


Abstract
Nuclear factor κB (NF-κB) is an important transcription factor in inflammation that has obtained a great interest as a drug target for the treatment of various allergic conditions. In this study, we show that the histamine H1 receptor, which is also an important player in allergic and inflammatory conditions, activates NF-κB in both a constitutive and agonist-dependent manner. Moreover, the observed constitutive NF-κB activation is inhibited by various H1-receptor antagonists, suggesting that inverse agonism may account, at least in part, for their ascribed antiallergic properties. Investigation of the H1 receptor-mediated NF-κB activation in transfected COS-7 cells indicates that the level of the observed constitutive activity of the H1 receptor can be modulated by the expression levels of either Gα-proteins or Gβγ-heterodimers. Members of the Gαq/11-family of Gα-proteins are most effective in increasing H1constitutive activity. Also, coexpression of Gβ2 in combination with either Gγ1 or Gγ2 results in an increased constitutive activity of the H1 receptor, whereas scavenging of Gβγ-subunits by coexpression of Gαt completely neutralizes the constitutive, but not the agonist-induced, NF-κB activity. Our data suggest that both Gαq/11- and Gβγ-subunits play a role in the agonist-induced, H1 receptor-mediated NF-κB activation, but that constitutive NF-κB activation by the H1 receptor is primarily mediated through Gβγ-subunits.
The Gαq/11-coupled histamine H1 receptor (Gutowski et al., 1991; Leopoldt et al., 1997) is expressed in a variety of cells and is believed to mediate many of the histamine-induced symptoms of allergic reactions by coupling to different signaling pathways. Consequently, during the past 20 years, the H1-receptor antagonists have become one of the most prescribed drug families in Western countries (Woosley, 1996) to relieve the symptoms of allergic reactions.
NF-κB is a ubiquitous transcription factor that can be activated by many stimuli and is considered to play an important role in inflammatory processes (Barnes et al., 1998). The physiological roles, including lymphocyte activation and protection from apoptosis, for the heterogeneous family of NF-κB proteins and activators thereof have recently gained much interest (Newton and Decicco, 1999). Elevated levels of activated NF-κB are found in persons with asthma (Hart et al., 1998), and NF-κB is therefore thought to play a pivotal role in asthma (Barnes and Adcock, 1997). Elucidation of activation mechanisms of NF-κB proteins is expected to lead to significant advances in our understanding of a variety of inflammatory disorders, including asthma and allergy, as well as their treatment. Much progress has already been made in unraveling pathways activating NF-κB via receptor tyrosine kinases. However, several additional activation pathways have recently been identified, many of which can be activated by G-protein-coupled receptors (GPCRs; Shahrestanifar et al., 1999; Xie et al., 2000;Casarosa et al., 2001). An increasing number of GPCRs, including histamine H1 receptors (Aoki et al., 1998; Hu et al., 1999), have been shown to activate NF-κB. However, little is known about the signal-transduction pathways leading to GPCR-mediated NF-κB activation.
We have shown in a previous study constitutive activation of phospholipase C (PLC) by the human histamine H1receptor (Bakker et al., 2000). Moreover, we could show that the agonist-independent accumulation of inositol phosphates is selectively inhibited by several therapeutics formerly known as H1 antagonists, which has led to the reclassification of various H1 antagonists as inverse H1 agonists. In this report, we show that H1 agonists induce NF-κB activation via the H1 receptor through activation of Gαq/11-proteins. Furthermore, in H1 receptor-expressing cells, we observed constitutive activation of NF-κB via a pathway that involves the liberation of Gβγ-subunits that is independent of PLC activation, indicating that constitutive GPCR signaling may not represent the same signaling mechanisms that are deployed in agonist-induced GPCR signaling. All tested H1 antagonists, including those that are used clinically to relieve symptoms of allergy, inhibited the constitutive activation of NF-κB, which means that these drugs act as inverse H1 agonists. The observed inhibition of the constitutive NF-κB activation by inverse H1 agonists may therefore represent a new mechanism of action of this important class of antiallergic drugs.
Experimental Procedures
Materials.
pNF-κB-Luc was obtained from Stratagene (La Jolla, CA). Doxepine hydrochloride, mepyramine (pyrilamine maleate), (S)-(+)-α-fluoromethylhistidine hydrochloride, and tripelennamine hydrochloride were obtained from Sigma/RBI (Natick, MA). ATP disodium salt, bovine serum albumin, chloroquine diphosphate, cholera toxin, DEAE-dextran (chloride form), histamine dihydrochloride, pertussis toxin, phorbol 12-myristate 13-acetate, polyethyleneimine, triprolidine hydrochloride, and Tween-20 were purchased from Sigma Chemical (St. Louis, MO). d-Luciferin was obtained from Duchefa Biochemie BV (Haarlem, The Netherlands), glycerol from Sigma-Aldrich Laborchemikalien (Seelze, Germany), and Triton X-100 from Fluka (Buchs, Switzerland). Cell culture media, penicillin, and streptomycin were obtained from Invitrogen (Merelbeke, Belgium). Fetal calf serum (FCS) was obtained from Integro BV (Dieren, The Netherlands), dialyzed fetal calf serum was obtained from HyClone Laboratories (Logan, UT). [3H]Mepyramine (30 Ci/mmol),myo-[2-3H]inositol (17 Ci/mmol), and the ECL reagents were from Amersham Pharmacia Biotech UK, Ltd. (Little Chalfont, Buckinghamshire, UK). Mouse anti-Gαt-1-subunit (bovine) was obtained from Calbiochem (San Diego, CA), goat anti-mouse IgG (H+L)-horseradish peroxidase conjugate and 30% acrylamide mix from Bio-Rad (Hercules, CA), and the protein molecular mass marker [7708S, prestained protein marker, broad range (6–175 kDa); Beverly, MA]. Clobenpropit, 4-methyldiphenhydramine, and 4,4-dimethyldiphenhydramine were taken from our own stock.
Gifts of 2-(3-trifluoromethyl)phenylhistamine dihydrogenmaleate [Dr. Schunack (Leschke et al., 1995)], acrivastine (The Wellcome Foundation Ltd., London, UK), (R)- and (S)-cetirizine hydrochloride (UCB Pharma, Brussels, Belgium), diphenhydramine hydrochloride (DSM Fine Chemicals, Sittard, The Netherlands), ebastine (Almirall Prodesfarma, Barcelona, Spain), epinastine hydrochloride (Roche Molecular Biochemicals, Mannheim, Germany), KW 4679 (Pharmaceutical Research Laboratories, Shizuoka, Japan), loratadine (Schering Plough, Kenilworth, NJ), mianserine hydrochloride (Organon NV, Oss, The Netherlands), mizolastine (Synthelabo Recherche, Bagneux, France), pcDEF3 (Dr. J. Langer), and (+)-terfenadine carboxylate and (−)-terfenadine carboxylate (Sepracor, Marlborough, MA), ranitidine dihydrochloride (GlaxoSmithKline, Uxbridge, Middlesex, UK), and of the cDNAs encoding mouse Gα11 and Gα11Q209L (Dr. H. Umemori), mouse Gαq (Dr. B. Conklin), bovine Gαt (Dr. B. Defize), Gα12Q229L and Gα13Q226L (Dr. Dhanasekaran), GαoQ205L (Dr. Iyengar), bovine Gγ1 and Gγ2, human Gβ1 and Gβ5 (Dr. M. Lohse), Gβ2, bovine GRK2 and GRK2K220R (Dr. S. Cotecchia), and the human histamine H1 receptor [Dr. Fukui (Fukui et al., 1994)] are greatly acknowledged.
Cell Culture and Transfection.
COS-7 African green monkey kidney cells were maintained at 37°C in a humidified 5% CO2/95% air atmosphere in either Dulbecco's modified Eagle's medium containing 2 mM l-glutamine, 50 IU/ml penicillin, 50 μg/ml streptomycin, and 5% (v/v) FCS or in Opti-MEM I medium with GlutaMAX I (both from Invitrogen) containing 50 IU/ml penicillin, 50 μg/ml streptomycin, and 0.5% (v/v) dialyzed FCS. COS-7 cells were transiently transfected using the DEAE-dextran method. The total amount of DNA transfected was maintained constant by addition of either pcDEF3 or pcDNA3. In coexpression experiments with Gα11 or Gα11Q209L, the H1 receptor-expression level was diminished by 10, 20, or 40%, respectively, whereas coexpression of Gαt did not affect H1receptor expression, as monitored by [3H]mepyramine binding experiments.
[3H]Inositol Phosphate Formation.
Cells were seeded in 24-well plates and 24 h after transfection labeled overnight in inositol-free culture medium supplemented with 1 μCi/mlmyo-[2-3H]inositol. Subsequently, the medium was aspirated and cells were incubated with drugs for 1 h at 37°C in Dulbecco's modified Eagle's medium containing 25 mM HEPES, pH 7.4, and 20 mM LiCl. Incubations were stopped by aspiration of the culture medium and the addition of cold 10 mM formic acid. After 90 min of incubation at 4°C, [3H]inositol phosphates were isolated by anion exchange chromatography and counted by liquid scintillation.
Reporter-Gene Assay.
Cells transiently cotransfected with pNFκB-Luc (125 μg/1 × 107 cells) and either pcDEF3 or pcDEF3hH1 (25 μg/1 · 107 cells) were seeded in 96-well blackplates (Costar, Cambridge, MA) in serum-free culture medium and incubated with drugs. After 48 h, cells were assayed for luminescence by aspiration of the medium and the addition of 25 μL/well luciferase assay reagent [0.83 mM ATP, 0.83 mM d-luciferin, 18.7 mM MgCl2, 0.78 μM Na2H2P2O7, 38.9 mM Tris, pH 7.8, 0.39% (v/v) glycerol, 0.03% (v/v) Triton X-100, and 2.6 μM dithiothreitol]. After 30 min, luminescence was measured for 3 s/well in a TopCount (Packard Instrument Co., Meriden, CT) or a Victor2 (PerkinElmer Wallac, Gaithersburg, MD).
Western Blot Analysis.
To assay the expression of Gαt, transfected COS-7 cells were washed twice with cold phosphate-buffered saline (PBS) and lysed with lysis buffer (PBS containing 1% Nonidet P40, 1 mM phenylmethylsulfonyl fluoride, 0.1% SDS, 0.5% sodiumdeoxycholate, 2 μg/ml aprotinin, and 2 μg/ml leupeptin). Insoluble material was pelleted by centrifugation, and supernatants were removed. Samples were prepared for electrophoresis by mixing 25-μg cell protein extracts with loading buffer [350 mM Tris-HCL, pH 6.8, 30% glycerol (v/v), 10% SDS (w/v), 600 mM dithiothreitol, and 0.01% bromphenol blue] and denatured (5 min, 100°C), followed by centrifugation for 3 min at 15,000g. Proteins were separated by SDS-polyacrylamide (12%) electrophoresis according to Laemmli (1970) and analyzed by Western blotting. Western blotting was carried out as described (Navon and Fung, 1988). Briefly, the gel was blotted onto a nitrocellulose filter, after which the washed blot (1 h at room temperature in PBS-T [PBS containing 0.1% Tween-20, 3% dry milk (w/v)]) was subsequently incubated overnight at 4°C with the mouse anti-Gαt-1-subunit in PBS-T (1:2000); after washing the blot with PBS containing 0.1% Tween-20, it was incubated for 1 h with the goat anti-mouse IgG (H+L)-horseradish peroxidase conjugate (1:7500) at room temperature. Protein bands were detected by film exposure using ECL chemoluminescence and subsequently quantified using Image Quant (version 1.1; Molecular Dynamics, Sunnyvale, CA).
Histamine H1 Receptor Binding Studies.
The transfected COS-7 cells used for radioligand binding studies were harvested after 48 h and homogenized in ice-cold H1-binding buffer. The COS-7 cell2homogenates were incubated for 30 min at 25°C in 50 mM Na2/K-phosphate buffer, pH 7.4, in 400 μL with 1 nM [3H]mepyramine. The nonspecific binding was determined in the presence of 1 μM mianserin. The incubations were stopped by rapid dilution with 3 ml of ice-cold 50 mM Na2/K-phosphate buffer, pH 7.4. The bound radioactivity was separated by filtration through Whatman GF/C filters that had been treated with 0.3% polyethyleneimine. Filters were washed twice with 3 ml of buffer, and radioactivity retained on the filters was measured by liquid scintillation counting. Binding data were evaluated by a nonlinear, least-squares curve-fitting procedure using GraphPad Prism (GraphPad Software, Inc., San Diego, CA).
Analytical Methods.
Proteins concentrations were determined according to Bradford (1976), using bovine serum albumin as a standard. All data shown are expressed as means ± S.E.M., statistical analyses were carried out by Student's t test. pvalues < 0.05 were considered to indicate a significant difference (*, p < 0.05; **, p < 0.01; and ***, p < 0.001).
Results
NF-κB Activation by the Histamine H1 Receptor.
In COS-7 cells expressing the H1 receptor at a density of 3.2 ± 0.4 pmol/mg of protein (n = 6), histamine stimulated NF-κB activation 3.6 ± 0.3-fold over basal with a pEC50 value of 6.8 ± 0.1 (n = 37; Table 1). Histamine did not increase NF-κB activation in mock-transfected COS-7 cells (n = 3). Also 2-[3-(fluoromethyl)phenyl]histamine, an H1selective agonist (Leschke et al., 1995) with a relatively high affinity (Table 1), increased InsP3 accumulation as well as NF-κB-driven luciferase-expression in H1-expressing cells (Table 1).
The pK i and pEC50 values of the tested agonists for the human H1 receptor and their intrinsic activities (α)
As found for the accumulation of [3H]InsP3 (Bakker et al., 2000), the basal NF-κB activation was increased upon expression of the human H1 receptor. As can be seen in Fig.1A, expression of the H1 receptor at a density of 1.4 ± 0.1 pmol/mg of protein resulted in a 2.6-fold increase of the basal luciferase expression compared with mock-transfected cells. This constitutive H1-receptor activity was inhibited by the H1 antagonist mepyramine, whereas mepyramine had no effect on NF-κB activation in transfected COS-7 cells that did not express the human H1 receptor (n = 5; Fig. 1A). Mepyramine inhibited the constitutive NF-κB activation by 78 ± 1% (α = −0.97 ± 0.01) with a pIC50 value of 7.9 ± 0.1 (n = 55; Table 2). Whereas increased H1-receptor expression led to a further rise in the basal response, the pIC50value of mepyramine was unaffected by varying the H1-receptor density from 1 to 4 pmol/mg of protein (Fig. 1A). Furthermore, the constitutive activity of the human H1 receptor was unaffected by the inverse H2 agonist ranitidine and the H3 receptor antagonist clobenpropit (Fig. 1B).

A, effects of mepyramine on the basal luciferase activity in mock-transfected COS-7 cells (●) or in COS-7 cells transiently expressing the human histamine H1 receptor at 1 ± 0.1 (○) or 4.2 ± 0.2 (♦) pmol/mg of protein. B, effects of mepyramine (●), ranitidine (○), and clobenpropit (♦) on NF-κB activation in COS-7 cells transiently expressing the human histamine H1 receptor (3.2 ± 0.4 pmol/mg of protein). C, inhibition of constitutive H1-receptor activity, as measured by the reporter-gene assay, by (R)-cetirizine (○) and (S)-cetirizine (●) in COS-7 cells transiently expressing the human histamine H1 receptor (3.2 ± 0.4 pmol/mg of protein). Representative experiments performed in triplicate are shown, each experiment was repeated at least three times.
The pK i and pIC50 values of the tested inverse agonists for the human H1 receptor and their intrinsic activities (α)
The H1 receptor is known to exhibit stereospecificity toward the enantiomers of H1antagonists, such as cetirizine (Moguilevsky et al., 1995). Indeed, the inhibition of NF-κB activation by the enantiomers of cetirizine was found to be stereospecific (Fig. 1C); (R)-cetirizine inhibits the constitutive H1 receptor activity for 59 ± 3% (α = −0.73 ± 0.04) with a pIC50 value of 8.2 ± 0.1 (n= 13; Table 2), whereas (S)-cetirizine shows an inhibition of 62 ± 3% (α = −0.77 ± 0.04) with a pIC50 value of 6.6 ± 0.1 (n= 13; Table 2).
The constitutive activation was also inhibited by a variety of other H1 ligands, including classical H1 antagonists such as diphenhydramine, tripelennamine, and triprolidine and other second-generation antihistamines such as acrivastine, ebastine, epinastine, mizolastine, and terfenadine carboxylate (Table 2), some of which are or have been in clinical use for the treatment of allergic conditions. The negative intrinsic activities of the tested compounds varied between −1.09 ± 0.06 for loratadine and −0.71 ± 0.06 for triprolidine (Table2), indicating partial inverse agonism for some of the compounds in the reporter-gene assay. In general, the potencies of the inverse agonists to inhibit constitutive H1 receptor-mediated NF-κB activation are in good agreement with their respective potencies to inhibit constitutive InsP3accumulation, although some differences are noted (Table 2).
Competitive Antagonism of Mepyramine.
Mepyramine competitively antagonized both the histamine-induced [3H]InsP3 accumulation (Fig. 2A) and NF-κB activation (Fig.2C). Schild plot analysis of the competitive antagonism by mepyramine of the histamine-induced [3H]InsP3 accumulation resulted in a pA2 value for mepyramine of 8.9 (slope = 0.86 ± 0.1, r 2 = 0.93; Fig. 2B), whereas analysis of the competitive antagonism by mepyramine of the histamine-induced NF-κB activity yielded a pA2 value for mepyramine of 8.0 (slope = 0.94 ± 0.1, r 2 = 0.93; Fig. 2D). The obtained pA2 values for mepyramine differ somewhat, but are in good agreement with the obtained pIC50 values of mepyramine as inverse agonist in the two assays (Table 2).

A, competitive antagonism of [3H]InsP3 accumulation in response to histamine (▪) in COS-7 cells expressing the human H1receptor (4.5 ± 0.5 pmol/mg of protein) by increasing concentrations of mepyramine [1 nM (■), 3 nM (▴), 10 nM (▵), 30 nM (●), 100 nM (○), 300 nM (▾), or 1 μM (▿)]. Shown is a representative example of two independent experiments, each performed in triplicate. B, Schild plot analysis of the combined data set of two independent experiments of the competitive antagonism by mepyramine of the histamine-induced [3H]InsP3 accumulation. C, competitive antagonism of luciferase activity in response to histamine (▪) in the reporter-gene assay in COS-7 cells expressing the human H1 receptor (3.2 ± 0.4 pmol/mg of protein) by increasing concentrations of mepyramine [10 nM (■), 30 nM (●), 100 nM (○), or 300 nM (▵)]. Shown is a representative example from three independent experiments, each performed in triplicate. D, Schild plot analysis of the competitive antagonism by mepyramine of the histamine-induced luciferase activity.
pKi Values for H1 receptor Antagonists.
To correlate the inverse agonist potencies with H1-receptor affinities, all tested H1 antagonists were evaluated in [3H]mepyramine displacement studies using whole cell homogenates of transfected COS-7 cells expressing the human H1 receptor. As shown in Table 2, the inverse H1 agonists display a pharmacological profile that is expected for the H1 receptor, including the known stereospecificity for the stereoisomers of cetirizine (Moguilevsky et al., 1995). Whereas the potencies as inverse agonists determined in the [3H]InsP3 assay correlate well with their respective pKi-values (Fig.3A; slope = 0.97,r 2 = 0.97, n = 6), the pKi values correlate less well with their respective potencies as inverse agonists determined in the NF-κB reporter-gene assay (Fig. 3B; slope = 0.74,r 2 = 0.68, n = 17).

Correlation graphs of the pIC50 values obtained for the inverse agonists in the [3H]InsP3 assay (A) and reporter-gene assay (B) versus the pK i values obtained by [3H]mepyramine displacement in COS-7 cells expressing the human H1 receptor (see Table 2).
Constitutive H1 Receptor Activity Is Not Caused by Endogenous Histamine.
Despite initial concern about the presence of residual agonist (Baxter and Tilford, 1995) the existence of constitutively active GPCRs and inverse agonists has now been widely accepted (Milligan and Bond, 1997). This is largely because of the identification of true antagonists (or neutral antagonists), ligands that do not affect constitutive GPCR signaling and compete with both agonists and inverse agonists for the GPCR binding site [e.g., burimamide for the H2 receptor (Smit et al., 1996; Alewijnse et al., 1998)]. Because all H1antagonists tested so far display (partial) inverse agonistic efficacy, we performed additional experiments to exclude contamination of our assays with endogenous histamine. Culturing COS-7 cells for several weeks in Opti-MEM I medium supplemented with 0.5% dialyzed serum did not affect the inverse agonistic activity of mepyramine after cotransfection of the cells and seeding them in serum-free Opti-MEM I medium. Under these conditions, mepyramine still inhibited constitutive luciferase expression with a pIC50 value of 8.0 ± 0.1 (n = 2) in H1receptor-expressing cells.
To address a putative endogenous synthesis of histamine by histidine decarboxylase (HDC) in COS-7 cells, we treated the cells with (S)-(+)-α-fluoromethylhistidine (FMH), a selective irreversible inhibitor and suicide substrate of HDC (Watanabe et al., 1990). Prolonged treatment with FMH did not affect constitutive activity of the H1 receptor. Treatment of transfected COS-7 cells expressing the human H1receptor with concentrations up to 100 μM FMH for 48 h did not affect constitutive activity of the H1 receptor. Under these conditions mepyramine inhibited luciferase expression with a pIC50 value of 8.0 ± 0.1 (n = 2). Furthermore, mepyramine also inhibited the constitutive activity of the H1 receptor in COS-7 cells that had been cultured for 5 days in the presence of 100 μM FMH (pIC50 = 8.2 ± 0.3, n = 4).
Mechanism of H1 Receptor-Mediated NF-κB Activation.
It is known that the human histamine H1 receptor is coupled to PTX- and CTX-insensitive Gq/11 proteins leading to the activation of PLC in various cell types (Leurs et al., 1994). As an initial approach to address the coupling specificity of the human histamine H1 receptor to the NF-κB pathway, we coexpressed the H1 receptor with various Gα-subunits and examined the effects of the bacterial toxins PTX and CTX, which covalently modify specific Gα-subunits. Pretreatment of the cells with either PTX or CTX or coexpression of the GTPase-deficient mutants of Gα12(Gα12Q229L), Gα13(Gα13Q226L), or Gαo(GαoQ205L) did not increase basal signaling or alter responsiveness of the H1 receptor to histamine or mepyramine (data not shown), indicating that members of Gαi/o-, Gα12-, and Gαs-families are not involved in the activation of the NF-κB pathway in COS-7 cells by the H1 receptor. Cotransfection of cDNA encoding the H1 receptor together with expression vectors carrying cDNA inserts for Gαq or Gα11 resulted in a Gα expression level-dependent increase in (constitutive) H1receptor-mediated NF-κB activation (Table3 and Fig. 4). In control experiments using cells that do not express the human H1 receptor, transfection with cDNA encoding Gα11 does not significantly alter basal activation of NF-κB (basal activation is 85 ± 5% of control). Whereas an elevated constitutive H1-receptor activity results in an increased basal signaling, the actual -fold increase upon agonist-stimulation is reduced. Thus, whereas coexpression of Gα11 results in an 2.3 ± 0.5-fold increase of constitutive H1-receptor activity (Table 3 and Fig. 4A), the histamine-induced stimulation of NF-κB activation is reduced to 1.0 ± 0.5-fold over basal levels (Table 3 and Fig. 4B). Mepyramine inhibited the Gα11-induced raise of constitutive H1 receptor-mediated activation of NF-κB completely (Fig. 4B). Expression of the constitutively active mutant Gα11Q209L resulted in an activation of NF-κB in the absence or presence of the H1 receptor. In control experiments using cells that do not express the human H1 receptor, transfection with cDNA encoding Gα11Q209L resulted in a 1.9 ± 0.1-fold increase of basal NF-κB activation. In cells coexpressing the H1 receptor and the constitutively active mutant Gα11Q209L, basal NF-κB activation is further increased, and the agonist-induced stimulation is reduced to 1.0 ± 0.5-fold over basal (Table 3). Moreover, the inverse H1 agonist mepyramine is not capable to inhibit the H1 receptor-independent activation of NF-κB by Gα11Q209L.
The pEC50 values of histamine and the pIC50 values of mepyramine for the human H1 receptor

A, effects of mepyramine on the basal luciferase activity in COS-7 cells transiently expressing the human histamine H1 receptor (3.2 ± 0.4 pmol/mg of protein) that had been cotransfected in the absence (●) or presence of 1.0 μg (○), 5.0 μg (▪), or 25 μg (■) of cDNA coding for Gα11/107 cells. B, effects of histamine (square symbols) and mepyramine (round symbols) on NF-κB activation in COS-7 cells transiently expressing the human histamine H1 receptor that had been cotransfected in the absence (filled symbols) or presence of 25 μg of cDNA coding for Gα11/107 cells (empty symbols).
Similarly, transfection of cDNA encoding the human H1 receptor together with expression vectors carrying cDNA inserts for Gα11 or Gα11Q209L resulted in an increase in constitutive H1 receptor-mediated InsP3 production (Fig.5). Cotransfection with cDNA encoding Gα11 induces a 77% increase in basal InsP3 accumulation in cells expressing the human H1 receptor compared with a 30% increase in basal InsP3 accumulation in cells not expressing the human H1 receptor. Expression of G11Q209L greatly affects basal InsP3 accumulation; the basal level of InP3 in G11Q209 l-expressing cells is 3.6-fold the level of that of human H1-expressing cells and is similar to cells expressing both G11Q209L and the human H1 receptor (Fig. 5). The elevated constitutive H1-receptor activity results in an increased basal but not agonist-induced signaling. Mepyramine inhibited the Gα11-induced but not the Gα11Q209 l-induced rise of constitutive H1 receptor-mediated activation of PLC (Fig. 5). Moreover, expression of the constitutively active mutant Gα11Q209L resulted in a similar activation of PLC in the absence (Fig. 5, inset) or presence of the human H1 receptor.

Effects of coexpression of the histamine H1 receptor with Gα11 or the constitutively active mutant Gα11Q209L (Gα11QL) subunits on constitutive and agonist-induced H1 receptor-mediated activation of PLC. COS-7 cells were cotransfected with pcDEF3H1 encoding the human histamine H1 receptor (25 μg/107 cells) together with equal amounts of cDNA coding for Gα11 or Gα11Q209L; the total amount of transfected DNA was kept constant using pcDEF3. Forty-eight hours after transfection, cells were incubated with medium to determine basal activity (■) or with 100 μM histamine (▪) or 100 μM mepyramine (░), and inositol phosphates were determined as described underExperimental Procedures. Inset, effects of expression of Gα11 or constitutively active mutant Gα11Q209L (Gα11QL) subunits on the activation of PLC in cells not expressing the human H1receptor.
Role of Gβ- and Gγ-Subunits.
Previous studies (Shahrestanifar et al., 1999; Xie et al., 2000; Casarosa et al., 2001) have indicated a role for Gβγ-subunits in the activation of NF-κB. To address the role of Gβγ-subunits in the H1 receptor-mediated NF-κB activation we coexpressed the H1 receptor either with various Gβγ-scavengers or with Gβγ-subunits. When over-expressed, Gαt is known to scavenge Gβγ-subunits and to inhibit Gβγ-mediated signaling (Clapham and Neer, 1997). Coexpression of Gαt neutralized constitutive activation of NF-κB by the H1 receptor in an expression level-dependent manner (Fig.6, A–C). When equal amounts of the cDNAs encoding the H1 receptor and Gαt were used for cotransfection of the cells, Gαt neutralized the constitutive activation of NF-κB by the H1 receptor without altering H1-receptor expression (Table 3 and Fig. 6, A and D). Depletion of free Gβγ-subunits did not prevent histamine-induced NF-κB activation (Table 3 and Fig. 6D), although the absolute increase in NF-κB activation was reduced. Although we cannot exclude the possibility of an incomplete scavenging of released Gβγ-subunits, these data suggest that signaling pathways other than those using free Gβγ-subunits are involved in the agonist-induced NF-κB activation. Similar results have been obtained in coexpression experiments using either GRK2 or the kinase deficient mutant GRK2K220R (Fig. 6A).

Effects of coexpression of the histamine H1 receptor with Gαt-subunits on constitutive H1 receptor-mediated NF-κB activation. A, COS-7 cells were cotransfected with either pcDEF3 (mock, ■) or pcDEF3H1 encoding the human histamine H1 receptor (25 μg/107 cells) together with various amounts cDNA coding for Gαt (0–25 μg/107 cells; ▪). The total amount of transfected DNA was kept constant using either pcDEF3 or pcDNA3. Inset, the effect of coexpression of the H1 receptor together with either GRK2 or the kinase deficient GRK2K220R on the constitutive H1-mediated NF-κB activation. The asterisks indicate significant differences from the H1 receptor-mediated NF-κB activation in the absence of Gαt-subunits (Gαt = 0 μg). B, Western blotting of COS-7 cells that had been cotransfected with various amounts of cDNA (0, 1, 3, 5, 15, or 25 μg/107 cells) coding for Gαt with a Gαt-specific antibody resulted in the detection of immunoreactive bands corresponding to Gαt-subunits. C, quantification of the Western blot indicates a linear relationship between the amount of transfected cDNA coding for Gαt and the expression of Gαt. D, effects of histamine (squares) and mepyramine (circles) on NF-κB activation in COS-7 cells transiently expressing the human histamine H1 receptor that had been cotransfected in the absence (filled symbols) or presence of 25 μg of cDNA coding for Gαt/107 cells (open symbols).
In coexpression studies with Gβγ-subunits, cells were cotransfected with a combination of expression vectors carrying various cDNA inserts for G-protein β- and γ-subunits. NF-κB activation is restricted to cells in which β- and γ-subunits are coexpressed together with the H1 receptor (data not shown). Therefore, a complex of free Gβγ-subunits act together to elevate constitutive H1-mediated NF-κB activation, in which specificity of the Gβγ-subunits is retained in the Gβ-subunit because both Gβ2γ1-and Gβ2γ2-subunits, but none of the other tested Gβγ-subunit combinations, elevate constitutive H1-receptor activity (Table4).
Effect of co-expression of various Gβγ-combinations on the constitutive H1 receptor-mediated NF-κB activation
We also examined the possible role of Gβγ-subunits in the constitutive H1 receptor-mediated InsP3 accumulation in transfected COS-7 cells. In contrast to the observed effects on the constitutive H1 receptor-mediated activation of NF-κB, coexpression of Gαt did not affect the constitutive H1 receptor-mediated InsP3 accumulation. The basal InsP3 level in COS-7 cells cotransfected with cDNAs encoding the human H1 receptor, and Gαt was 82 ± 13% of the basal InsP3 level in control cells expressing the same level of the human H1 receptor (100 ± 16%).
Discussion
The actual therapeutic importance of constitutive GPCR activity has not yet been fully clarified. However, for a proper evaluation of drug action this new aspect in GPCR pharmacology cannot be ignored. In view of the widespread therapeutic use of H1antagonists in allergic conditions (Woosley, 1996; Zhang et al., 1997;Handley et al., 1998), we investigated the constitutive H1-receptor activity and inverse agonistic activity of several H1 antagonists. In this study, we show that the H1 receptor activates the important pro-inflammatory transcription factor NF-κB in both a constitutive and agonist-dependent manner. Moreover, a developed NF-κB reporter-gene assay proved to be a very sensitive discriminator between positive and negative ligand efficacy for the H1 receptor. The observed pharmacological differences for mepyramine in the two assays might be due to phenomena such as receptor trafficking (Berg et al., 1998) and is part of our ongoing investigations.
As observed for other GPCRs (Samama et al., 1993; Burstein et al., 1995; MacEwan and Milligan, 1996; Smit et al., 1996), constitutive H1-receptor activity increases as the receptor or G-protein expression level increases; the inverse agonist mepyramine reduces this constitutive activity with unchanged potency. The inhibition of constitutive H1 receptor-mediated NF-κB activation is specific for H1antagonists, because an inverse H2 agonist or an H3 antagonist has no effect. Furthermore, in accordance with the known H1-receptor stereospecificity (Moguilevsky et al., 1995), the enantiomers of cetirizine display stereospecific inhibition of constitutive H1-receptor NF-κB activation. The constitutive H1 receptor-mediated NF-κB activation is inhibited by all tested, clinically used H1antagonists—including cetirizine (Zyrtec), ebastine (Kestine), epinastine (Flurinol), loratadine (Claritin), mizolastine (Mizolen), and fexofenadine (Allegra)—indicating that these drugs in fact all act as inverse H1 agonists. Previously there has been some debate about whether the presence of endogenous agonists may account for the observed constitutive GPCR signaling (Baxter and Tilford, 1995). Yet we show that in experiments with cells cultured in histamine-free medium or after treatment with FMH, a suicide inhibitor of HDC (Watanabe et al., 1990), constitutive H1-receptor activity and inverse agonism of H1-receptor antagonists is still observed. Our observations therefore show that the negative intrinsic activity of the tested H1 antagonists is a true property of the various drugs and might contribute to their proven successful therapeutic application (Zhang et al., 1997).
The discovery of inverse agonism at GPCRs has also raised the general question of potential differences in therapeutic outcome upon treatment with inverse agonists or neutral antagonists (Milligan et al., 1995;Smit et al., 1996; Milligan and Bond, 1997). An important issue that has been considered in this respect is the modulation of GPCR expression levels (Milligan and Bond, 1997; Leurs et al., 1998). Inverse agonists, but not neutral antagonists, can up-regulate constitutively active GPCRs, an effect that might not be beneficial under all circumstances. Yet, because all of the tested H1 antagonists are inverse agonists, we cannot currently address this issue experimentally. Moreover, the availability of a neutral antagonist would offer the opportunity to investigate whether inverse agonism at the H1 receptor is truly beneficial in allergic diseases. The developed NF-κB reporter-gene assay will be a useful tool to identify future neutral H1 antagonists.
The observed constitutive H1-receptor activation of NF-κB gene transcription is another very interesting feature, because both histamine acting at H1 receptors and NF-κB are known to be involved in inflammatory conditions, such as atherosclerosis (Takagishi et al., 1995; Bourcier et al., 1997) and allergy (Zhang et al., 1997; Barnes et al., 1998; Handley et al., 1998). It is attractive to speculate, therefore, that the observed (constitutive) coupling of the H1 receptor to the NF-κB pathway in transfected COS-7 cells can also be of physiological importance. By activating NF-κB, H1 receptors may participate in the regulation of gene expression under physiological and pathological conditions. In the nasal mucosa of patients suffering from allergic rhinitis (Iriyoshi et al., 1996;Hamano et al., 1998), H1-receptor mRNA is significantly up-regulated. It is tempting to consider that in these conditions the constitutive H1 receptor signaling is also increased and is at least partly responsible for some of the symptoms. Thus, constitutively active H1receptors may regulate basal NF-κB-mediated transcriptional activity.
Because a detailed characterization of the histamine-stimulated NF-κB-mediated gene transcription will extend our understanding of the molecular actions of histamine and the H1antagonists, we investigated the mechanisms of NF-κB activation by the H1 receptor to some extent. In agreement with previous findings that the H1 receptor is a Gq/11-coupled GPCR (Gutowski et al., 1991;Leopoldt et al., 1997), coexpression of either wild-type Gαq or Gα11 increases H1 receptor-mediated signaling, confirming the involvement of Gαq/11 heterotrimers in H1 agonist-mediated responses (Fig.7). Furthermore, coexpression of the constitutively active mutant of Gα11(Gα11Q209L) results in an H1 receptor-independent activation of NF-κB and PLC (Table 3 and Fig. 5), which cannot be inhibited by an inverse H1 agonist (Table 3). These data show that active Gα11-subunits can stimulate cellular signaling partners leading to NF-κB activation, possibly via protein kinase C (PKC; Shahrestanifar et al., 1999).

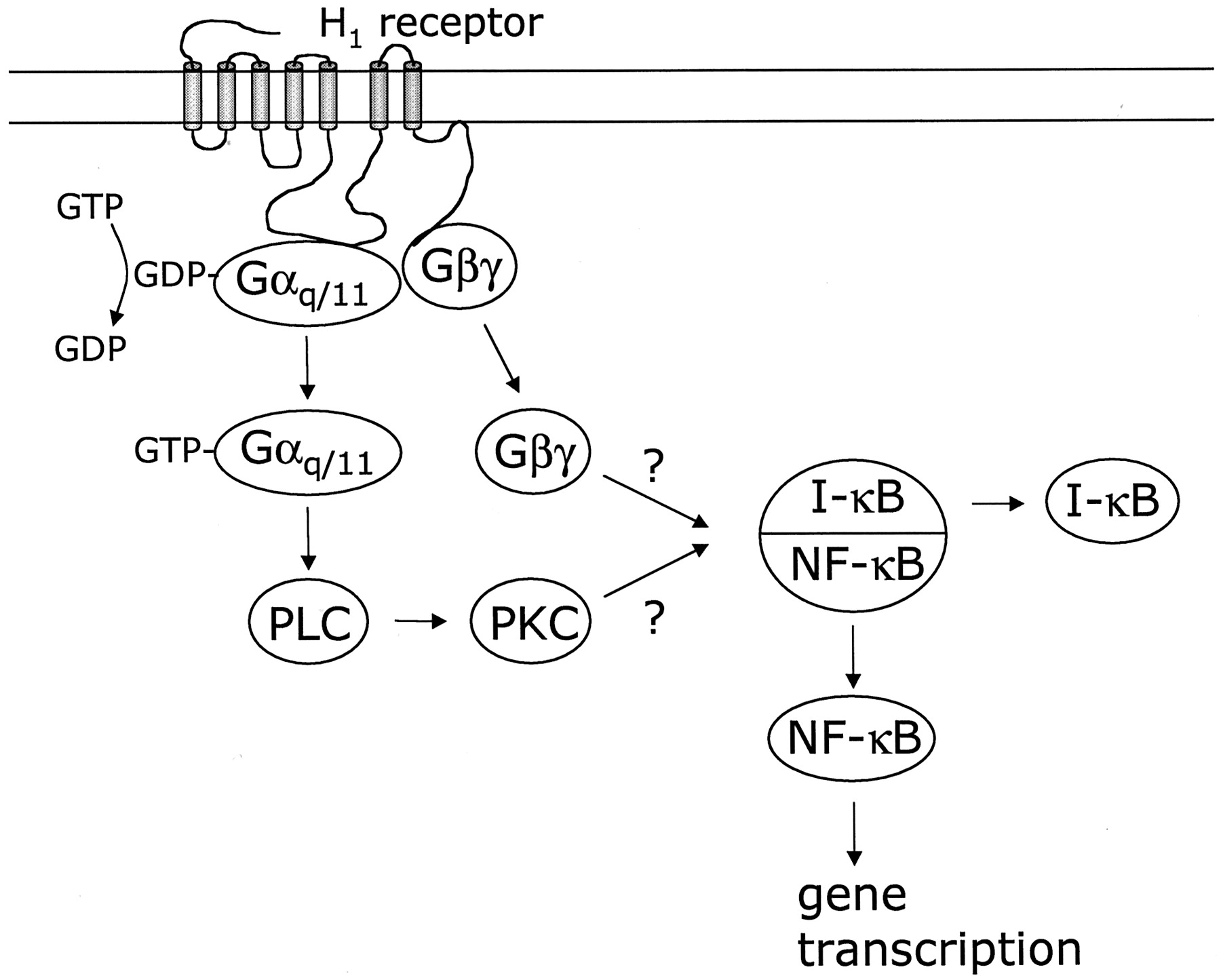
Proposed mechanism of NF-κB activation via Gα and Gβγ-subunits in COS-7 cells transiently transfected with the human histamine H1 receptor. Activation of the H1receptor induces GDP/GTP exchange on the Gα-subunit, upon which the activated Gαq/11Gβγ heterotrimer dissociates into its corresponding Gα11-subunit and Gβγ heterodimer, which can both activate cellular effectors. Activated Gα11(GTP-Gα11) activates PLC to induce the formation of InsP3 and release diacyl glycerol to activate PKC, which in turn activates the NF-κB/I-κB complex (Rothwarf and Karin, 1999) and NF-κB is translocated to the nucleus to induce gene transcription of a wide variety of genes. Also, free Gβγ-subunits activate the NF-κB/I-κB complex to induce gene transcription by the released NF-κB via an as-yet-unidentified mechanism. Our data suggest that the signal transduction pathway of agonist-mediated activation is via the Gα-subunit, whereas the constitutive H1 receptor-mediated activation is via the free Gβγ-subunits (see text).
However, several studies have indicated that GPCR-linked signal transduction pathways other than Gα-mediated PLC activation can also result in NF-κB activation, possibly via Gβγ-subunits (Xie et al., 2000; Casarosa et al., 2001). Specific Gβγ-heterodimer combinations (Gβ2γ1 and Gβ2γ2) coexpressed together with the H1 receptor indeed elevate NF-κB-activation, indicating that signal transduction pathways other than the Gα-mediated PLC activation may mediate NF-κB activation. The lack of any effect of Gβγ-dimers containing Gβ-subunits other than Gβ2 is not attributable to differences in their expression levels, because previous studies in our laboratory have shown all tested Gβ-subunits to be expressed to a similar extent upon transfection of COS-7 cells (Casarosa et al., 2001).
Likewise, by competing for free Gβγ-subunits, Gβγ-scavengers completely neutralize constitutive H1receptor-mediated NF-κB activation, confirming the involvement of Gβγ-subunits in the activation of NF-κB by GPCRs (Xie et al., 2000; Casarosa et al., 2001). Whereas the studies with coexpressed Gβγ-scavengers suggest that Gβγ-subunits may be responsible for the H1-mediated constitutive activation of NF-κB, Gβγ-scavengers do not affect the (constitutive) H1 receptor-mediated activation of PLC or the agonist-induced activation of NF-κB. The difference in basal signaling that is observed upon expression of Gβγ-scavengers may arise from a varying sensitivity of activation of the two pathways. Possibly, the pathway of activation of NF-κB activation is much less sensitive to the Gq/11 subunits and highly sensitive to Gβγ-subunits, whereas activation of PLC is mostly sensitive to Gq/11 subunits. Activation of PLC by these Gq/11 subunits could thus mask possible effects of Gβγ-subunits; therefore, no effects on PLC activation are detected upon Gβγ-subunit scavenging. A low sensitivity of PLC for Gβγ-subunits may be explained by the lack of expression of the βγ-sensitive PLC-β2 isoenzyme in COS-7 cells (Wu et al., 1993). The observation that Gβγ-scavengers completely neutralize basal signaling to NF-κB but do not totally reduce the agonist-induced response suggests that signaling pathways other than those using free Gβγ-subunits are primarily involved in the agonist-induced NF-κB activation. Although we cannot exclude the possibility that not all free Gβγ-subunits are scavenged after agonist-induced G-protein activation, these data suggest that Gαq/11-subunits are likely to be involved in this response. This suggestion is corroborated by the observations that expression of constitutively active Gαq/11Q209L results in NF-κB activation and that coexpression of the H1 receptor with Gα11strongly enhances the agonist-induced NF-κB activation. The complete reduction of basal H1-receptor activity by Gβγ-scavengers is surprising in view of the fact that some level of activated Gαq/11-subunits will be present under these conditions as well. At the moment, we have no explanation for these observations. One can speculate that for activation of the NF-κB pathway, the level of basal H1receptor-mediated PKC stimulation via Gαq/11-dependent PLC activation is insufficient. Only at higher levels of diacylglycerol production (e.g., after agonist stimulation) would the activation of PKC become important and result in the activation of NF-κB. Previously, mechanistic differences have also been reported for basal and agonist-induced activation for the phosphatidylinositide-3-OH-kinase-dependent activation of p42/p44 mitogen-activated protein kinase in a variety of different cell types (Versteeg et al., 2000).
In conclusion, we have shown that the histamine H1 receptor constitutively activates NF-κB-mediated transcription in COS-7 cells. The various tested, clinically used H1 antagonists act as inverse agonists, suggesting that inverse agonism might be part of their mechanism of action. Moreover, we have identified the initial signaling events in the H1 receptor-mediated NF-κB activation in COS-7 cells. Our data indicate that both Gαq/11- and Gβγ-subunits play a role in the H1 receptor-mediated NF-κB activation similar to NF-κB activation mediated via the bradykinin B2 receptor or US28 (Xie et al., 2000; Casarosa et al., 2001). We suggest that the constitutive H1 receptor-mediated activation of NF-κB is mediated via free Gβγ-subunits from Gαq/11-proteins, whereas histamine-mediated NF-κB activation is most likely regulated via Gαq/11-subunits. This study shows for the first time a role for Gβγ-subunits in the signal transduction of the histamine H1 receptor. Although PKC is a likely candidate for the αq/11-mediated NF-κB activation (Shahrestanifar et al., 1999) and both phosphatidylinositol 3-kinase and Akt have been shown to be involved in the Gβγ-mediated activation (Xie et al., 2000), the actual target(s) of the Gα- and Gβγ-subunits in the H1 receptor-mediated responses is at present not known and is currently under investigation.
Footnotes
- Received March 16, 2001.
- Accepted July 16, 2001.
-
Supported in part by UCB Pharma (Belgium) and the EU BIOMED 2 program Inverse Agonism: Implications for Drug Research and by the Royal Dutch Academy of Arts and Sciences (M.J.S.).
Abbreviations
- NF-κB
- nuclear factor κB
- GPCRs
- G-protein coupled receptors
- PLC
- phospholipase C
- FCS
- fetal calf serum
- PBS
- phosphate-buffered saline
- InsP3
- inositol trisphosphate
- HDC
- histidine decarboxylase
- FMH
- (S)-(+)-α-fluoromethylhistidine
- PKC
- protein kinase C
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}