydropyridines (DHPs) and phenylalkylamines (PAs), but it has unique properties that distinguish it from these other drug classes. Wild-type L-type channels containing α1CII subunits, wild-type P/Q-type channels containing α1A subunits, and mutants of both channel types were transiently expressed in tsA-201 cells with β1B and α2δ subunits. Whole-cell, voltage-clamp recordings showed that diltiazem blocks L-type Ca2+ channels approximately 5-fold more potently than it does P/Q-type channels. Diltiazem blocked a mutant P/Q-type channel containing nine amino acid changes that made it highly sensitive to DHPs, with the same potency as L-type channels. Thus, amino acids specific to the L-type channel that confer DHP sensitivity in an α1A background also increase sensitivity to diltiazem. Analysis of single amino acid mutations in domains IIIS6 and IVS6 of α1CII subunits confirmed the role of these L-type-specific amino acid residues in diltiazem block, and also indicated that Y1152 of α1CII, an amino acid critical to both DHP and PA block, does not play a role in diltiazem block. Furthermore, T1039 and Y1043 in domain IIIS5, which are both critical for DHP block, are not involved in block by diltiazem. Conversely, three amino acid residues (I1150, M1160, and I1460) contribute to diltiazem block but have not been shown to affect DHP or PA block. Thus, binding of diltiazem to L-type Ca2+channels requires residues that overlap those that are critical for DHP and PA block as well as residues unique to diltiazem.
L-type voltage-gated Ca2+ channels play a critical role in the initiation of muscle contraction in the cardiovascular system and also contribute to the timing of the cardiac action potential (Bers and Perez-Reyes, 1999). Therefore, drugs that inhibit Ca2+ flux through L-type Ca2+ channels are used to treat hypertension, angina pectoris, and some cardiac arrhythmias. Three major classes of Ca2+ channel blockers are currently in clinical use: dihydropyridines (DHP), phenylalkylamines (PA), and benzothiazepines (BZP). Ca2+ channels are composed of a large, pore-forming α1 subunit consisting of four homologous domains (I–IV), each containing six transmembrane segments (S1–S6), in a complex with auxiliary β, γ, and α2δ subunits (Takahashi et al., 1987;Tanabe et al., 1987; Catterall, 1995). L-type Ca2+ channels in the heart and vascular smooth muscle contain α1c subunits, which are highly sensitive to Ca2+ channel blocking drugs, whereas the drug-insensitive N-type and P/Q-type Ca2+channels contain α1B and α1A subunits, respectively (reviewed in Hofmann et al., 1999).
Drugs from all three of these classes bind to L-type Ca2+ channels, and their binding sites are closely linked but not identical (Gould et al., 1983). Biochemical experiments utilizing photoreactive derivatives of DHPs and PAs indicated interaction of DHPs with domains IIIS6 and IVS6 (Nakayama et al., 1991; Striessnig et al., 1991), and PAs with domain IVS6 of the α1 subunit (Striessnig et al., 1990). Subsequent experiments using site-directed mutagenesis and electrophysiological recordings of recombinant channels expressed in various cell types have led to a detailed picture of the amino acid residues required for modulation of L-type Ca2+channels by DHPs and PAs (Hockerman et al., 1997b; Mitterdorfer et al., 1998; Hofmann et al., 1999).
The site of action for BZPs has been less extensively characterized. Diltiazem, a clinically relevant member of the BZP class of drugs, has several unique properties that suggest that its binding site is distinct from that of DHPs or PAs. Diltiazem stimulates the binding of DHPs but inhibits the binding of PAs to L-type Ca2+ channels (DePover et al., 1982; Ferry and Glossmann, 1982) suggesting that there are elements of the DHP binding site that are distinct from the BZP binding site. In addition, block of L-type Ca2+ channels by diltiazem is frequency-dependent, similar to but less pronounced than the frequency-dependent block by PAs (Lee and Tsien, 1983). This suggests that both diltiazem and PAs bind preferentially to the open and/or inactivated states of the channel, and may therefore share some common binding determinants within the channel. Finally, diltiazem shows lower selectivity for L-type versus non-L-type Ca2+channels than DHPs or PAs (Diochot et al., 1995; Ishibashi et al., 1995). This suggests that many of the binding determinants for diltiazem are identical across Ca2+ channel types, or that differences at these positions are conservative enough to allow diltiazem binding.
Biochemical experiments using a photoreactive BZP reported derivatization of transmembrane segments IIIS6 and IVS6 of L-type Ca2+ channels (Kraus et al., 1996). Subsequent studies using chimeric Ca2+ channels with IIIS6 and IVS6 segments from α1C spliced into an α1A background found that many of the same amino acid residues critical for high affinity PA block (Hockerman et al., 1995, 1997a) were also involved in diltiazem block (Hering et al., 1996; Kraus et al., 1998). To date, no studies have systematically examined the role of each amino acid residue in the IIIS6 and IVS6 segments. Therefore, we have constructed single amino acid mutations to alanine along the entire IIIS6 and IVS6 segments of α1C and have analyzed each mutant for sensitivity to block by diltiazem. In addition, we have measured the sensitivity to diltiazem block of both the wild-type α1A (P/Q-type) channel and an α1A mutant (α1A/DHPS) that contains the DHP binding site. To assess the contribution to diltiazem block of amino acid residues in transmembrane domain IIIS5 known to be critical for DHP block (Grabner et al., 1996; He et al., 1997), we have constructed a double amino-acid mutation in IIIS5 (α1C/DHPI) and analyzed it for sensitivity to both the DHP PN200–110 and diltiazem. Our results reveal several amino acid residues that contribute to block by diltiazem but not by DHPs and/or PAs; conversely, several key amino acid residues contribute to block by DHPs and/or PAs but do not contribute to block by diltiazem.
Experimental Procedures
Construction of Mutants.
Single amino acid mutations in domains IIIS6 and IVS6 of the α1C subunit (Snutch et al., 1991) were constructed using oligonucleotide-directed mutagenesis, as described previously (Kunkel, 1985). The IIIS6 mutations were inserted into full-length subunit constructs in the expression vector Zem 229 (Dr. Eileen Mulvihill, University of Washington, Seattle, WA) using the 1.5-kilobaseSpeI/DraIII fragment and the 272 bpDraIII/DraIII fragment in a three-way ligation. Construction of the α1A/DHPS mutant was described previously (Hockerman et al., 1997c). The α1C/DHPI mutant contains two mutations in domain IIIS5 (T1039 to Y and Q1043 to M) and was constructed using the splice overlap extension method (Horton et al., 1989). The mutant 600-bp DNA fragment was cut with SpeI and BglII and inserted into the full-length subunit construct in the expression vector Zem 229 using the 900-bp BglII//DraIII fragment and the 272-bp DraIII/DraIII fragment in a four-way ligation. All mutations were confirmed by cDNA sequencing.
Cell Culture.
tsA201 cells, a subclone of the human embryonic kidney cell line 293 that expresses simian virus 40 T antigen (a gift of Dr. Robert Dubridge, Cell Genesis, Foster City, CA) were maintained in monolayer culture in Dulbecco's modified Eagle's medium/Ham's F-12 medium (Life Technologies, Inc., Gaithersburg, MD), supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), and incubated at 37°C in 10% CO2.
Expression.
Wild-type and mutant α1Cchannel subunits (Snutch et al., 1991) were expressed with β1B (Pragnell et al., 1991) and α2δ channel (Ellis et al., 1988) subunits and either CD8 antigen (EBO-pCD-Leu2, American Type Culture Collection, Manassas, VA) or enhanced green fluorescent protein (CLONTECH, Palo Alto, CA) in tsA201 cells by transfection with either Ca2+ phosphate (Margolskee et al., 1993) or the transfection reagent GenePorter (Gene Therapy Systems, San Diego, CA). Transfectants were selected by labeling with anti-CD8 antibodies conjugated to polystyrene beads (Dynal, Oslo, Norway) or by fluorescence at 510 nm with excitation at 480 nm (enhanced green fluorescent protein transfected cells).
Electrophysiology.
Ba2+ currents through Ca2+ channels were measured using the whole-cell, patch-clamp configuration. Pipettes were pulled from VWR micropipettes (VWR, West Chester, PA) and fire polished to produce an inner tip diameter of 4 to 6 μm. Currents were recorded using an Axon Instruments Axopatch 200B amplifier and filtered at 1 or 2 kHz (six-pole Bessel filter, −3 dB). Voltage pulses were applied and data were acquired using pClamp6 software (Axon Instruments, Foster City, CA). Voltage-dependent currents have been corrected for leak using an online P/−4 subtraction paradigm. (+)-cis-Diltiazem (RBI, Natick, MA) dissolved in bath saline was applied to cells under voltage clamp using a fast perfusion system with constant exchange of the bath solution. PN200–110 was added to the bath without background perfusion as a 3× stock. The bath saline contained 150 mM Tris, 10 mM BaCl2, and 4 mM MgCl2. The intracellular solution contained 130 mMN-methyl-d-glucamine, 10 mM EGTA, 60 mM HEPES, 2 mM MgATP, 1 mM MgCl2. The pH of both solutions was adjusted to 7.3 with methanesulfonic acid. All experiments were done at room temperature (20–23°C).
Results
Block of Ca2+ Channels Containing Wild-Type α1C and α1A Subunits by (+)-cis-Diltiazem.
The α1Csubunit (Snutch et al., 1991) was expressed in tsA201 cells along with the β1B (Pragnell et al., 1991) and α2δ (Ellis et al., 1988) subunits. Ba2+ currents through the resulting L-type channels were blocked by (+)-cis-diltiazem with an IC50 value of 33.3 ± 4.6 μM (Fig.1A, F). For L-type channels, a holding potential of −60 mV with a 100-ms test pulse to +10 mV at a frequency of 0.05 Hz was used. Under these conditions, block developed rapidly, and reached equilibrium within 200 s (Fig. 1B). Little (<10%) frequency-dependent block accumulated using this low stimulation frequency. These conditions were used for the assay of diltiazem sensitivity of all the α1C mutants.

Diltiazem block of wild-type α1C, α1A, α1C/DHPI, and α1A/DHPS. A, representative Ba2+ current records from a tsA201 cell expressing wild-type α1C in the absence (control) or presence of the indicated concentrations of diltiazem. B, time course of diltiazem block of α1C Ba2+ current expressed in a tsA201 cell. (+)-cis-diltiazem was applied at the pulse number and concentrations indicated. Pulses were to +10 mV from a holding potential of −60 mV every 20 s. C, Representative current records from a tsA 201 cell expressing wild-type α1A in the absence (control) or presence of the indicated concentrations of diltiazem. D, representative current records from a tsA201 cell expressing the mutant α1A/DHPS in the absence (control) or presence of the indicated concentrations of diltiazem. E, time course of block of wild-type α1C (○) and α1C/DHPI (●) by the DHP PN200–110 (10 μM). F, summary of the IC50 values of wild-type α1C, wild-type α1A, α1A/DHPS, and α1C/DHPI for (+)-cis-diltiazem. The IC50 values (shown ± S.E.) were determined as described in the legend to Fig. 2. The asterisks denote that the IC50 values for α1C, α1A/DHPS,and α1C/DHPI are significantly different from the IC50 value for α1A (Student'st test, * P < .05, **P < .01). Note that α1C/DHPI has the same sensitivity to diltiazem as wild type α1C even though it is virtually insensitive to PN200–110.
In addition to the L-type Ca2+ channel containing wild-type α1C, we also measured the concentration dependence of diltiazem block of the P/Q-type Ca2+ channel containing wild-type α1A (Stea et al., 1994). These channels have been reported to be blocked by 2- to 10-fold higher concentrations of diltiazem than those required to block L-type Ca2+ channels (Diochot et al., 1995; Ishibashi et al., 1995). We expressed the α1A subunit along with the β1B and α2δ subunits in tsA201 cells and applied increasing concentrations of (+)-cis-diltiazem to cells under voltage clamp using a holding potential of −80 mV and 100-ms test pulses to 0 mV at a frequency of 0.033 Hz. A more negative holding potential (−80 mV) was used for α1A because the fraction of inactivated channels at this holding potential is similar to that of α1C at −60 mV (Hockerman et al., 1997c). The lower frequency of stimulation was used to prevent accumulation of inactivated channels. We found that the wild-type α1A channel was blocked by diltiazem under these conditions with an IC50 value of 169 ± 10 μM, only five times the IC50 value for wild-type α1C (Fig. 1C, F).
Block of Mutant Ca2+ Channels Containing α1A/DHPS and α1C/DHPI by (+)-cis-Diltiazem.
We hypothesized that the difference in sensitivity to diltiazem block between the wild-type α1C and α1A channels might be caused by changes in relatively few amino acid residues. As an initial experiment, we tested the diltiazem affinity of a mutant α1A channel in which nine amino acids from α1C had been substituted. These substitutions had been shown previously to confer nearly full sensitivity to DHPs (Hockerman et al., 1997c). We expressed this mutant, α1A/DHPS, along with the β1B and α2δ subunits in tsA201 cells and applied increasing concentrations of (+)-cis-diltiazem to cells under voltage clamp using a holding potential of −120 mV and 100-ms test pulses to 0 mV at a frequency of 0.033 Hz. The strongly negative holding potential was necessary to compensate for the very negative voltage dependence of inactivation of the α1A/DHPS mutant (Hockerman et al., 1997c). Diltiazem blocked Ba2+ currents through α1A/DHPS with an IC50 value of 35.5 ± 13.6 μM, a concentration virtually identical with that of wild-type α1C (Fig. 1, D and F).
The nine L-type specific amino acids substituted in α1A/DHPS are in domains IIIS5, IIIS6, and IVS6 of α1A. Amino acid residues in transmembrane segments IIIS6 and IVS6 had been previously implicated in diltiazem block (Kraus et al., 1996), so we focused on segment IIIS5. α1A/DHPS contains substitutions of two L-type-specific amino acid residues in IIIS5. We made a mutant L-type channel, α1C/DHPI, containing the converse changes by substituting T1039 with Y and Q1043 with M in domain IIIS5 of α1C. Mutation of these amino acids dramatically decreases DHP affinity for α1C(Mitterdorfer et al., 1996; He et al., 1997). We expressed the α1C/DHPI mutant channel in tsA201 cells along with the β1B and α2δ subunits and added increasing concentrations of (+)-cis-diltiazem to cells under voltage clamp using a holding potential of −60 mV and test pulses to +10 mV for 100 ms at 0.05 Hz. The wild-type α1C and α1C/DHPI could be compared at the same holding potential because their V 1/2 values for inactivation are not significantly different (data not shown). Although Ba2+ currents through the resulting channels were not blocked by PN200–110 at 10 μM (Fig. 1E), the IC50 value for diltiazem was not different from wild-type α1C (Fig. 1F). Thus, T1039 and Q1043 are critical for DHP block but unnecessary for diltiazem block of α1C.
Effects of Single Amino Acid Alanine Substitution Mutants in Transmembrane Segment IIIS6 on Block by (+)-cis-Diltiazem.
Because photoaffinity labeling studies suggest that diltiazem interacts with both transmembrane domains IIIS6 and IVS6 (Kraus et al., 1996), we screened a battery of single amino acid mutations to alanine spanning both the IIIS6 and IVS6 transmembrane domains of α1C for concentration-dependent block by diltiazem. At positions where the native amino acid is alanine, we substituted the corresponding amino acid in non-L-type channels (A1157P, A1467S). We also made more conservative changes at tyrosines 1152 and 1463, mutating them to phenylalanine. We expressed the α1C mutant channels in tsA201 cells along with the β1B and α2δ subunits and added increasing concentrations of (+)-cis-diltiazem to cells under voltage clamp using a holding potential of −60 mV and test pulses to +10 mV for 100 ms at 0.05 Hz. Of the single amino acid mutations tested, only A1157P did not express current. The functional properties of the mutant channels that were expressed at low level have been described previously (Hockerman et al., 1997a). Of the 21 single amino acid mutants in segment IIIS6 that we screened, eight had no significant change in diltiazem affinity, and six had <2-fold change in the IC50 value for diltiazem (Fig.2). In contrast, mutants I1150A, I1153A, I1156A, and M1160A in the central portion of the segment had substantially reduced sensitivity to diltiazem block compared with wild-type α1C (I1150A, IC50 = 97.1 ± 20.6 μM; I1153A, IC50 = 93.1 ± 24.4 μM; I1156A, IC50 = 83.6 ± 10.9 μM; M1160A, IC50 = 107.9 ± 7.7 μM). In addition, mutation of two amino acid residues near the intracellular end of this transmembrane segment, F1164A and V1165A, also resulted in substantially decreased potency of diltiazem block of Ba2+ current (F1164A, IC50= 90.8 ± 9.4 μM; V1165A, IC50 = 131 ± 40 μM).

Diltiazem block of wild-type α1Cchannels and α1C channels with single-amino-acid mutations in IIIS6. A, L-type Ba2+ currents were recorded from wild-type and mutant α1C L-type channels as described under Experimental Procedures. Representative traces are shown in the absence (control) or presence of the indicated concentrations of (+)-cis-diltiazem for wild-type amino acids (left), the mutant I1150A (center), and the mutant M1160A (right). B, dose-response relationships for the indicated wild type and mutant channels. In each case, the averaged, normalized current amplitudes at 5, 10, 50, 100, and 500 μM diltiazem (symbols ± S.E.) were plotted against the corresponding drug concentration, and the IC50 value was determined by fitting the averaged relative current values at each diltiazem concentration to the equation, relative current = 1− {1/[1+(IC50/[diltiazem])]} (smooth lines). C, the IC50 values of the indicated mutations in IIIS6 for diltiazem are shown ± S.E. Asterisks denote that the IC50 value for diltiazem of the indicated mutant channel is significantly different from that of wild-type α1C(Student's t test: * P < .05, **P < .01, *** P < .001).
Effects of Block of Single-Amino-Acid Alanine Substitution Mutants in Transmembrane Segment IVS6 by (+)-cis-Diltiazem.
We also screened single-amino-acid mutations across transmembrane IVS6 for concentration dependence of diltiazem block as described above. The functional properties of these mutant Ca2+channels have been described previously (Hockerman et al., 1995). Of the 13 amino acids in IVS6 that were mutated, only mutant channels I1460A, Y1463F, and M1464A showed decreased sensitivity to diltiazem (I1460A, IC50 = 69.5 ± 7.5 μM; Y1463F, IC50 = 114.4 ± 19 μM; M1464A, IC50 = 91.8 ± 8.9 μM; Fig.3).

Diltiazem block of wild-type α1Cchannels and α1C channels with single-amino-acid mutations in IVS6. A, L-type Ba2+ currents were recorded from wild-type and mutant α1C L-type channels as described under Experimental Procedures. A, representative traces are shown in the absence (control) or presence of the indicated concentrations of (+)-cis-diltiazem for wild-type (left), the mutant I1460A (center), and the mutant M1464A (right). B, dose-response relationships are shown for the indicated wild-type and mutant channels and are as described in the legend to Fig. 2. C, the IC50 values of the indicated mutations in IVS6 for diltiazem are shown ± S.E. and were determined as described in the legend to Fig. 2. An asterisk denotes that the IC50 value for diltiazem of the indicated mutant channel is significantly different from that of wild-type α1C(Student's t test, * P < .05, ***P < .001).
Because diltiazem binds with higher affinity to L-type channels in the inactivated state, we measured the effect of the single-amino-acid mutations found to decrease diltiazem sensitivity on the voltage-dependence of inactivation. In IVS6, I1460A (V 1/2 = −18.2 mV), and Y1463F (V 1/2= −19.2 mV), both of which significantly decreased potency of diltiazem block, were not different from wild-type α1C(V 1/2 = −17.0 mV), whereas M1464A (V 1/2 = −13.5 mV) was only slightly positively shifted compared with wild-type. In IIIS6, I1150A (V 1/2= −13.0 mV), I1153A (V 1/2= −12 mV), and V1165A (V 1/2= −14.5 mV) were also slightly shifted from wild-type α1C, whereas I1156A (V 1/2= −10.0 mV) and F1164A (V 1/2= −9.1 mV) had greater positive shifts in voltage dependence of inactivation. Nevertheless, the holding potential used in our experiments (−60 mV) is sufficiently negative to prevent inactivation of either wild-type α1C or mutant channels, so that positive shifts inV 1/2 will not result in a smaller fraction of inactivated channels. Therefore, the reduced sensitivity of the mutant channels to diltiazem is most likely caused by changes in drug affinity and not by changes in inactivation properties.
In addition to the single amino acid mutations, we also examined the effect of a triple mutation in IVS6 (YAI, Y1463I+A1467S+I1470A) that had previously been shown to greatly decrease block by the high-affinity phenylalkylamine (−)-D888 (Hockerman et al., 1995). Transfer of the L-type amino acids to the corresponding positions in IVS6 of the α1A subunit has been shown to increase the sensitivity of P/Q-type Ca2+channels to diltiazem (Hering et al., 1996). We found that the YAI mutant is less sensitive to diltiazem than wild-type α1C (IC50 = 134.5 ± 13.8 μM), but only to an extent similar to the much more conservative mutation Y1463F (Fig. 3). Furthermore, we found no appreciable increase in the IC50 value of diltiazem for the single-amino-acid mutants A1467S and I1470A.
Discussion
Distinct but Overlapping Binding Sites for BZPs and DHPs on α1C.
Using the α1A/DHPSchannel, we have shown that insertion of a DHP binding site into a channel that is normally insensitive to DHP increases the sensitivity to block by diltiazem by 5-fold, making the IC50value of diltiazem for this mutant α1A channel virtually identical with the wild-type α1C(Fig. 1, D and F). Because α1A/DHPS contains amino acid substitutions in three transmembrane segments, IIIS5, IIIS6, and IVS6, we assessed the contribution of amino acid residues in these three regions to diltiazem block. Mutation of both T1039 and Q1043 in IIIS5 of α1C (mutant α1C/DHPI), removes DHP block (Mitterdorfer et al., 1996; He et al., 1997) (Fig. 1E) and virtually eliminates DHP binding (He et al., 1997), but does not affect block by diltiazem (Fig.1F). Therefore, T1039 and Q1043 in IIIS5 are clearly involved in DHP block, but not diltiazem block, of α1C. Similarly, Y1152, F1158, F1159, and M1161 in transmembrane segment IIIS6 as well as I1470, I1471, and N1472 in segment IVS6 are required for DHP binding and block but not for BZP block (Fig.4). Thus, nine amino acid residues in these three transmembrane segments are involved in the DHP receptor site but not the BZP receptor site.
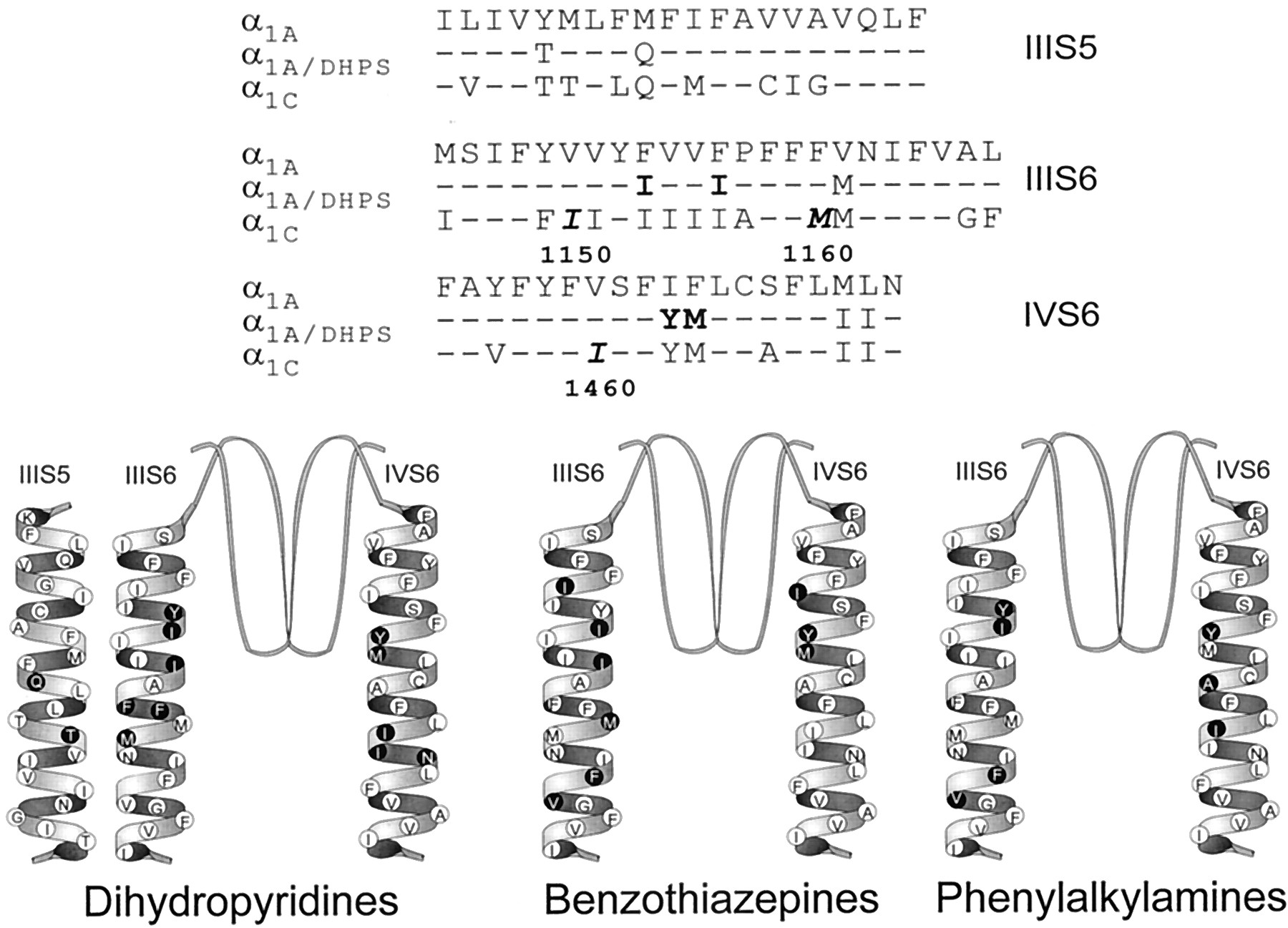
Amino acid residues required for binding of calcium antagonist drugs. Top, amino acid sequences of transmembrane segments IIIS5, IIIS6, and IVS6 of α1A, α1C, and α1A/DHPS. Amino acid residues in bold type are required for high-affinity BZP binding. Amino acid residues in bold italic type are required for high-affinity BZP binding, but not included in α1A/DHPS. Bottom, transmembrane α-helical segments, with amino acid residues required for high-affinity binding of DHPs, BZPs, and PAs indicated as ●.
Other amino acid residues in transmembrane segments IIIS6 and IVS6 are required for BZP block but not for block by DHPs (Fig. 4). I1150, F1164, and V1165 in segment IIIS6 and I1460 in segment IVS6 are important for BZP block but not for DHP binding and block. These results also support the conclusion that the BZP and DHP sites are separate.
In contrast, four amino acid residues in segments IIIS6 and IVS6 contribute to DHP binding and block and to BZP block (Fig. 4). Y1463 and M1464 in segment IVS6 as well as I1153 and I1156 in segment IIIS6 all contribute to DHP action (Peterson et al., 1997). These residues were included in α1A/DHPS and two other similar chimeric mutants resulting in gain of DHP binding and modulation (Hockerman et al., 1997c; Ito et al., 1997; Sinnegger et al., 1997). These four amino acids also play an essential role in BZP block (Fig.4). Thus, a distinct but overlapping set of amino acid residues in IIIS6 and IVS6 comprise portions of the DHP and BZP binding sites.
The positions of the determinants of BZP block relative to those of DHP block are notable for two reasons. First, I1150 and I1460 lie more toward the extracellular ends of their respective transmembrane segments than other amino acids interacting with DHPs or PAs (Fig. 4). This location is consistent with studies using quaternary amine BZPs indicating that BZPs approach their binding site from the extracellular side of the membrane (Hering et al., 1993). We did, however, find that F1164 and V1165, located toward the intracellular end of IIIS6, also affect diltiazem block. Intracellular ends of S6 segments are thought to undergo conformational changes during K+channel gating (Holmgren et al., 1998). Therefore, the effects of these mutant residues may be long-distance allosteric ones secondary to effects on channel gating. Second, the aliphatic side chains of I1150, I1153, I1156, and M1160, which are required for BZP block, lie primarily on one face of the IIIS6 helix. It is possible that BZPs and DHPs bind simultaneously to different faces of the IIIS6 helix, with both drugs contacting I1153 and I1156 from opposite sides (Fig. 4).
BZPs and DHPs interact allosterically but not competitively when binding to the α1C subunit of the L-type Ca2+ channel. Diltiazem reduces both the association and dissociation rates of DHP binding, without increasing the apparent affinity for DHPs (Brauns et al., 1997). Thus, binding of diltiazem does not prevent access of DHPs to the determinants within α1C that contribute the bulk of the binding energy. These previous studies are consistent with a model in which DHPs and diltiazem bind to distinct faces of the IIIS6 and IVS6 transmembrane segments at distinct but overlapping sites.
Similar but Nonidentical Binding Sites on α1C for Diltiazem and Phenylalkylamines.
Diltiazem and PAs block L-type Ca2+ channels in a similar manner. They are less potent than DHPs and, unlike DHPs, they exhibit striking frequency-dependent block (Lee and Tsien, 1983). Previous studies have implicated transmembrane segments IIIS6 and IVS6 in block of L-type Ca2+ channels by the high-affinity PA (−)-D888 (Hockerman et al., 1995, 1997a; Doring et al., 1996; Hering et al., 1997). Of the seven amino acid residues that may interact directly with BZPs, only I1153 in segment IIIS6 and Y1463 in segment IVS6 are also important for block by PAs (Fig. 4). I1150, I1156, and M1160 in segment IIIS6 as well as I1460 and M1464 in segment IVS6 are required for BZP binding but not PA binding (Fig. 4). Interestingly, F1164 and V1165, which are not implicated in DHP block, have been proposed as key determinants in the frequency dependence of both PA and BZP block (Hering et al., 1997; Kraus et al., 1998). Both F1164 and V1165 are conserved between α1C and α1A, and are therefore present in the α1A/DHPS mutant, where they most likely contribute substantially to diltiazem action. As noted above, it is likely that these residues are indirect allosteric effectors of state-dependent binding of PA and BZPs.
Other amino acid residues in the PA and BZP binding sites in IIIS6 and IVS6 do not overlap. Amino acids Y1152 in IIIS6 and A1467 and I1470 in IVS6 are key residues for (−)-D888 block but do not affect block by diltiazem. Indeed, α1A/DHPS has L-type sensitivity to diltiazem block despite the absence of the L-type specific residue at position 1467. Conversely, I1150, I1156, and M1160 in IIIS6 as well as I1460 and M1464 in IVS6 appear to exclusively affect diltiazem block. Our results differ from those of Hering et al. (1996) in which Y1463, A1467, and I1470 were inserted into a P/Q-type channel and increased diltiazem sensitivity. Also, M1464A had no effect on diltiazem block, but increased block by the phenylalkylamine (−)-D600 in the otherwise P/Q-type channel. It is possible that the different P/Q-type channel subtypes used in these experiments or the different expression and recording conditions in Xenopus laevis oocytes (Hering et al., 1996) mediate the different effects on drug binding.
Amino Acid Residues Involved in Diltiazem Block of α1C Are Conservatively Substituted in α1A.
A surprising finding of this study is the relatively small difference between the IC50values for diltiazem block of wild-type α1C and α1A. Diltiazem has been considered a relatively specific blocker of L-type Ca2+ channels, although block of other channel types at higher concentrations has been noted (Diochot et al., 1995; Ishibashi et al., 1995). Our findings suggest that the selectivity of diltiazem for L-type channels over P/Q-type channels is less than an order of magnitude. Most of the determinants of diltiazem block reported here are not conserved between α1C and α1A. How then, can the P/Q-type channel retain its relatively high degree of sensitivity to diltiazem?
A clue is provided by our results with the α1A/DHPS mutant, which contains nine L-type specific amino acids that render it highly sensitive to block by DHP (Hockerman et al., 1997c) and diltiazem. The alanine scans of IIIS6 and IVS6 identified several amino acids that are important for diltiazem block and are not included in α1A/DHPS (i.e., I1150, M1160, I1460; Fig. 4). However, the differences between the α1A and α1C protein sequences at these positions are very conservative (i.e., V for I at 1150, F for M at 1160, and V for I at 1460; Fig. 4). It is likely that these hydrophobic amino acids in α1A can substitute efficiently for the nonidentical but similar hydrophobic amino acids in α1C, and that the importance of these positions in diltiazem block comes to light only when the more drastic substitution of alanine is made. We have previously emphasized the importance of systematic mutagenesis for identifying functional properties of amino acids conserved between channel subtypes (Hockerman et al., 1995, 1997a; Peterson et al., 1996; Peterson et al., 1997). This result emphasizes the utility of systematic alanine substitutions in locating binding determinants where subtype differences are conservative enough to support the same function.
Further analysis of amino acid differences between the α1A and α1C in positions that we found to be important for diltiazem block reveals that three of the remaining four nonconserved residues are relatively conservative aliphatic-to-hydrophobic aromatic substitutions (i.e., I1153 to F, I1156 to F, M1464 to F). One nonconserved position, Y1463, is replaced by isoleucine in α1A, a substantial difference in side chain character. In fact, the removal of the tyrosine hydroxyl in the Y1463F mutation has nearly the same effect as substitution to alanine in the context of the YAI mutation (Fig. 3). Thus, at the positions critical for diltiazem block, only one amino acid is substantially different between α1A and α1C whereas several positions contain conservative amino acid substitutions. These conservative substitutions along with conserved residues F1164 and V1165 allow relatively potent block of α1A by diltiazem.
Acknowledgments
We thank Lonnie Yeung for excellent technical help with cell culture and molecular techniques.
Footnotes
- Received April 27, 2000.
- Accepted September 7, 2000.
-
Send reprint requests to: Dr. Gregory H. Hockerman, Department of Medicinal Chemistry and Molecular Pharmacology, 1333 RHPH, Purdue University, West Lafayette, Indiana. E-mail:gregh{at}pharmacy.purdue.edu
-
This work was supported by National Institutes of Health Grant PO1-HL44948 and by a research grant-in-aid from the American Heart Association (W.A.C.) and Scientist Development Grant 9930016N from the American Heart Association (G.H.H.).
Abbreviations
- DHP
- dihydropyridine
- PA
- phenylalkylamine
- BZP
- benzothiazepine
- bp
- base pairs
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}