


Abstract
We attempt to identify the plasma membrane transporter involved in the uptake of 5′-deoxy-5-fluorouridine (5′-DFUR), an intermediate metabolite of capecitabine. This novel oral fluoropyrimidine is used in cancer treatments and is a direct precursor of the cytostatic agent 5′-fluorouracil. We also examine the role of the transporter in 5′-DFUR cytotoxicity. The human concentrative nucleoside transporter (hCNT1) was cloned from human fetal liver and expressed in Xenopus laevis oocytes. The two-electrode voltage-clamp technique was used to demonstrate that 5′-DFUR, but not capecitabine or 5′-FU, is an hCNT1 substrate. Then, hCNT1 was heterologously expressed in the mammalian cell line Chinese hamster ovary-K1. Functional expression was demonstrated by monitoring transport of radiolabeled substrates and by using a monospecific polyclonal antibody generated against the transporter. hCNT1-expressing cells were more sensitive to 5′-DFUR than vector-transfected or wild-type cells. The sensitivity of the three cell types to other agents such as cisplatin or 5′-FU was identical. In conclusion, this study shows that 1) the pharmacological profile of a nucleoside transporter can be determined by an electrophysiological approach; 2) the hCNT1 transporter is involved in 5′-DFUR uptake; and 3) hCNT1 expression may increase cell sensitivity to 5′-DFUR treatment. This study also reports for the first time the generation of an antibody against hCNT1, which may be useful in the elucidation of the relationship between hCNT1 expression and tumor response to capecitabine treatment.
Capecitabine (Xeloda; N4-pentyloxycarbamyl-5′-deoxy-5-fluorocytidine) is a novel oral fluoropyrimidine derivative that is active against a variety of solid tumors (Cassidy 1999; Villalona-Calero et al., 1999;Van Cutsem et al., 2000). Capecitabine treatment results in 5′-fluorouracil (5′-FU) synthesis inside tumor cells as a consequence of metabolic activation, which involves three sequential reactions (Verweij, 1999). The first is catalyzed by a hepatic enzyme, carboxylesterase, and yields 5′-deoxy-5-fluorocytidine, which is then converted to 5′-deoxy-5-fluorouridine (5′-DFUR) by cytidine deaminase. This enzyme is also highly expressed in liver, although it can be present in various solid tumors. 5′-DFUR is the direct precursor of the cytotoxic drug 5′-FU. This conversion occurs mostly in tumors, which express thymidine phosphorylase (dThdPase) activity at different levels (Yoshimura et al., 1990). In fact, this enzyme may limit 5′-FU production as suggested by the fact that heterologous expression of dThdPase in a variety of tumor cell lines increases 5′-DFUR cytotoxicity (Patterson et al., 1995; Kato et al., 1997; Evrard et al., 1999). However, 5′-FU cytotoxicity also depends on the activity of dihydropyrimidine dehydrogenase, which inactivates 5′-FU. The dThdPase/dihydropyrimidine dehydrogenase ratio is variable among cancer types but it is even more variable among patients, which indicates that it may be a useful criterion for selecting patients for chemotherapy (Mori et al., 2000).
In this scenario of metabolic activation of capecitabine, plasma membrane transporters may play an accessory or even a key role, thus modulating the uptake into tumor cells of the active intermediate metabolite 5′-DFUR. Recent evidence suggests that most nucleoside-derived drugs are substrates of transporters responsible for the uptake of natural nucleosides (Yao et al., 1996; Schaner et al., 1997; Mackey et al., 1998; 1999; Pastor-Anglada et al., 1998; Baldwin et al., 1999). So far, two families of transporters have been identified in kinetic and molecular terms: the equilibrative family (ENT), for which two isoforms, hENT1 and hENT2, have been cloned, and the concentrative nucleoside transporter family (CNT) for which two isoforms have also been characterized at the cDNA level, hCNT1 and hCNT2. Whereas hCNT1 may be involved in the uptake of pyrimidine-derivatives, including fluoropyrimidines with anticancer properties, hCNT2 is a purine nucleoside-preferring transporter. The CNT transporters show higher affinity for their substrates than the ENTs (for recent reviews, see Wang et al., 1997; Pastor-Anglada et al., 1998; Baldwin et al., 1999).
Here we have cloned and expressed the hCNT1 transporter inXenopus laevis oocytes and in CHO-K1 cells. The expression of a functional hCNT1 transporter in oocytes was demonstrated by an electrophysiological approach, which was used to determine the pharmacological profile of the transporter (Lostao et al., 2000). Neither capecitabine nor 5′-FU is an hCNT1 substrate, although 5′-DFUR is. The expression of a functional hCNT1 transporter in CHO-K1 cells was demonstrated by flux measurements of radiolabeled natural nucleosides and by immunocytochemistry using a monospecific polyclonal antibody generated against an N-terminal oligopeptide of the hCNT1 protein. To our knowledge, this is the first antibody against a human concentrative nucleoside transporter to be reported in the literature. Interestingly, according to the pharmacological profile of hCNT1, CHO-K1 cells expressing this transporter are more sensitive to 5′-DFUR, which suggests that its uptake contributes to drug cytotoxicity and thus tumor sensitivity. If so, the effectiveness of capecitabine may depend not only on the activities of the enzymes but also on the transport process.
Materials and Methods
Cloning of the Human Fetal Liver hCNT1 Transporter.
The hCNT1 clone was obtained by amplification of an oligo(dT)-primed cDNA library from human fetal liver (Marathon Ready cDNA, CLONTECH, Palo Alto, CA), using a PCR approach with Advantage 2 Polymerase Mix (CLONTECH), as described previously (Lostao et al., 2000). Primers HC1E (5′-GCTGCACTGCATG GTTGCTGCTGGATGTGTTG-3′) and HC1D (5′-GGGAAGGGGCATGTGA GGGACCCCAGGAGA-3′), derived from the reported sequence for hCNT1 (GenBank accession number U62966), were used in a touchdown PCR that produced a unique DNA band of approximately 2 kilobase pairs. This PCR product was gel purified, used in an additional reaction of dATP added to the 3′ end with TaqPolymerase (Promega, Madison, WI), again purified in a DNA clean-up column (Wizard DNA Cleanup System; Promega), and finally ligated to the plasmid pGEM-Teasy (pGEM-Teasy Vector System II; Promega) with T4 DNA Ligase (New England Biolabs). The construct pGEMT-hCNT1, after Taq DyeDeoxy terminator cycle sequencing of both strands (DNA sequencing kit, dRhodamine Terminator Cycle Sequencing; PerkinElmer Life Science Products, Norwalk, CT) and aligning with published sequence, confirmed the nature of the hCNT1 cDNA. The hCNT1 cDNA was then inserted into the pBSII(KS) vector, which contained a poly(A) tail of 54 bp from the original plasmid pBSII(KS)-hSGLT1 (Hediger et al., 1987) as follows. pBSII(KS)-hSGLT1 was digested with NsiI, blunted with Klenow fragment, purified in a DNA clean-up column, digested withSalI, and gel-purified. The construct pGEMT-hCNT1 was digested in parallel. The two fragments were ligated with T4 DNA Ligase as described above and the product, pBSII(KS)-hCNT1-poly(A) tail-, was used to transform Escherichia coli XL-1 Blue. Plasmid pBSII(KS)-hSGLT1 was kindly donated by Dr. E. M. Wright (University of California Los Angeles, Los Angeles, CA).
Expression of hCNT1 in X laevis Oocytes and Electrophysiological Studies.
The plasmid containing the hCNT1 cDNA was linearized with XbaI and cRNA synthesized using the T3 MEGAscript kit (Ambion, Austin, TX) in the presence of m7G(5′)ppp(5′)G. cRNA (50 ng ) was injected into X laevis oocytes, which were then maintained at 18°C in Barth's medium for 3 to 5 days. Electrophysiology experiments were performed to examine substrate-induced Na+ inward currents using the two-electrode voltage-clamp method (Hirayama et al., 1996;Lostao et al., 2000). The oocyte membrane potential (V m) was held at −50 mV and continuous current data were recorded using Axoscope V 1.1.1.14 (Axon Instruments, Foster City, CA).
The apparent affinity constant (K 0.5) and the maximal current for saturating cytidine and 5′-DFUR (Imax) were obtained by fitting the steady-state currents at each membrane potential to the equation: I = Imax. [S]/(K 0.5 + [S]), where [S] is the substrate concentration, using the nonlinear fitting method in SigmaPlot 4 (SPSS, Chicago, IL). Natural nucleosides used in these experiments were purchased from Sigma (St. Louis, MO). Capecitabine and 5′-DFUR were kindly donated by Roche (Fukuoka, Japan).
Expression of hCNT1 in CHO-K1 Cells.
The hCNT1 cDNA was also subcloned into a pCDNA3 vector and stably transfected into CHO-K1 cells using Fugene (Roche). Putative hCNT1-expressing clones were selected using geneticin treatment and several independent clones were checked for hCNT1 activity, mRNA, and protein. Wild-type (nontransfected) CHO-K1 cells and cells transfected with the empty pCDNA3 vector were used as control cells in these experiments.
hCNT1-related activity was measured in several independent clones by monitoring the uptake of 1 μM uridine (Sigma), as described previously (del Santo et al., 1998). Transport measurements were performed by incubating cell monolayers in an uptake buffer, supplemented with either sodium or choline chloride, in which [3H]uridine was added at a specific activity of 1 μCi/nmol. This method allows the calculation of Na+-dependent uptake rates. Incubations were stopped by rapidly aspirating the uptake buffer and immediate washing with a cold stop buffer, as described previously (del Santo et al., 1998). The specificity of uridine transport was further studied by inhibiting 1 μM uridine uptake by pyrimidine nucleosides and nucleoside-derived drugs at a concentration of 100 μM.
Generation of a Monospecific Polyclonal Antibody against the hCNT1 Protein.
Expression of hCNT1 was also assessed by confocal microscopy using a monospecific polyclonal antibody generated in our laboratory. The primary structure of hCNT1 protein was analyzed on the basis of the prediction of transmembrane domains, the position of the α-helix in the predicted secondary structure using a program that combines the algorithms of Chou-Fasman and Rose, the position of hydrophobicity peaks in the hydrophilicity plot using the Kyte-Doolitle method with a window size of 9 amino acids, and, finally, the Surface Probability and Flexibility Index plots according to the algorithms of Boger and Karplus and Schulz, as described previously (Felipe et al., 1998). An oligopeptide corresponding to residues 48 to 69 was chosen on the basis of its putative antigenicity and the unique nature of this sequence. This oligopeptide was coupled to keyhole limpet hemocyanin, emulsified with an equal volume of Freund's adjuvant, and injected into rabbits. The IgG fraction of the immunoreactive antisera was purified using a protein A-Sepharose CL-4B column and used for further immunoassays. Cells were plated onto glass coverslips. Forty-eight hours after plating, the cells were washed in phosphate-buffered saline (PBS) and fixed with a 2% p-formaldehyde solution supplemented with 0.05% saponin for 15 min. After fixation, the cells were washed twice in 20 mM glycine-PBS for 10 min. After blocking with 1% bovine serum albumin in 20 mM glycine/PBS for 15 min, the coverslips were incubated with a 1:40 diluted hCNT1 primary antibody for 1 h. After the incubation, the cells were washed three times in PBS, incubated with an anti-rabbit IgG antiserum coupled to fluorescein (1:50) as a second antibody, washed in PBS, and then mounted onto glass slides using immunofluor mounting solution. A confocal microscope was used for observation and photography.
Antibody Specificity Assay.
Briefly, frozen tissue was air-dried, then fixed with 3% paraformaldehyde/PBS for 5 min. After washing in PBS, samples were permeabilized with 0.1% Triton X-100/PBS. Sections were incubated in 3% H2O2/methanol for 30 min to block endogenous peroxidases before blocking unspecific bindings with normal blocking serum (0.1 M glycine/1% bovine serum albumin/5% goat normal serum/PBS) for 20 min. Slides were incubated overnight at 4°C in a humidified chamber with a 1:125 diluted hCNT1 primary antibody incubated previously with hCNT1 peptide (residues 48–69) at a final concentration of 25 μg/μl for 15 h at room temperature in a shaker. After two washes in PBS, a 1:200 diluted second antibody Anti-rabbit IgG biotin conjugate (Sigma) was added for 30 min at room temperature. Slides were then incubated with a 1:250 dilution of Streptavidin horseradish peroxidase-conjugated (Zymed Laboratories, South San Francisco, CA)/PBS for 30 min. Samples were revealed with aminoethyl carbazole substrate (Zymed). Finally, specimens were counterstained with Mayer's hematoxylin and mounted the coverslips with an aqueous mounting medium.
Cytotoxicity Assays.
The cytotoxicity assays were performed by seeding the three cell lines, CHO-K1 wild-type (CHO-K1 WT), CHO-K1 transfected with the vector alone (CHO-K1 PC), and CHO-K1 cells transfected with the hCNT1 transporter cDNA (CHO-K1 HC1), at a density of 2500 cells/cm2. Twenty-four hours after seeding, cultures were exposed for 90 min to the cytotoxic agents (capecitabine, 5′-DFUR, 5′-FU or cisplatin). Viability was assessed 24, 48, 72, and 96 h after the addition of the putative cytotoxic drugs by counting the cells in the monolayer after trypsinization of the cultures (Multisizer; Beckman Coulter, Inc., Fullerton, CA). Viability determinations using trypan blue exclusion gave identical results (not shown). Viability was then calculated on a percentage basis by comparing the number of viable cells after treatment with that of a culture that had not been exposed to any cytotoxic drug.
Results
Cloning of the hCNT1 cDNA from Human Fetal Liver.
A 2.1-kilobase pair fragment was amplified and cloned from a human fetal liver cDNA library, as indicated under Materials and Methods. This fragment contained the hCNT1 cDNA as deduced from sequencing of both ends of the fragment. Amplification did not result in the incorporation of any mutation inside the cDNA sequence and, as indicated below, resulted in a functional protein when expressed heterologously. However, this cDNA contained a 116-bp segment at the 5′ end before the ATG start codon that is not present in the original reported sequence, which was obtained from adult human small intestine. This new sequence (GenBank number AF309632) may be the result of different RNA processing or splicing or alternate use of promotors in the hCNT1 gene, which may be either tissue-specific or developmentally regulated, as long as this cDNA was indeed isolated from a different tissue (liver instead of small intestine) but also from fetuses instead of adults.
Heterologous Expression of the hCNT1 cRNA in Oocytes fromX laevis.
hCNT1 expression in X laevisoocytes was monitored by the two-electrode voltage-clamp technique, and representative currents generated by cytidine, a natural pyrimidine nucleoside and nucleoside derivative, are shown in Fig.1. Cytidine and 5′-DFUR induced inward currents, whereas neither capecitabine nor 5′-FU induced any significant current, in the hCNT1-expressing oocytes. Similar results were obtained with oocytes from two different frogs. The concentration dependence of Na+-dependent induced currents generated by cytidine and 5′-DFUR was also studied. A representative kinetic analysis is shown in Fig. 2. Although V max values varied among hCNT1-expressing oocytes, the K m values were reproducible. Apparent K m values derived from these measurements were lower for cytidine than for 5′-DFUR, but the current intensity generated by 5′-DFUR was markedly higher.
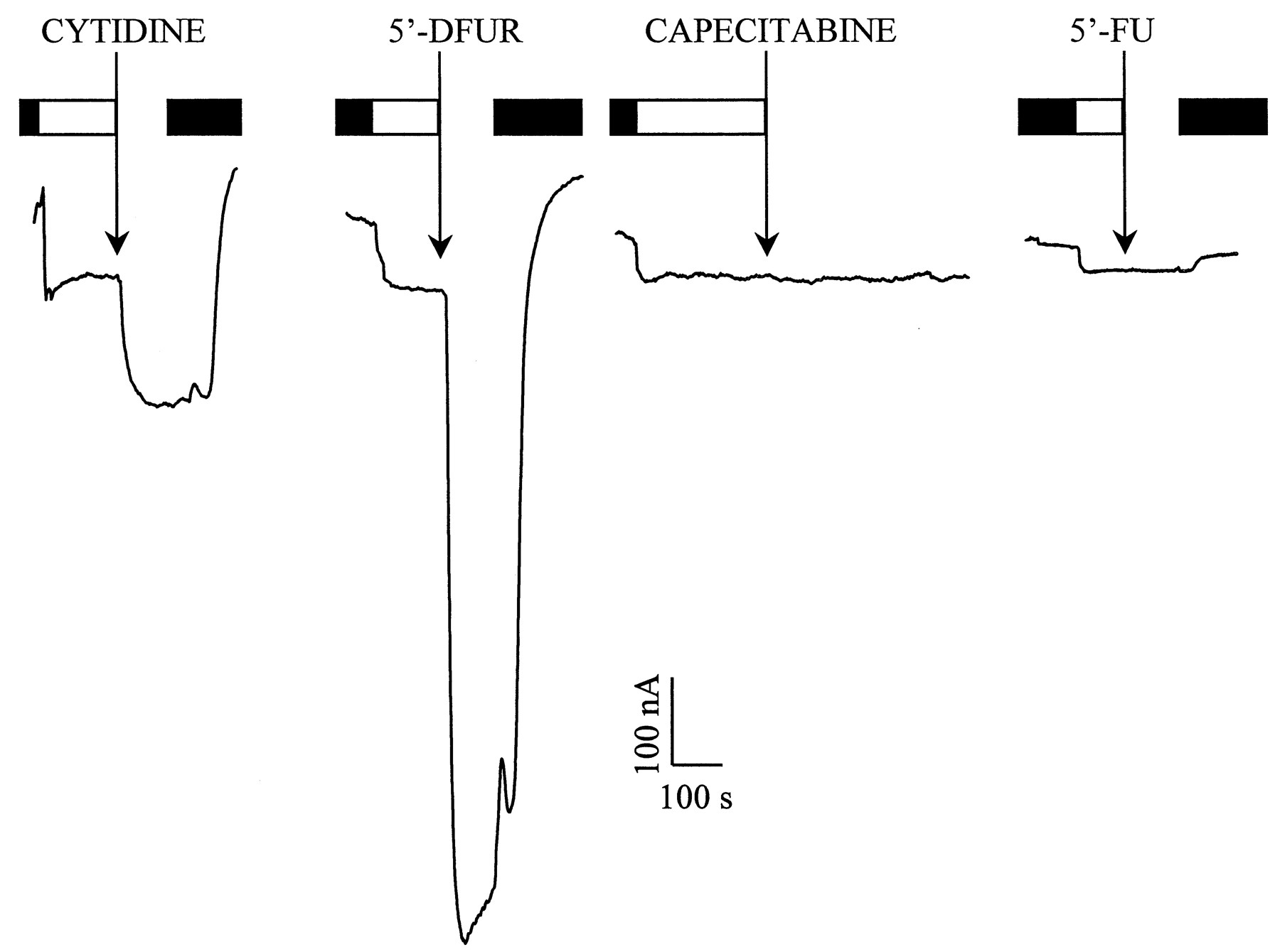
Na+ currents generated by hCNT1 in the presence of cytidine and nucleoside derivative drugs. Oocyte was continuously perfused with Na+ buffer (open box). The addition of 0.5 mM cytidine or 5′-DFUR induced an inward current of 151 and 764 nA, respectively. Capecitabine or 5′-FU (0.5 mM) did not induce any current. After the perfusion of each substrate, the oocyte was washed out in Na+-free medium (filled box). 5′-FU was tested in a different oocyte in which 0.5 mM uridine induced 52 nA of inward Na+ current. Both oocytes were used 5 days after injection with hCNT1 cRNA and the membrane potential was held at −50 mV.

Na+ inward current generated by 5′-DFUR and cytidine as a function of their concentrations. Experiment was performed on a single oocyte, 6 days after injection with hCNT1 cRNA. The membrane potential was held at −50 mV. Kinetic constants were obtained by fitting the currents obtained at 6 to 7 different concentrations (from 10 to 1000 μM) to the equation underMaterials and Methods. Dashed lines correspond to the predicted curves using the kinetic constants.
Expression of hCNT1 in CHO-K1 Cells.
The hCNT1 transporter was also expressed in CHO-K1 cells. Selected clones were checked for Na+-dependent nucleoside transport activity as a functional measurement of hCNT-1 expression, because wild-type CHO-K1 cells only express equilibrative nucleoside transport system(s). Up to six independent clones were analyzed. All had significant Na+-dependent uridine transport activity (not shown), but clone 1 was chosen for further experiments because of its high Na+-dependent uridine transport activity (Fig. 3A). CHO-K1 cells transfected with the pCDNA3 vector alone (without hCNT1) showed similar uptake rates to the wild-type (Fig. 3A.). Nevertheless, to check for the pyrimidine-preferring nature of the expressed activity, the classical feature of the hCNT1 transporter, a panel of putativecis-inhibitors of Na+-dependent nucleoside transport was analyzed (Fig. 3B.). Cytidine completely inhibited Na+-dependent uridine uptake, whereas Formycin-B (somehow a specific substrate for hCNT2, a purine-preferring transporter) did not exert any significant inhibition on Na+-dependent uridine transport. In agreement with the electrophysiological data, 5′-DFUR, but not capecitabine, inhibited hCNT1-mediated uptake. The Na+-independent components of nucleoside transport were also analyzed in all three cell lines, (Fig.4). This was done to determine whether the heterologous expression of the hCNT1 transporter modified the endogenous transport activity. Wild-type CHO-K1 cells only express an equilibrative transport activity, which, according to the inhibition pattern, is of the es type, because it is mostly inhibited by low concentrations of nitrobenzylthioinosine and dipyridamole (not shown). es-Type transport activity was the same in the three cell lines (wild-type, CHO-K1 cells transfected with the vector alone, and clone 1) expressing the hCNT1 transporter (Fig. 4).

Characterization of uridine transport activity in CHO-K1 cell line stably transfected with hCNT1 gene. CHO-K1 cell types (WT, wild-type; PC, vector alone; HC1, hCNT1) cultured as described under experimental procedures were incubated with 1 μM uridine either in a NaCl or a choline chloride medium for 90 s. Values are the mean ± S.E. of three independent experiments. A, characterization of uridine transport activity in the different cell types. The transfection of hCNT1 gene into CHO-K1 induces a previously nonexistent Na+- dependent uridine uptake. B, inhibition profile of hCNT1 induced Na+-dependent transport activity by 100 μM putative cis-inhibitors. Na+-dependent transport activity was calculated by subtracting the rates measured in choline chloride medium from those obtained in the Na+-medium.
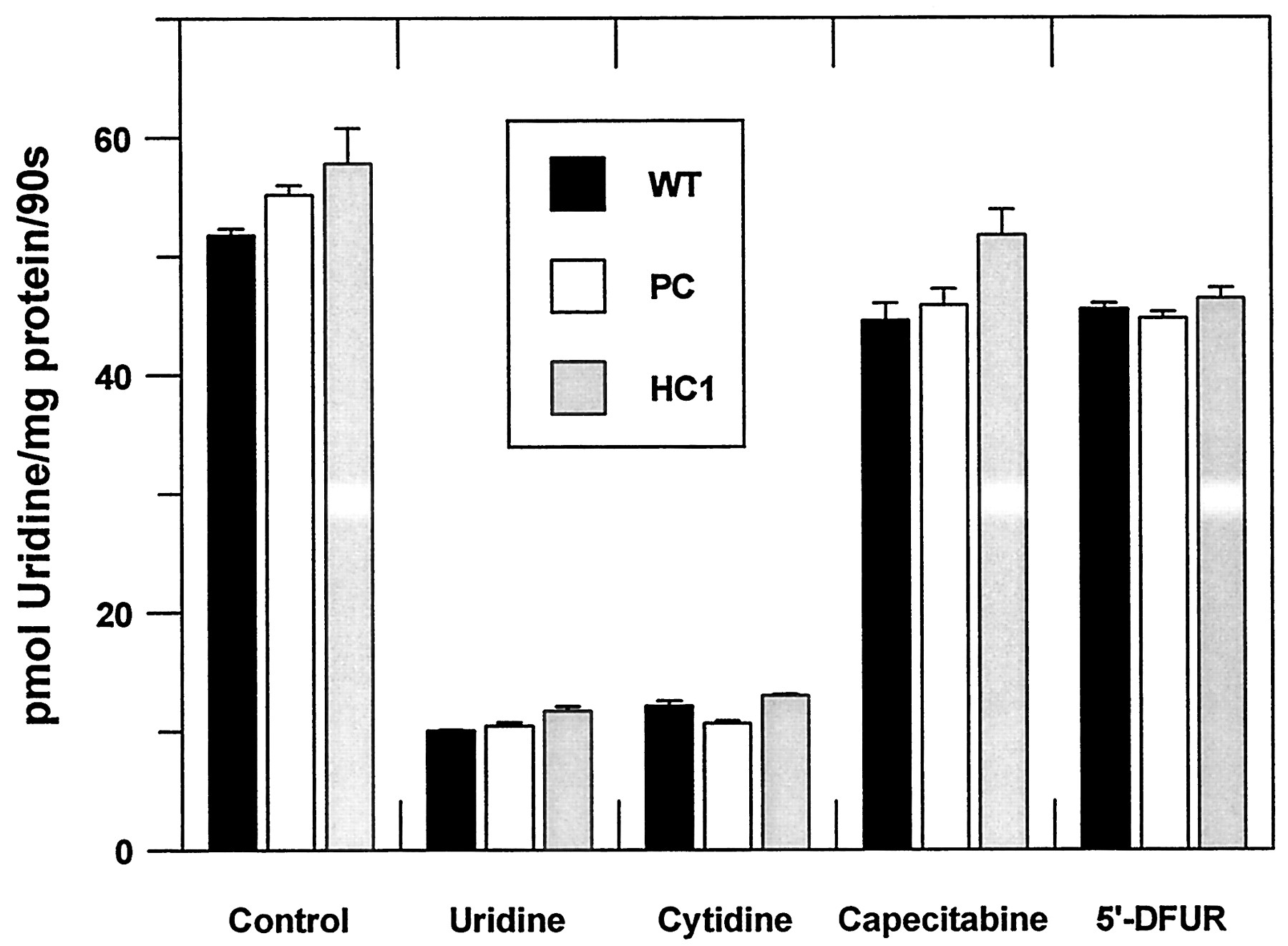
Inhibition profile of Na+-independent transport activity in the three cell types. CHO-K1 cells (WT, wild-type; PC, vector alone; HC1, hCNT1) were incubated, as indicated under Materials and Methods, in the presence of 1 μM uridine in a Na+-free transport medium (choline chloride). Values are the mean ± S.E. of three independent experiments.
Further demonstration of hCNT1 expression in CHO-K1 cells came from the analysis of hCNT1 mRNA by Northern blot using the previously cloned hCNT1 gen as probe (not shown) and from immunocytochemistry (Fig.5). Only cells functionally expressing an hCNT1-type of transporter reacted with the antibody generated. Significant staining inside the cells may be the result of the overexpression of the transporter protein in this stable cell model. Wild-type cells and cells transfected with the empty vector did not show significant immunoreactivity (Fig. 5), nor did the preimmune antiserum from the same rabbit (data not shown). Obviously, the hCNT1 mRNA was not detected in WT and PC cells. To provide independent evidence of the selectivity of the antibody raised against the hCNT1 transporter, biopsies from human intestine were stained in either the absence or the presence of an excess of the synthetic peptide, which successfully blocked the immunoreactivity, further demonstrating the specificity of this monospecific polyclonal antibody (Fig.6).

Immunocytochemistry of hCNT1 protein in the three independent clones. In this representative result, no positive signal is present in wild-type (A) or in vector transfected (B) cell clone. In contrast, the antibody raised against the protein hCNT1 specifically recognizes it in the clone HC1 (C), which presents the stable expression of the hCNT1 gene.

Immunohistochemistry assay in tissue samples from human intestine, counterstained with Mayer's hematoxylin. A, histochemical staining for hCNT1. B, blockade of specific hCNT1 antibody immunoreactivity by competition with an excess of pure hCNT1 peptide.
Effect of Heterologous hCNT1 Expression on Cell Sensitivity to 5′-DFUR.
The effect of heterologous hCNT1 expression on the sensitivity of CHO-K1 cells to 5′-DFUR was analyzed. Cells expressing hCNT1 were more sensitive to 5′-DFUR (Fig.7) than the wild-type cells or cells transfected with the empty vector. In independent experiments, the expression of hCNT1 always resulted in sensitization of cells to 5′-DFUR treatment by a factor of 2.5- to 5-fold, depending on the experiment. The relative role of the endogenous equilibrative nucleoside transporters in 5-DFUR cytotoxicity was assessed by inhibiting these transport activities with 10 μM dipyridamole (Table1). Dipyridamole induced significant resistance to 5′-DFUR in wild-type CHO-K1 and empty vector-transfected cells, but it barely modified the IC50 value for 5′-DFUR in cells expressing the hCNT1 transporter (Table 1). Obviously, the hCNT1-mediated sensitivity was specific for hCNT1 substrates, because cell death induced by other genotoxic agents, such as 5′-FU and cisplatin, was independent of the expression of the hCNT1 transporter (not shown). Capecitabine had no toxic effect on either cell line (not shown).

Cytotoxicity of 5′-DFUR in the three different clones of the cell line CHO-K1. Cell viability was assessed 96 h after 90-min exposure to the drug. IC50 values of 648 μM ± 66, 509 μM ± 32, and 207 μM ± 10 are from wild-type (WT), vector alone (PC), and hCNT1(HC1)-transfected cells, respectively. These results are the mean of three independent experiments.
Relationship between hCNT1 expression and cytotoxic effect of 5′-DFUR
Discussion
This study identifies the plasma membrane transporter responsible for 5′-DFUR transport. 5′-DFUR is the immediate precursor of 5′-FU and is taken up by solid tumors after being endogenously synthesized in patients treated with the prodrug capecitabine. As indicated above, capecitabine is one of the most novel chemotherapeutical agents rationally designed to generate the drug 5′-FU in a relatively tumor-specific manner (Cassidy 1999; Verweij, 1999; Villalona-Calero et al., 1999; Van Cutsem et al., 2000). This is achieved because the enzyme converting 5′-DFUR into 5′-FU, thymidine phosphorylase, is highly expressed in tumors (Yoshimura et al., 1990). Nevertheless, the activity of this enzyme is highly variable, which may have some treatment prognostic relevance, especially when it is referred to that of the 5′-FU inactivating enzyme dihydropyrimidine dehydrogenase (Mori et al., 2000).
Advantage was taken of the ability of the human pyrimidine-preferring nucleoside transporter 1 (hCNT1) to generate substrate-evoked currents, when heterologously expressed in X laevis oocytes, to determine whether capecitabine or its endogenously synthesized metabolites are hCNT1 substrates. The use of electrophysiology in the elucidation of the pharmacological profile of the CNT transporters has been developed very recently (Mackey et al., 1999; Lostao et al. 2000;Dresser et al., 2000) and opens the possibility for rapid and massive screening for uptake of newly developed nucleoside-derived drugs, as shown here.
This study suggests that the transporter involved in 5′-DFUR uptake also plays a role in determining drug sensitivity. Although extensive recent work has reported the positive effects of heterologously expressing thymidine phosphorylase on promoting sensitivity to 5′-DFUR (Patterson et al., 1995; Kato et al., 1997; Evrard et al., 1999), here it is shown that the expression of a specific transporter protein also promotes increased sensitivity to this prodrug. Stable expression of hCNT1 confers CHO-K1 cells with increased sensitivity to 5′-DFUR. This effect is significant and specific to this compound, because other nucleoside-derived and non–nucleoside-derived genotoxics do not show increased therapeutic indexes associated with hCNT1 expression. Moreover, this effect is also specific to the hCNT1 transporter because its heterologous expression has no effect on the endogenous transport activity, which is mostly of the es-type. However, the endogenous equilibrative transporter is also likely to contribute to the cytotoxic action of 5′-DFUR because 1) cells not expressing hCNT1 also die after treatment and 2) inhibition of this transport by dipyridamole results in strong resistance to the prodrug. Nevertheless, the evidence provided here, which demonstrates that blocking of the endogenous nucleoside transport activity has little effect on 5′-DFUR cytotoxicity in hCNT1-expressing cells, is consistent with the concept that in cells coexpressing equilibrative and concentrative nucleoside transporters, the CNT proteins (in this specific case the hCNT1 isoform) are key determinants of nucleoside bioavailability and cytotoxicity. This is in agreement with the observation that the IC50 value for the cytotoxic action of 5′-DFUR on hCNT1-expressing CHO-K1 cells closely resembles the reported apparentK m value of the hCNT1 transporter for this compound, irrespective of the endogenous equilibrative nucleoside transport activity. On the other hand, the lack of effect of capecitabine on the three cell lines tested may be a result of the need for metabolic activation or, more likely, the inability of CHO-K1 cells to take up this compound efficiently. This is based upon the evidence that capecitabine is not an hCNT1 substrate and the fact that capecitabine did not significantly inhibit the endogenous equilibrative nucleoside transport activity. In fact, the identification of the capecitabine transporter(s) is still an open question that is currently being addressed in our laboratory.
The evidence that 5′-DFUR-evoked currents are greater than those evoked by the natural substrate cytidine, suggests that, whereas the binding of the prodrug to the transporter may be less efficient, once bound, translocation occurs more rapidly than when cytidine is the substrate.
The reported cytotoxic effect of 5′-DFUR may indeed be much lower in CHO-K1 cells than in human tumor cells, because this cell line, although easy to transfect, select, and grow, is neither human nor tumoral in origin and may show very low thymidine phosphorylase activity. This suggests that the role of the transporter in drug bioavailability and cytotoxicity may be even greater in cells expressing high thymidine phosphorylase-specific activity.
hCNT1 may determine cell toxicity to a variety of fluoropyrimidines, because it has also been demonstrated that gemcitabine is an hCNT1 substrate (Mackey et al., 1998; 1999; Lostao et al., 2000). It is probable that the expression levels of this protein may determine drug bioavailability and cytotoxicity. In this context, the present study also provides the characterization of a monospecific polyclonal antibody against the human isoform of CNT1, which may prove useful in further clinical studies using human biopsies from patients treated with capecitabine. We have shown that CNT expression, at least in liver parenchymal cells, depends on the differentiation status of the cell (del Santo et al., 1998) and in a transgenic rat model of hepatocarcinogenesis, we have demonstrated a selective loss of CNT1 expression in adenomas and hepatocarcinomas (Dragan et al., 2000). Unfortunately, the antibody used in the present study did not cross-react with human tissue and so the provision of a new antibody against the human isoform of this drug transporter is of interest. Evidence that CNT expression depends on differentiation and/or proliferation has also been provided in other cell models, such as human B lymphocytes (Soler et al., 1998, 2000) and cell lines derived from human pancreatic tumors (J. Garcı́a-Manteiga, J. Calbó, A. Felipe, F. J. Casado, A. Mazo, and M. Pastor-Anglada, submitted).
In summary, this study demonstrates that the human concentrative nucleoside transporter hCNT1 is the 5′-DFUR transporter and its expression confers sensitivity to the drug. We also provide the characterization of an hCNT1 antibody, which may be useful in further clinical studies.
Footnotes
- Received October 11, 2000.
- Accepted January 31, 2001.
-
Send reprint requests to: Dr. Marçal Pastor-Anglada Department of Biochemistry and Molecular Biology University of Barcelona Diagonal 645, E-08028 Barcelona, Spain. E-mail:mpastor{at}porthos.bio.ub.es
-
This study was mainly funded by Grants SAF99–0115 and 2FD1997–1268 (Plan Nacional de I+D, Plan Nacional de Salud) (M.P.-A.) and partially supported by Programa Praxis XXI-F.C.T. (Portugal) (J.F.M) and PIUNA (Universidad de Navarra, Spain) (M.P.L.).
Abbreviations
- 5′-FU
- 5′-fluorouracil
- 5′-DFUR
- 5′-deoxy-5-fluorouridine
- dThdPase
- thymidine phosphorylase
- ENT
- equilibrative nucleoside transporter
- CNT
- concentrative nucleoside transporter
- CHO
- Chinese hamster ovary
- PCR
- polymerase chain reaction
- bp
- base pair(s)
- PBS
- phosphate-buffered saline
- WT
- wild-type
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}