


Abstract
In superior cervical ganglion neurons,N-(piperidiny-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (SR141716A) competitively antagonizes the Ca2+ current effect of the cannabinoid (CB) agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (WIN55212-2), and behaves as an inverse agonist by producing opposite current effects when applied alone. In contrast, in neurons expressing CB1 with a K→A mutation at residue 3.28(192) (i.e., K3.28A), SR141716A competitively antagonizes the effects of WIN55212-2, but behaves as a neutral antagonist by producing no current effects itself. Receptor modeling studies suggested that in the CB1 inactive (R) state, SR1417A16A stabilizes transmembrane helix 6 in its inactive conformation via aromatic stacking with F3.36/W6.48. In this binding site, SR141716A would exhibit higher affinity for CB1 R due to a hydrogen bond between the SR141716A C3 substituent and K3.28(192), a residue available to SR141716A only in R. To test this hypothesis, a “mutant thermodynamic cycle” was constructed that combined the evaluation of SR141716A affinity at WT CB1 and K3.28A with an evaluation of the wild-type CB1 and K3.28A affinities of an SR141716A analog, 5-(4-chlorophenyl)-3-[(E)-2-cyclohexylethenyl]-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole (VCHSR), that lacks hydrogen bonding potential at C3. Binding affinities suggested that K3.28 is involved in a strong interaction with SR141716A in WT CB1, but does not interact with VCHSR. Thermodynamic cycle calculations indicated that a direct interaction occurs between the C3 substituent of SR141716A and K3.28 in WT CB1. Consistent with these results, VCHSR acted as a neutral antagonist at WT CB1. These results support the hypothesis that hydrogen bonding of the SR141716A C3 substituent with K3.28 is responsible for its higher affinity for the inactive R state, leading to its inverse agonism.
To date, two subtypes of the cannabinoid receptor, CB1 (Gerard et al., 1991) and CB2 (Munro et al., 1993), have been identified. These receptors belong to the rhodopsin family of G protein-coupled receptors. CB1 and CB2 receptor agonists inhibit forskolin-stimulated adenylyl cyclase by activation of a pertussis toxin-sensitive G protein (Felder et al., 1995). In heterologous cells, CB1 but not CB2 receptors inhibit N-, P-, and Q-type calcium channels and activate inwardly rectifying potassium channels (Felder et al., 1995; Mackie et al., 1995; Pan et al., 1996). Inhibition of calcium channels and enhancement of inwardly rectifying potassium currents is pertussis toxin-sensitive, but independent of cAMP inhibition, suggestive of a direct G protein mechanism (Mackie et al., 1995).
The CB1 antagonist SR141716A (1) displays nanomolar CB1 affinity, but very low affinity for CB2. SR141716A antagonizes the pharmacological and behavioral effects produced by CB1 agonists after intraperitoneal or oral administration (Rinaldi-Carmona et al., 1994).
SR141716A (1) has been shown to act as a competitive antagonist and inverse agonist in host cells transfected with exogenous CB1 receptor, as well as in biological preparations endogenously expressing CB1. Bouaboula et al. (1997) reported that Chinese hamster ovary cells transfected with hCB1 receptor exhibit high constitutive activity at both levels of mitogen-activated protein kinase and adenylyl cyclase. Guanine nucleotides enhanced the binding of SR141716A, a property of inverse agonists. Lewis and coworkers (Pan et al., 1998) demonstrated constitutive activity of CB1 receptors in inhibiting Ca2+ currents that was not due to endogenous agonist. These investigators reported that SR141716A antagonized the Ca2+ current inhibition induced by the cannabinoid agonist WIN55212-2, in neurons heterologously expressing either rat or hCB1 receptors. Furthermore, when applied alone, SR141716A increased the Ca2+ current, with an EC50 value of 32 nM, via a pertussis toxin-sensitive pathway, indicating that SR141716A can act as an inverse agonist by reversal of tonic CB1 receptor activity (Fig. 1). Meschler et al. (2000) demonstrated that constitutive activity is demonstrable in neuronal cells that endogenously express CB1 (N18TG2 cells) and that SR141716A acts as a competitive antagonist and reduces basal activity in the manner of an inverse agonist in these cells.

A, WIN decreased and SR increased the Ca2+ current in a neuron expressing human CB1 receptors after cDNA microinjection. The double-pulse protocol was used to elicit control (○) and facilitated (●) Ca2+ currents, and current amplitudes were plotted over the time course of the experiment. Application of 1 μM WIN55,212-2 decreased the Ca2+current, which slowly recovered to a greater amplitude. A subsequent application of 1 μM SR141716A rapidly increased the Ca2+current. Inset, superimposed current traces recorded at the beginning of the experiment (control), in the presence of 1 μM WIN55,212-2, and in the presence of 1 μM SR141716A. B, summary of the changes in Ca2+ current amplitudes in the presence of 1 μM WIN55,212-2 (negative axis) or 1 μM SR141716A (positive axis) in neurons microinjected with hCB1 or mutant K3.28(192)A receptor cDNA is shown. In neurons micro-injected with hCB1 receptor cDNA (hCB1), WIN55,212-2 inhibited the Ca2+ current and 1 μM SR141716A increased the Ca2+ current. In neurons microinjected with mutant K3.28(192)A receptor cDNA (K192A), 1 μM WIN55,212-2 inhibited the Ca2+ current, but 1 μM SR141716A did not increase the Ca2+ current. In neurons microinjected with hCB1 cDNA and recorded in the absence of Ca2+, using Ba2+ as the charge carrier and BAPTA to chelate intracellular Ca2+(hCB1 Bapta), SR141716A increased the current. SR141716A also increased the current in neurons microinjected with hCB1 cDNA and preincubated with 300 nM anandamide (hCB1 Anand). This figure is reprinted with permission from Pan et al. (1998).
In some experiments, SR141716A has been found to be more potent in blocking the actions of CB1 agonists than in eliciting inverse responses by itself. For example, in their study that focused upon rat brain membrane and brain sections, Sim-Selley et al. (2001) suggested that SR141716A may bind to two sites on the cannabinoid receptor, a high-affinity site at which it exerts its competitive antagonism and a lower affinity site at which it exerts its inverse agonism.
The present study was prompted by results reported by Pan et al. (1998). These investigators found that Ca2+current was tonically inhibited in neurons expressing a mutant CB1 K3.28(192)A receptor. Surprisingly, SR141716A had no effect on the Ca2+ current in these neurons, but SR141716A could still antagonize the effect of WIN55212-2. The authors concluded that the K3.28(192) site is critical for the inverse agonist activity of SR141716A and that SR141716A seemed to become a neutral antagonist at the K3.28(192)A mutant receptor. Prompted by these intriguing SR141716A/K3.28(192)A results, we assessed through modeling studies, the proximity of K3.28(192) to SR141716A in CB1. Our modeling suggested that SR141716A has equivalent aromatic stacking interactions in both the inactive and active states of CB1, but only in the inactive state of the CB1 receptor can SR141716A (via its carboxamide oxygen) hydrogen bond with K3.28(192).
The work presented herein was designed to test the hypothesis that a direct interaction between K3.28(192) and the C3 substituent region of SR141716A is responsible for the inverse agonist activity of SR141716A at CB1. An SR141716A analog, 5-(4-chlorophenyl)-3-[(E)-2-cyclohexylethenyl]-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole (VCHSR; 2) was designed and synthesized in which the C3 substituent carboxamide trans-geometry was preserved, but in which all hydrogen bonding capability was removed. A mutant cycle was constructed in which the binding affinities of SR141716A and VCHSR at both CB1 WT and K3.28(192)A receptors were analyzed. Results of this study suggest that K3.28 is a direct interaction site for the C3 substituent of SR141716A in CB1. Furthermore, consistent with the neutral antagonism displayed by SR141716A at CB1 K3.28(192)A in a Ca2+ current assay, VCHSR, which lacks the ability to interact with K3.28(192), behaved as a neutral antagonist at WT CB1 in this same assay. This result lends further support to the hypothesis that a K3.28(192) direct interaction with the C3 substituent of SR141716A is crucial for the inverse agonist activity of SR141716A at CB1.
Materials and Methods
Molecular Modeling
A recent crystal structure of SR141716A confirms that the carboxamide of the C3 substituent is in a trans-geometry (C. George, personal communication). Complete conformational analyses of SR141716A and VCHSR were performed using the semiempirical method, AM1 within the Spartan molecular modeling program (Wavefunction, Inc., Irvine, CA). AM1 six-fold Conformer Searches were performed for the rotatable bonds in SR141716A (Fig. 2; C3 substituent: C3-C1′ and N2′-N3′; C5 substituent: C5-C1‴; N-1 substituent: N1-C1") and in VCHSR (C3 substituent: C3-C1′ and C2′-C3′; C-5 substituent: C5-C1‴; N-1 Substituent: N1-C1"). The energy separation between a smaller set of conformers was recalculated using an ab initio Hartree Fock calculation at the 6-31G* level as encoded in Jaguar (Schrödinger, Inc., Portland, OR).

Structures of SR141716A (1) and VCHSR (2).
Receptor Model Construction
Amino Acid Numbering System.
In the discussion of receptor residues that follows, the amino acid numbering scheme proposed byBallesteros and Weinstein (1995) is used. In this numbering system, the most highly conserved residue in each transmembrane helix (TMH) is assigned a locant of 0.50. This number is preceded by the TMH number and may be followed in parentheses by the sequence number. All other residues in a TMH are numbered relative to this residue. In this numbering system, for example, the most highly conserved residue in TMH 2 of the CB1 receptor is D2.50(163). The residue that immediately precedes it is A2.49(162).
Model of R Form of CB1.
A model of the R form of CB1 was created using the 2.8-Å crystal structure of bovine rhodopsin (Rho) (Palczewski et al., 2000). First, the sequence of the human CB1 receptor (Gerard et al., 1991) was aligned with the sequence of bovine Rho using the same highly conserved residues as alignment guides that were used initially to generate our first model of CB1 (Bramblett et al., 1995). TMH 5 in CB1 lacks the highly conserved proline in TMH 5 of Rho. The sequence of CB1 in the TMH 5 region was aligned with that of Rho as described previously using its hydrophobicity profile (Bramblett et al., 1995). Helix ends for CB1 were chosen in analogy with those of Rho (Palczewski et al., 2000); TMH 1: N1.28(112)-R1.61(145); TMH 2: R2.37(150)-H2.68(181); TMH 3: S3.21(185)-R3.56(220); TMH 4: T4.38(229)-C4.66(257); TMH 5: H5.34(270)-K5.64(300); TMH 6:R6.28(336)-K6.62(370); TMH 7: K7.32(376)-S7.57(401); and intracellular extension of TMH 7: D7.59(403)-C7.71(415). With the exception of TMH 1, these helix ends were found to be within one turn of the helix ends originally calculated by us and reported in Bramblett et al. (1995). Changes to the general Rho structure that were necessitated by sequence divergences included the absence of helix kinking proline residues in TMH 1 and TMH 5, the lack of a GG motif in TMH 2, as well as the presence of extra flexibility in TMH 6.
Because TMH 6 figures prominently in the R-to-R* transition (i.e., activation), we have studied the conformations accessible to TMH 6 in CB1 and CB2 using conformational memories (CMs) (Barnett-Norris et al., 2002a). These studies revealed that TMH 6 in CB1 has high flexibility due to the small size of residue 6.49 (a Gly) immediately preceding Pro 6.50. Two families of conformers were identified by CM for TMH 6 in CB1. Cluster 1 showed a pronounced proline kink (40 members of 100, 71.2° average kink angle). Cluster 2 contained helices with less pronounced kinks (51 members of 100, 30.1° average kink angle). A conformer from the more kinked CM family of CB1 TMH 6 (cluster 1) was used in our model of the R state of CB1. This conformer was selected (Pro kink angle of 53.1°) so that R3.50(214) and D6.30(338) could form a salt bridge at the intracellular ends of TMHs 3 and 6 in the CB1 TMH bundle. An analogous salt bridge has been shown to be an important stabilizer of the inactive state of the β2-adrenergic receptor (Ballesteros et al., 2001) and to be present in Rho (Palczewski et al., 2000).
Model of R* Form of CB1.
An R* CB1 model was created by modification of our rhodopsin-based model of the R form of CB1. This R* model construction was guided by the biophysical literature on the R-to-R* transition in Rho and the β2-adrenergic receptor. This literature has indicated that for the β2-adrenergic receptor, a salt bridge between R3.50 and E6.30 at its intracellular end stabilizes this receptor in its inactive state (Ballesteros et al., 2001). Biophysical studies of Rho and/or the β2-adrenergic receptor have indicated that rotation of both TMH 3 and 6, as well as a conformational change in TMH 6 occurs upon activation (Farrens et al., 1996; Lin and Sakmar, 1996; Javitch et al., 1997; Jensen et al., 2001).Jensen et al. (2001) recently demonstrated through fluorescence studies in the β2-adrenergic receptor that P6.50 in the highly conserved CWXP motif of TMH 6 can act as a flexible hinge that mediates the transition from R to R*. In the R state, these investigators proposed that TMH 6 is kinked at P6.50 such that its intracellular end is nearly perpendicular to the membrane and close to the intracellular end of TMH 3. The transition to the R* state is accomplished by the straightening of TMH 6 such that the intracellular part of TMH 6 moves away from the receptor core and upwards toward the lipid bilayer (Jensen et al., 2001). All of these experimental findings were used to create the R* model of CB1 described herein.
Our conformational memories study of CB1 TMH 6 revealed two distinct conformational families for TMH 6 that differed in the degree of kinking at CWGP (Barnett-Norris et al., 2002a). These conformationally distinct TMH 6s were used to create the R and R* states depicted herein. In the R* bundle, a TMH 6 conformer from the second major conformational family (less kinked: 21.8° kink angle) identified by CM (Barnett-Norris et al., 2002a) was substituted for the TMH 6 conformer used in the inactive model of CB1. This conformer was chosen so that the salt bridge in the inactive state between R3.50(214) and D6.30(338) would be broken due to the movement of the intracellular end of TMH 6 away from that of TMH 3 and out into lipid (Ballesteros et al., 2001). In addition, the R* CB1 bundle was created by rotating TMH 3 so that residue 3.41 changes environments (Lin and Sakmar, 1996). This was accomplished by a 20° counterclockwise (extracellular view) rotation of TMH 3 from its orientation in the R bundle. TMH 6 was also rotated (counterclockwise from extracellular view) so that Cys 6.47 became accessible from inside the binding site crevice (Javitch et al., 1997).
Preparation of Helices.
Each helix of the model was capped as the acetamide at its N terminus and as the N-methyl amide at its C terminus. Ionizable residues in the first turn of either end of the helix were neutralized, as were any lipid-facing charged residues. Ionizable residues were considered charged if they appeared anywhere else in the helix.
Ligand-Receptor Complex.
Each ligand was docked in the aromatic, residue-rich TMH 3-4-5-6 region of the CB1 R or R* TMH bundle using interactive computer graphics. The energy of the ligand/CB1 R or R* TMH bundle complex was minimized using the AMBER* united atom force field in Macromodel 6.5 (Schrödinger Inc.). A distance-dependent dielectric, 8.0 Å, extended nonbonded cutoff (updated every 10 steps), 20.0-Å electrostatic cutoff, and 4.0-Å hydrogen bond cutoff were used. The first stage of the calculation consisted of 2000 steps of Polak-Ribier conjugate gradient (CG) minimization in which a force constant of 225 kJ/mol was used on the helix backbone atoms to hold the TMH backbones fixed, while permitting the side chains to relax. The second stage of the calculation consisted of 100 steps of CG in which the force constant on the helix backbone atoms was reduced to 50 kJ/mol to allow the helix backbones to adjust. Stages 1 and 2 were repeated with the number of CG steps in stage two incremented from 100 to 500 steps until a gradient of 0.001 kJ/(mol · Å2) was reached.
Assessment of Aromatic Stacking Interactions.
Burley and Petsko (1985) have reported that aromatic-aromatic (π-π) stacking interactions in proteins operate at distances (d) of 4.5 to 7.0 Å between ring centroids. The angle (α) between normal vectors of interacting aromatic rings typically is between 30° and 90°, producing a “tilted-T” or “edge-to-face” arrangement of interacting rings. Hunter et al. (1991) have reported that π-π parallel stacking interactions (α < 30°) between phenylalanine residues in proteins are favorable if the rings are offset from each other. Residues and/or ligand regions were designated herein as participating in an aromatic stacking interaction if they had centroid-to-centroid distances between 4.5 and 7.0 Å. These interactions were further classified as tilted-T arrangements if 30° ≤ α ≤ 90° and as parallel arrangements for α < 30°. Parallel arrangements were considered favorable only if the interacting rings were offset from each other (Hunter et al., 1991). All measurements were made using Macromodel 6.5 (Schrödinger Inc.).
Synthesis
The conformationally constrained compound VCHSR (2), a vinylcyclohexyl analog of SR141716, was synthesized by Wittig olefination. The route started with the reported pyrazole ester ethyl 1-(2,4-dichlorophenyl)-5-(4-chlorophenyl)-4-methylpyrazole-3-carboxylate (Barth et al., 1995), an intermediate in the synthesis of SR141716, which was reduced with lithium aluminum hydride to the alcohol [5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl]methanol. The latter was converted with CBr4 and triphenylphosphine to the benzylic bromide 3-(bromomethyl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole and then to the phosphonium salt, 5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazol-3-yl-methyl-triphenylphosphonium bromide with triphenylphosphine.
Deprotonation of this phosphonium salt with lithium diisopropylamide, to the corresponding stabilized phosphorus ylide, and treatment with cyclohexanecarboxaldehyde afforded the putative olefin 2. The trans-geometry of the olefin was proven by an NMR shift reagent experiment that resolved the overlapping vinyl resonances to reveal their 16-Hz coupling constant, indicative of trans(E)-geometry.
Ligand Binding Assay
Materials.
Dulbecco's modified Eagle's medium, fetal bovine serum, penicillin/streptomycin, l-glutamine, trypsin, and geneticin were purchased from BioWhittaker (Walkersville, MD). Enzymes and reagents used for recombinant DNA experiments were purchased from Invitrogen (Carlsbad, CA) or Promega (Madison, WI). Adenovirus-transformed 293 cells were obtained from American Type Culture Collection (Rockville, MD). Glass tubes used for diluting cannabinoid drugs and for ligand binding assays were silanized through exposure to dichlorodimethylsilane (Sigma-Aldrich, St. Louis, MO) vapor while under vacuum for 3 h. [3H]SR141716A was purchased from Amersham Biosciences (Piscataway, NJ).
Expression and Mutagenesis of CB1 Cannabinoid Receptor Gene.
A 1.5-kilobase SstI/XbaI fragment of the human CB1 gene containing the entire coding region was subcloned into expression vector pRC/CMV (Invitrogen) to construct the expression plasmid pHCB1-RC/CMV (Song and Bonner, 1996). A lysine-to-alanine mutation at the position 192 of the CB1 cannabinoid receptor was made by site-directed mutagenesis (Song and Bonner, 1996).
Cell Transfection and Culture.
Expression plasmids containing wild-type and mutant cannabinoid receptors were purified with QIAGEN plasmid kit (QIAGEN, Chatsworth, CA) and then transfected into human embryonic kidney 293 cells using the calcium phosphate precipitation method (Chen and Okayama, 1987). Transfected cells were selected in culture medium containing 500 μg/ml geneticin, and cell lines stably expressing wild-type and mutant cannabinoid receptors were established according to a method established previously (Chen and Okayama, 1987). Cells were grown as monolayers in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 2 mM glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere consisting of 5% CO2 and 95% air at 37°C.
Ligand Binding Assay.
For membrane preparations, cells were washed twice with cold phosphate-buffered saline and scraped off the tissue culture plates. Subsequently, cells were homogenized in binding buffer (50 mM Tris-HCl, 5 mM MgCl2, and 2.5 mM EDTA, pH 7.4) with a Tissumizer (Tekmar-Dohrmann, Mason, OH). The homogenate was centrifuged at 32,000g for 20 min at 4°C. The pellet was resuspended in binding buffer and stored at −80°C. Protein concentrations were determined by the use of a bicinchoninic acid protein reagent kit (Pierce Chemical, Rockford, IL).
For binding assays, cannabinoid ligand dilutions were made in binding buffer containing 25 mg/ml bovine serum albumin and then added to assay tubes. [3H]SR141716A was used as a labeled ligand for ligand binding assays. Binding assays were performed in 0.5 ml of binding buffer containing 5 mg/ml bovine serum albumin for 60 min at 30°C. Free and bound radioligands were separated by rapid filtration through polyethylenimine-treated GF/B filters (Whatman, Maidstone, UK) that had been soaked in cold wash buffer (50 mM Tris-HCl, pH 7.4, and 1 mg/ml bovine serum albumin). The filters were washed three times with 3 ml of cold wash buffer. The bound [3H]SR141716A was determined by liquid scintillation counting after overnight equilibration of the filter in 5 ml of scintillation cocktail (Hydrofluor; National Diagnostics, Manville, NJ). The assays were performed in duplicate, and the results represent the combined data from three independent experiments.
Data Analysis.
Data from ligand binding assays were analyzed, and curves were generated with use of the Prism program (GraphPad Software, San Diego, CA). IC50 values were determined through nonlinear regression analysis performed with the Prism program. K d andB max values were estimated from competition binding experiments with the following equations:K d = IC50 −L and B max = (B 0IC50)/L, where L is the concentration of free radioligand andB 0 is specifically bound radioligand (DeBlasi et al., 1989). The K i values were calculated based on the Cheng-Prusoff equation:K i = IC50/(1 +L/K d) (Cheng and Prusoff, 1973).
Mutant Cycle Calculations
To assess whether a direct interaction occurs between K3.28(192) and the C3 substituent of SR141716A, we performed a mutant cycle analysis using K d andK i data for WT CB1/SR141716A, WT CB1/VCHSR, K3.28(192)A/SR141716A, and K3.28(192)A/VCHSR displacement of [3H]SR141716A. Mutant cycles have commonly been used in the literature to analyze whether indirect or direct interactions occur between certain amino acid residues in a protein (Faiman and Horovitz, 1996) and also between an amino acid residue and a ligand (Ambrosio et al., 2000). In the mutant cycle, two nonidentical perturbations (WT CB1→K3.28(192)A and SR141716A→VCHSR) are applied to the system. As described by Ambrosio et al. (2000), the effects on the binding energy produced by the two perturbations should obey the principle of free energy conservation and allow us to consider the thermodynamic cycle below:
Herein, ΔGs indicate free energy changes measured in binding experiments using the equation
Thermodynamic cycle for removal of hydrogen bond potential at residue 3.28 from CB1 receptor

Ca2+ Channel Assay
Cannabinoid Receptor Expression and Electrophysiology.
Mammalian expression plasmids pCI (Promega), containing the human brain hCB1 cannabinoid receptor cDNA (from Dr. Tom I. Bonner, Laboratory of Cell Biology, National Institute of Mental Health, Bethesda, MD), were injected (100 ng/μl) into nuclei of isolated rat superior cervical ganglion (SCG) neurons as described previously (Pan et al., 1998;Vásquez and Lewis, 1999). The pEGFP-N1plasmid (10 ng/μl) containing the coding sequence of enhanced green fluorescent protein (CLONTECH, Palo Alto, CA) was used as a coinjection marker. After an overnight incubation, Ca2+ currents from injected neurons were recorded at room temperature (24–26°C) with an Axopatch 200A patch-clamp amplifier (Axon Instruments, Union City, CA). The cell membrane capacitance and series resistance were electronically compensated to >80%. Whole-cell currents were low pass-filtered at 5 kHz using the Bessel filter of the clamp amplifier.
Voltage-clamp protocols were generated with a Power Macintosh 8600 computer (Apple Computer, Cupertino, CA) equipped with a PCI-16 Host Interface card connected to an ITC-16 Data Acquisition Interface (InstruTECH Corporation, Port Washington, NY) using Pulse Control 5.0 XOPs (Richard J. Bookman, Jack D. Herrington, and Kenneth R. Newton, University of Miami, Miami, FL) with Igor software (WaveMetrics, Lake Oswego, OR). Ca2+ currents were elicited by voltage steps from a holding potential of −80 mV and digitized at 180 μs/point. A double pulse protocol consisting of two 25-ms steps to +5 mV was used to elicit Ca2+ currents. The first step to +5 mV was followed by a 50-ms step to + 80 mV to reverse G protein-dependent inhibition of the Ca2+ current. Current amplitudes were measured isochronally 10 ms after the voltage step to +5mV and current traces show the current elicited by the first voltage step to +5 mV.
Ca2+ currents were isolated with an external solution that contained 140 mM tetraethylammonium methanesulfonate, 10 mM HEPES, 15 mM glucose, 10 mM CaCl2, and 0.0001 mM tetrodotoxin, pH 7.4 (adjusted with methanesulfonic acid). The intracellular solution contained 120 mMN-methyl-d-glucamine, 20 mM tetraethylammonium chloride, 10 mM HEPES, 11 mM EGTA, 1 mM CaCl2, 4 mM MgATP, 0.1 mM Na2GTP, and 14 mM phosphocreatine, pH 7.2 (adjusted with methanesulfonic acid). Stock solutions of 10 nM WIN55,212-2 mesylate (Sigma/RBI, Natick, MA) and 10 mM VCHSR were prepared in dimethyl sulfoxide. On the day of the experiment, stock solutions were diluted to 1 μM in external solution and briefly sonicated to facilitate dispersion. WIN55,212-2 and VCHSR were applied by a fast-switching device (SF-77B Perfusion Fast-Step; Warner Instrument, Hamden, CT).
Results
Chemistry
X-ray crystal structure analysis of SR141716A has revealed that the carboxamide in the C3 substituent of SR141716A is in atrans-geometry (C. George, personal communication). VCHSR (2) was designed to mimic this carboxamidetrans-geometry in the C3 substituent of SR141716A, but to lack hydrogen bonding potential in this substituent region. NMR spectroscopy was used to confirm the trans geometry of 2. Compound 2 was further characterized by thin layer chromatography, gas chromatography, high-pressure liquid chromatography, and high-resolution mass spectrometry. The high-resolution mass spectral data confirm the empirical formula for2 as C24H23N2Cl3: Calculated m/z = 444.0927; observed high-resolution mass spectra, 444.0927. Further details of the synthesis and characterization of 2 will be published elsewhere.
Conformational Analysis Results
SR141716A Global Minimum Energy Conformer.
The global minimum energy conformer of SR141716A (Fig. 3, top, left) has the carboxamide oxygen of the C3 substituent nearly in plane with the pyrazole ring and pointing in the direction of the C4 methyl group (O-C1′-C3-C4 = 9.2°). The piperidine ring is in a chair conformation with the nitrogen lone pair of electrons pointing in the same direction as the carboxamide hydrogen (LP-N3′-N2′-H = 0.2°). The monochlorophenyl ring is out of plane with the pyrazole ring (C4-C5-C1‴-C2‴ = −46.2°), and the dichlorophenyl ring is also out of plane with the pyrazole ring (N2-N1-C1"-C2" = −63.1°). In this position, the ortho-chloro is in the bottom face of the molecule (i.e., below the plane of the paper).
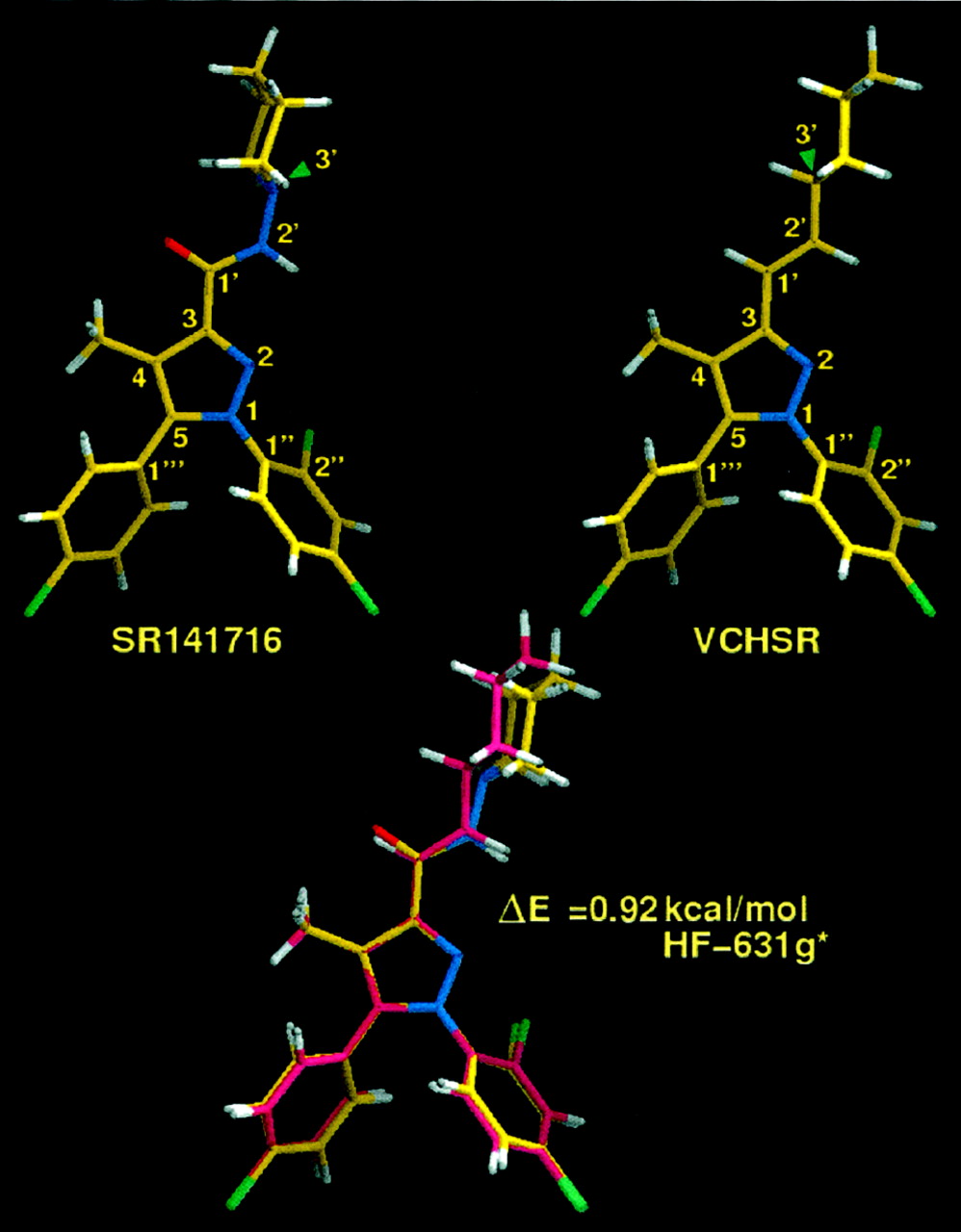
Global minimum energy conformer of SR141716A (1) (top, left) and global minimum energy conformer of VCHSR (2) (top, right) as determined by the AM1 semiempirical method. Bottom, superposition at their pyrazole rings of the global min of 2 (in yellow) and a higher minimum energy conformation of1 (ΔE = 0.92 kcal/mol; ab initio Hartree Fock 6-31G*; in pink). These are the conformations used for docking studies (Figs. 4and 5).
VCHSR Global Minimum Energy Conformer.
Fig. 3, top, right, illustrates the global minimum energy conformer of VCHSR. This conformer of VCHSR differs from that of SR141716A only in the orientation of its cyclohexyl ring compared with that of the piperidine ring in SR141716A. In the global minimum of VCHSR, thetrans-ethylene group is oriented such that the hydrogen attached to C1′ is nearly in the plane of the pyrazole ring, pointing toward the C4 methyl group (H-C1′-C3-C4 = −0.4°) and the hydrogen attached to C3′ (cyclohexyl ring) points in the opposite direction from the C2′ hydrogen (H-C3′-C2′-H = −179.7°).
Conformer Selection for Docking.
A molecular electrostatic potential (MEP) map calculated at the AM1 level (data not shown) indicated that the piperidine nitrogen of SR141716A generates an MEP minimum (i.e., negative potential region), second only to that generated by the carboxamide oxygen in SR141716A. AM1 conformational searches identified another minimum energy conformation of SR141716A in which the piperidine nitrogen's lone pair points in the same direction as the carboxamide oxygen (LP-N3′-N2′-H = 178.5°). An MEP of this conformer showed an enhanced negative potential region associated with the C3 substituent. Although AM1 calculations showed that this conformer was 4.83 kcal/mol higher in energy than the global minimum, ab initio Hartree Fock 6-31G* calculations indicated that the energy separation between these two conformers was only 0.92 kcal/mol. For docking studies, we chose the global minimum energy conformer of VCHSR and the minimum energy conformer of SR141716A that matches this conformation of VCHSR. Figure 3 (bottom) illustrates the superposition of these two conformers at their pyrazole rings. This conformer of SR141716A was chosen because it produces the highest negative electrostatic potential in the C3 substituent region, potentially enabling the C3 substituent to form the strongest hydrogen bond possible with a hydrogen bond donor.
Receptor Docking Studies
One of the significant features of the CB1 R TMH bundle is a salt bridge between K3.28(192) and D6.58(366) (N-O distance = 2.6 Å; N-H-O angle = 159°). Unlike the intracellular R3.50/E6.30 (or R3.50/D6.30) salt bridge shown to stabilize G protein-coupled receptors (GPCRs) in their inactive states (Ballesteros et al., 2001), the extracellular K3.28/D6.58 salt bridge in CB1 (present only in the inactive state of CB1) seems to be important for positioning K3.28 for ligand interaction in the inactive state, rather than for stabilizing the receptor in the inactive state. In fact, Pan et al. (1998) found that WT CB1 and the CB1 K3.28(192)A mutant exhibit the same level of constitutive activity. So, the absence of the K3.28/D6.58 salt bridge does not lead to greater ease of activation.
The K3.28/D6.58 salt bridge is made possible by two special structural features of the CB1 receptor, its EC-2 loop and the flexibility of TMH 6 in CB1. Despite the fact that the CB1 and CB2 receptors belong to the Rho family of GPCRs, there are important differences between CB1/CB2 and Rho that impact the ligand binding pocket in the TMH 3-4-5-6 region. The CB1 and CB2 extracellular loop 2 (E-2 loops) are shorter than that of Rho (CB1 15 residues in length; CB2 13 residues in length; Rho 25 residues in length) and there is no corresponding Cys residue in TMH 3 of CB1 or CB2 that would cause the E-2 loop to dip down into the binding site crevice as the E-2 disulfide bridge with Cys3.25(110) causes in Rho. However, there is a Cys residue at the extracellular end of TMH 4 and a Cys near the middle of the E-2 loop in the CB receptors. Mutation results of these residues (C174 and C179) in CB2 suggest that a disulfide bridge between these two Cys residues may exist, but protein expression problems have hampered attempts to prove the existence of this bridge in CB1 (Shire et al., 1999). As the result of this important difference between Rho and the CB receptors, the binding site crevice around TMHs 3-4-5-6 is likely to be different with the E-2 loop occupying less volume in the upper part of the binding pocket than does the E-2 loop in Rho.
The different spatial requirements of the E-2 loop are important because this difference permits the extracellular end of TMH 6 to occupy a different position in the TMH bundle than is seen in Rho. We have recently shown that the small size of residue 6.49 in CB1 (a Gly) results in pronounced flexibility of the CWXP motif in TMH 6 (Barnett-Norris et al., 2002a). This motif has been suggested to function as a flexible hinge, permitting agonist-promoted movement of the intracellular end of TMH 6 that occurs during activation (Jensen et al., 2001). In addition to permitting the intracellular end of TMH 6 to come close to the intracellular end of TMH 3 in the inactive state of CB1, this flexibility in CB1 TMH 6 permits the extracellular end of TMH 6 to bend toward TMH 3, resulting in the formation of a salt bridge between D6.58 (near the extracellular end of TMH 6) and K3.28 in TMH 3. In the R* TMH bundle, the K3.28(192) and D6.58(366) salt bridge is broken (N-O distance = 16.8 Å).
Both the CB1 inverse agonist SR141716A (SR) and its analog VCHSR are highly aromatic compounds. We hypothesized that aromatic stacking interactions might be important for the binding of these compounds at CB1. The CB1 TMH 3-4-5-6 region is rich in aromatic residues that face into the ligand binding pocket, including F3.25(189), F3.36(200), W4.64(255), Y5.39(275), W5.43(279), and W6.48(356). Shire et al. (1999)have shown in CB1/CB2 chimera studies that the TMH-4-E-2-TMH5 region of CB1 contains residues critical for the binding of SR141716A. In Monte Carlo/Stochastic Dynamics Simulations of the inactive state of WT CB1,McAllister et al. (2002) found a persistent aromatic stack between Y5.39(275) and W4.64(255) that seemed to be important for stabilizing the positions of TMHs 4 and 5 in the TMH bundle on the extracellular side and a second aromatic stack between F3.36(200), W5.43(279), and W6.48(356) that seemed to be open for additional interaction with ligand. We therefore pursued the aromatic residue-rich TMH 3-4-5-6 region as the binding site for SR and VCHSR. The R bundle in the TMH 3-4-5-6 region is characterized by a W6.48(356)/W5.43(279)/F3.36(200) and a Y5.39(275)/W4.64(255)/F5.42(278) aromatic cluster. In the R* TMH bundle, an aromatic cluster exists between W6.48(356)/W5.43(279)/Y5.39(275)/W4.64(255)/F5.42(278). We found that each ligand could insert itself into this aromatic residue-rich region to become an integral part of an extended aromatic cluster.
SR141716A/CB1 Complexes.
SR was docked in the TMH 3-4-5-6 region in a model of both the R and R* states of CB1. Figures4A and 5A illustrate SR141716A docking results.

SR141716A in a minimum energy conformation (ΔE = 0.92 kcal/mol) (A) and VCHSR in its global minimum energy conformation (B), each docked in the computer TMH bundle model of the R state of CB1. The R state is characterized by a salt bridge between K3.28(192) and D6.58(366) and two patches of aromatic residues that form clusters in the TMH 3-4-5-6 region of CB1, W5.43(279)/F3.36(200)/W6.48(356), and Y5.39(275)/W4.64(255)/F5.42(278). Residues with which ligands have direct interactions are yellow. Residues that form part of the aromatic cluster complex with ligand but that do not stack directly with ligand are green. Residues that have no direct or indirect interaction with ligand are cyan. In A, SR141716A has a hydrogen bonding interaction with K3.28(192) in the salt bridge. Because SR141716A directly stacks with both F3.36(200) and W5.43(279) and with Y5.39(275), the ligand joins the F3.36(200)/W5.43(279)/W6.48(356) and Y5.39(275)/W4.64(255)/F5.42(278) aromatic clusters into one large extended cluster in the minimized complex. In B, VCHSR has aromatic stacking interactions with W5.43(279), F3.36(200) and Y5.39(275). In binding, VCHSR bridges between the F3.36(200)/W5.43(279)/W6.48(356) and Y5.39(275)/W4.64(255)/F5.42(278) aromatic clusters and helps form one large extended cluster in the minimized complex. However, VCHSR has no interaction with K3.28(192) due the lack of hydrogen bonding potential in the C3 substituent region of VCHSR.
SR141716A/CB1 R Complex.
In the inactive CB1 bundle, the carboxamide oxygen of SR141716A forms a hydrogen bond with K3.28(192), which is part of the salt bridge with D6.58(366) (Fig. 4A). In this interaction, the nitrogen of K3.28(192) is central and provides a hydrogen to an oxygen of D6.58(366) (N-O distance = 2.6 Å; N-H-O angle = 159°) and to the carboxamide oxygen of SR141716A (N-O distance = 2.7 Å; O-H-N angle = 163°). The geometry of this interaction mimics that of complex salt bridges in proteins. In their statistical analysis of salt bridges in proteins, Musafia et al. (1995) documented that in 35.6% of the protein complex salt bridges analyzed that had the lysine nitrogen as the connecting position (central portion) of the salt bridge, a lysine N-H had an interaction with a single oxygen from each of two separate acidic residues (Fig. 3K in Musafia et al., 1995). Figure 4A illustrates the geometry of this D6.58/K3.28/SR141716A interaction in the R state.
Using the criteria described under Materials and Methods, we analyzed each minimized ligand/CB1 complex for the presence of aromatic stacking interactions. As described below, W5.43(279) and F3.36(200) formed the closest aromatic stacking interactions with SR141716A in CB1. In the SR141716A/CB1 R complex, both the monochlorophenyl (MC) and dichlorophenyl (DC) rings of SR were found to be involved in aromatic stacking interactions with W5.43(279) (MC d = 4.8 Å, α = 50°; DC d = 4.8 Å, α = 90°). In addition, the dichlorophenyl ring was found to have a stacking interaction with F3.36(200) (d = 5.0 Å, α = 80°) and the monochlorophenyl ring, an interaction with Y5.39(275) (MC d = 6.5 Å, α = 25°). Aromatic residues in the TMH 3-4-5-6 region form two networks or clusters that are bridged by SR141716A. In one cluster, F3.36(200) directly stacks with W6.48(356) (d = 4.9 Å, α = 50°) and with W5.43(279) (d = 5.9 Å, α = 60°). In a second cluster, Y5.39(275) directly stacks with W4.64(255) (d = 6.5 Å, α = 80°), which in turn stacks with F5.42(278) (d = 4.6 Å, α = 30°). Because SR141716A directly stacks with both F3.36(200) and W5.43(279) and with Y5.39(275), the ligand joins the F3.36(200)/W5.43(279)/W6.48(356) and Y5.39(275)/W4.64(255)/F5.42(278) aromatic clusters into one large extended cluster in the minimized complex.
Importance of the F3.36/W6.48 Interaction to the Inactive State.
In the minimized complex, SR141716A stabilizes the inactive (bent) conformation of TMH 6 by its interaction with F3.36/W6.48. Residues W6.48 [χ1 = g+ (−60°)] and F3.36 [χ1 = trans (180°)] in the inactive bundle are engaged in a direct stacking interaction. Rotations of TMH 3 and 6 concomitant with activation move F3.36 and W6.48 away from each other. We have recently proposed that in CB1, χ1 of F3.36 must change from trans to g+ during activation (Barnett-Norris et al., 2002b). Spectroscopic studies (Lin and Sakmar, 1996) have indicated that W6.48 undergoes a conformational change when Rho is activated. Visiers et al. (2002) have proposed that a χ1 change in W6.48 from g+ (−60°) totrans (180°) is part of the activation mechanism for the 5-hydroxytryptamine-2A receptor. In CM studies of 5-hydroxytryptamine-2A TMH 6, these investigators found a correlation between the proline kink angle of TMH 6 and the W6.48 χ1 torsion angle. More kinked (inactive state) TMH 6 conformers were found to have a g+ (−60°) χ1 torsion angle value for W6.48. Less kinked (active state) TMH 6 conformers were found to have atrans (180°) χ1 torsion angle value for W6.48. In agreement with the results reported by Ballesteros et al. (1998), CM studies of CB1 TMH 6 revealed that the kink angle of TMH 6 clusters and the χ1 of W6.48 were highly correlated in this same way (unpublished observations). Therefore, in our models, both W6.48 and F3.36 undergo a change in their χ1 values from R to R*. χ1 in W6.48 changes from g+ (−60°) totrans (180°) and χ1 of F3.36 changes from trans to g+. Because SR141716A in R stacks directly with F3.36, which in turn, has a direct stacking interaction with W6.48 in the CB1 inactive state, the binding of SR141716A to the R state of CB1 stabilizes this F3.36 (χ1 =trans)/W6.48 (χ1 = g+) aromatic stacking interaction, preventing any changes in the χ1 values of F3.36 and W6.48 and therefore stabilizing the bent (inactive state) conformation of TMH 6.
SR141716A/CB1 R* Complex.
The conformational changes that occur upon receptor activation result in rotations of TMHs 3 and 6 as well as a change in the conformation of TMH 6 (by moderation of its proline kink angle) (Fig. 5A). As a result, the position and accessibility of residues in the TMH 3-4-5-6 region to SR141716A are altered. Figures 4 and 5 illustrate the difference between the TMH bundle conformation in R versus R*. It is clear herein that activation has caused significant changes in the relative position of certain TMH 3 and TMH 6 residues, with K3.28(192) and D6.58(366) rotating away from each other in R*, breaking their salt bridge (O-N distance = 16.8 Å). In R*, K3.28 is no longer accessible in the TMH 3-5-6 region, but has shifted toward the TMH 2-3-7 region, having rotated away from the SR141716A binding pocket in the R* state. D6.58(366) has rotated toward the TMH 5-6 interface and is raised higher above the ligand binding pocket with the moderation of the TMH 6 proline kink angle. The carboxamide oxygen of SR141716A is now 11.4 Å from the nitrogen of the K3.28(192) side chain. As the result of these changes, K3.28(192) is no longer available for interaction with the C3 substituent of SR141716A. The F3.36(200)/W5.43(279)/W6.48(356) aromatic cluster also undergoes rearrangement, with F3.36(200) and W6.48(356) rotating away from each other in R*.

SR141716A in a minimum energy conformation (ΔE = 0.92 kcal/mol) (A) and VCHSR in its global minimum energy conformation (B), each docked in the computer TMH bundle model of the R* state of CB1. In the R* state, the K3.28(192)-D6.58(366) salt bridge is broken due to rotations of TMHs 3 and 6 and a conformational change in TMH 6. The aromatic cluster in this region has rearranged with F5.42(278)/W4.64(255)(175)/Y5.39(275)/W5.43(279)/W6.48(356), forming a cluster. Residues with which ligands have direct interactions are yellow. Residues that form part of the aromatic cluster complex with ligand but that do not stack directly with ligand are green. Residues that have no direct or indirect interaction with ligand are cyan. In A, SR141716A interacts directly with W5.43(279) and Y5.39(275). In binding, SR141716A becomes part of an aromatic cluster that includes W5.43(279)/Y5.39(275)/W4.64(175)/F5.42(278) in the minimized complex. In B, VCHSR interacts directly with W5.43(279) and W6.48(356). In binding, VCHSR becomes part of an F5.42(278)/W4.64(255)/Y5.39(275)/W5.43(279)/W6.48(356) extended aromatic cluster in the minimized complex.
In the ligand/receptor complex, W5.43(279) is positioned to interact with both phenyl rings (MC d = 4.8 Å, α = 70°; DC d = 4.9 Å, α = 80°) by tilted-T interactions that are characterized by centroid-to-centroid distances at the low end of the 4.5 to 7.0 Å range defined for a tilted-T aromatic stacking interaction (see Assessment of Aromatic Stacking Interactions under Materials and Methods; Hunter et al., 1991). The monochlorophenyl ring also has an offset parallel stack with Y5.39(275) (MC d = 5.7 Å, α = 0°). Aromatic residues in the TMH 3-4-5-6 region form a network or cluster with which SR141716A interacts. W5.43(279) directly stacks with Y5.39(275) (d = 6.0 Å, α = 40°); Y5.39(275) stacks both with W4.64(175) (d = 5.5 Å, α = 80°) and F5.42 (d = 5.7 Å, α = 30°); and W4.64(175) stacks with F5.42(275) (d = 5.7 Å, α = 80°). In binding, SR141716A becomes part of an aromatic cluster that includes W5.43(279)/Y5.39(275)/W4.64(175)/F5.42(278) in the minimized complex.
In evaluating the preference that SR141716A shows for R versus R*, the overall extent of aromatic stacking (both direct and indirect) created by ligand binding was assessed in each state. Results depicted herein in Figs. 4A and 5A show that although the extent of aromatic stacking is similar in both the R and R* states, SR141716A will have a preference for the R state, because only in this state is a hydrogen bonding interaction possible. Consequently, based on modeling results, SR141716A should compete with agonist, because it has affinity for the R* state and behave as an inverse agonist, because it has higher affinity for the R state.
VCHSR/CB1 Complexes.
VCHSR was docked in the TMH 3-4-5-6 region in a model of both the R and R* states of CB1. Figures 4B and 5B illustrate VCHSR docking results.
VCHSR/CB1 R Complex.
The interactions in which VCHSR engages are very similar to those described above for SR141716A with one important exception (Fig. 4B). Because the C3 substituent in VCHSR lacks hydrogen bonding potential, it cannot form a hydrogen bonding interaction with K3.28(192), despite its proximity to the K3.28(192)-D6.58(366) salt bridge. Similar to interactions found for the CB1/SR141716A complex, W5.43(279) and F3.36(200) formed the closest aromatic stacking interactions with VCHSR in the CB1 model. In the VCHSR/CB1 R complex, both the MC and DC rings of VCHSR were found to be involved in aromatic stacking interactions with W5.43(279) (MC d = 4.7 Å, α = 50°; DC d = 4.9 Å, α = 80°). In addition, the monochloro ring was found to have a stacking interaction with Y5.39(275) (MC d = 6.7 Å, α = 25°), and the dichlorophenyl ring was found to have a stacking interaction with F3.36(200) (d = 5.0 Å, α = 80°). Aromatic residues in the TMH 3-4-5-6 region form a network with which VCHSR interacts. F3.36(200) directly stacks with W6.48(356) (d = 4.8 Å, α = 50°) as well as with W5.43(279) (d = 5.8 Å, α = 60°), and Y5.39(275) directly stacks with W4.64(255) (d = 6.4 Å, α = 80°), which stacks with F5.42(278) (d = 4.7 Å, α = 40°). In binding, VCHSR bridges between the F3.36(200)/W5.43(279)/W6.48(356) and Y5.39(275)/W4.64(255)/F5.42(278) aromatic clusters and helps form one large extended cluster in the minimized complex.
VCHSR/CB1 R* Complex.
As illustrated in Fig. 5B, in CB1 R*, W5.43(279) is positioned to form an aromatic cluster by inserting itself between the monochloro- and the dichlorophenyl ring of VCHSR. In so doing, W5.43(279) forms tilted-T interactions with VCHSR that are characterized by centroid-to-centroid distances at the low end of the 4.5 to 7.0 Å range defined for a tilted-T aromatic stacking interaction (see Assessment of Aromatic Stacking Interactions under Materials and Methods; Hunter et al., 1991) (MC d = 4.9 Å, α = 60°; DC d = 4.7 Å, α = 80°). In addition, the dichloro ring of VCHSR has an off-set parallel aromatic stacking interaction with W6.48(356) (DC d = 6.5 Å, α = 30°). Aromatic residues in the TMH 3-4-5-6 region form a network with which VCHSR interacts. W5.43(279) has a stacking interaction with Y5.39(275) (d = 6.2 Å, α = 30°); Y5.39(275) has a stacking interaction with both F5.42(278) (d = 5.2 Å, α = 40°) and W4.64(255) (d = 5.6 Å, α = 70°); and W4.64(255) stacks with F5.42(278) (d = 5.3 Å, α = 80°). In binding, VCHSR becomes part of an F5.42(278)/W4.64(255)/Y5.39(275)/W5.43(279)/W6.48(356) extended aromatic cluster in the minimized complex.
In evaluating the preference that VCHSR will show for R versus R*, the overall extent of aromatic stacking (both direct and indirect) created by ligand binding was assessed in each state. Results depicted here in Figs. 4B and 5B show that the extent of aromatic stacking is similar in both the R and R* states. As a result, VCHSR should have nearly equal affinity for both the R and R* states, causing it to behave as a neutral antagonist.
Ligand Binding in hCB1 WT and K3.28(192)A
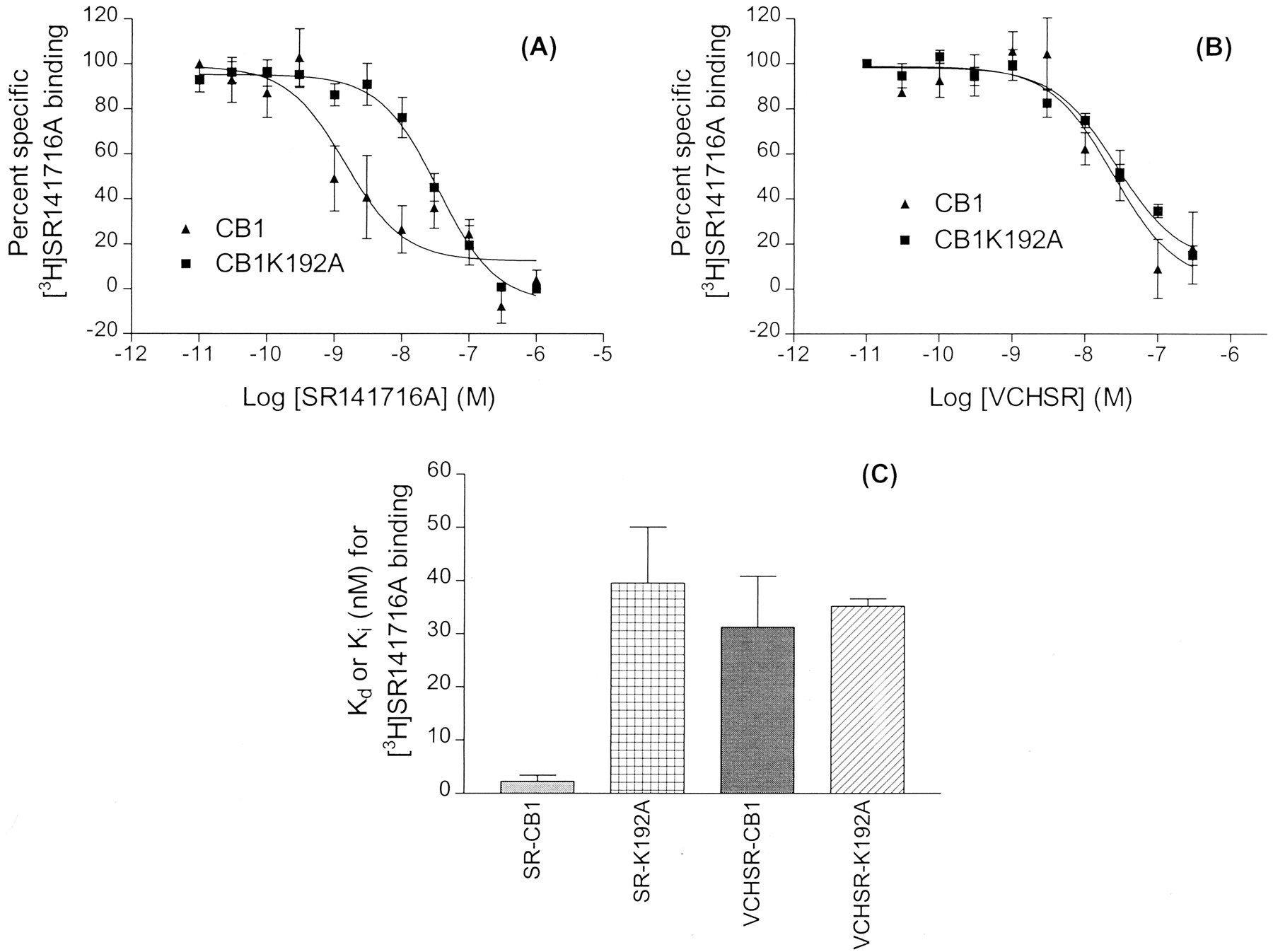
Scatchard analysis and ligand binding results for WT CB1 and the K3.28(192)A mutant cell lines are presented in Table1. To test whether K3.28(192) is an interaction site for SR141716A, we evaluated the binding of [3H]SR141716A in CB1 WT and K3.28(192)A mutant cell lines (Fig. 6A). Results in Table 1demonstrate the importance of K3.28(192) for SR141716A binding at CB1. In WT CB1, the K d value for SR141716A was 2.3 ± 1.1 nM, whereas in CB1 K3.28(192)A, theK d value for SR141716A was 39.6 ± 10.5 nM.
Ligand binding profile and free energy data for WT and K3.28A CB1

Competition of [3H]SR141716A binding in wild-type CB1 and CB1 K3.28(192)A receptors. Ligand binding assays were performed on membranes prepared from human embryonic kidney 293 cells stably expressing wild-type CB1 or CB1 K3.28(192)A receptor. The competition by SR141716A and VCHSR are shown in A and B, respectively. Points represent mean ± S.E. of three independent experiments performed in duplicate. Curves were generated as described underMaterials and Methods. The comparison ofK d or K i values for [3H]SR141716A binding is shown in C. Graphs represent mean ± S.E. of K i orK d values obtained from three independent experiments performed in duplicate.
As a further test of our hypothesis that K3.28(192) is an interaction site for SR141716A, VCHSR was used in competition binding assays (Fig.6B). The K i value for VCHSR binding in cloned human WT CB1 and CB1 K3.28(192)A cell lines versus [3H]SR141716A was 31.3 ± 9.6 and 35.2 ± 1.4 nM, respectively. Figure 6C presents a comparison ofK d orK i values for [3H]SR141716A binding. The bar graphs represent the mean ± S.E. of three independent experiments performed in duplicate. It is clear herein that theK i values for VCHSR binding to WT CB1 and to CB1 K3.28(192)A are comparable and that theseK i values, in turn, are comparable with the K d value of SR146716A for CB1 K3.28(192)A, but not to the K d value for SR141716A binding to WT CB1.
These results suggest that K3.28(192) is important to the binding of SR141716A at WT CB1, but this residue is not important to the binding of VCHSR at WT CB1. Residue K3.28(192) has previously been shown to be a very important residue for agonist binding at CB1 (Song and Bonner, 1996; Chin et al., 1998). The results reported herein are the first demonstration that this residue is also important for inverse agonist binding at CB1.
Mutant Cycle
To evaluate whether a direct interaction takes place between the C3 substituent of SR141716A and K3.28(192) in WT CB1, a mutant cycle was constructed herein. An analog of SR141716A, VCHSR (2; Fig. 2), was designed and synthesized to be used in this study. As illustrated in Fig. 3, VCHSR (2) can mimic a minimum energy conformation of SR141716A, but unlike SR141716A, VCHSR has no hydrogen bonding capability in its C3 substituent region. Table1 summarizes the values of ΔG calculated for each state in the thermodynamic cycle using eq. 1. Table 2summarizes the results obtained for the mutant cycle using eqs. 2 to 5.
In the mutant cycle, a set of complementary chemical groups was deleted from both ligand (SR141716A → VCHSR) and receptor (WT CB1 → K3.28(192)A). The resultant losses in binding energy for each of these deletions [ΔΔG(1) for SR/WT CB1 → SR/K3.28A and ΔΔG(2) for SR/WT CB1 → VCHSR/WT CB1, respectively] were found not to be statistically different from each other (p = 0.77, Student's paired t test; Table 2). However, even if the two deletions produce similar or identical losses in binding energy, this result is insufficient evidence to conclude that the two groups interact directly with each other. The ligand may interact in one receptor region and the receptor residue, although remote from the ligand, may be engaged in a network of interactions that are crucial to the binding process. The decrease in binding energy due to deletion of ligand functionality may result from a loss in binding energy, whereas the effects on the free energy due to receptor residue substitution may come from conformational contributions. These losses may have similar magnitudes, even if the deleted groups do not directly interact with each other (Ambrosio et al., 2000). Therefore, key to the determination of whether deletions have occurred between two groups that interact indirectly or directly is the effect produced by simultaneous deletion of both groups [i.e., K3.28(192)A/VCHSR]. If the modified groups do not interact directly with each other in the WT state then the effect of the two simultaneous changes will be additive [i.e., ΔΔG(1) ≈ ΔΔG(1/2), ΔΔG(2) ≈ ΔΔG(2/1) and δG1,2 ≈ 0]. This will be reflected in a higher K i value for the double deletion. If, on the other hand, the two groups interact directly, then the effect of the two simultaneous changes will be nonadditive [i.e., ΔΔG(1/2) ≈ ΔΔG(2/1) ≈ 0 and ΔΔG(1) ≈ ΔΔG(2) ≈ −δG1,2], and theK i value for the double deletion will be comparable in value with the K ivalues for each single deletion. Ambrosio et al. (2000) suggest that such “clear-cut” results may not be encountered frequently in proteins such as receptors, in which binding and conformational change are inextricably linked. However, these authors conclude that, in general, if the magnitude of the free energy of coupling (δG1,2) is comparable (even if not necessarily identical) with the magnitude of ΔΔG(1) or ΔΔG(2), it can be concluded that a direct interaction occurs. It is clear from Tables 1and 2 that such a direct interaction is the case herein, with the effect of the double change clearly being nonadditive (δG1,2 ≠ 0) and the effect of each single change being comparable [(ΔΔG(1) = 7.17 ± 1.37 kJ/mol) ≈ (ΔΔG(2) = 6.58 ± 1.43 kJ/mol; these values were shown not to be statistically different, p= 0.77, using Student's paired t test]. This result suggests, therefore, that there is a direct interaction between the C3 substituent of SR141716A and K3.28(192) in WT CB1. In addition, the magnitude of the free energy of coupling (δG1,2= −6.88 ± 2.55 kJ/mol) is comparable with the free-energy change associated with a hydrogen bonding interaction.
Calcium Current Effects of SR141716A and VCHSR in SCG Neurons
Figure 7 illustrates that in SCG neurons injected with hCB1 receptor cRNA, VCHSR attenuated the inhibition of the Ca2+ current by the cannabinoid agonist WIN55,212-2. Figure 7A shows the Ca2+current amplitude recorded over time during a patch-clamp experiment from an SCG neuron expressing the human CB1 cannabinoid receptor. Application of 1 μM WIN (shaded column) rapidly inhibited the Ca2+ current. The current slowly recovered during washout of WIN55,212-2. Application of 1 μM VCHSR slightly increased the Ca2+ current and attenuated the effect of WIN55,212-2. Figure 7B shows superimposed Ca2+current traces for the same neuron shown in Fig. 7A in the absence (control) and presence of WIN, VCHSR, and VCHSR + WIN. Figure 7C shows a graph of the percentage of change in Ca2+current amplitude in the presence of WIN, VCHSR, and VCHSR + WIN. VCHSR significantly attenuated the effect of WIN55,212-2 (p< 0.05). The number of neurons tested is indicated in each bar of Fig.7C.
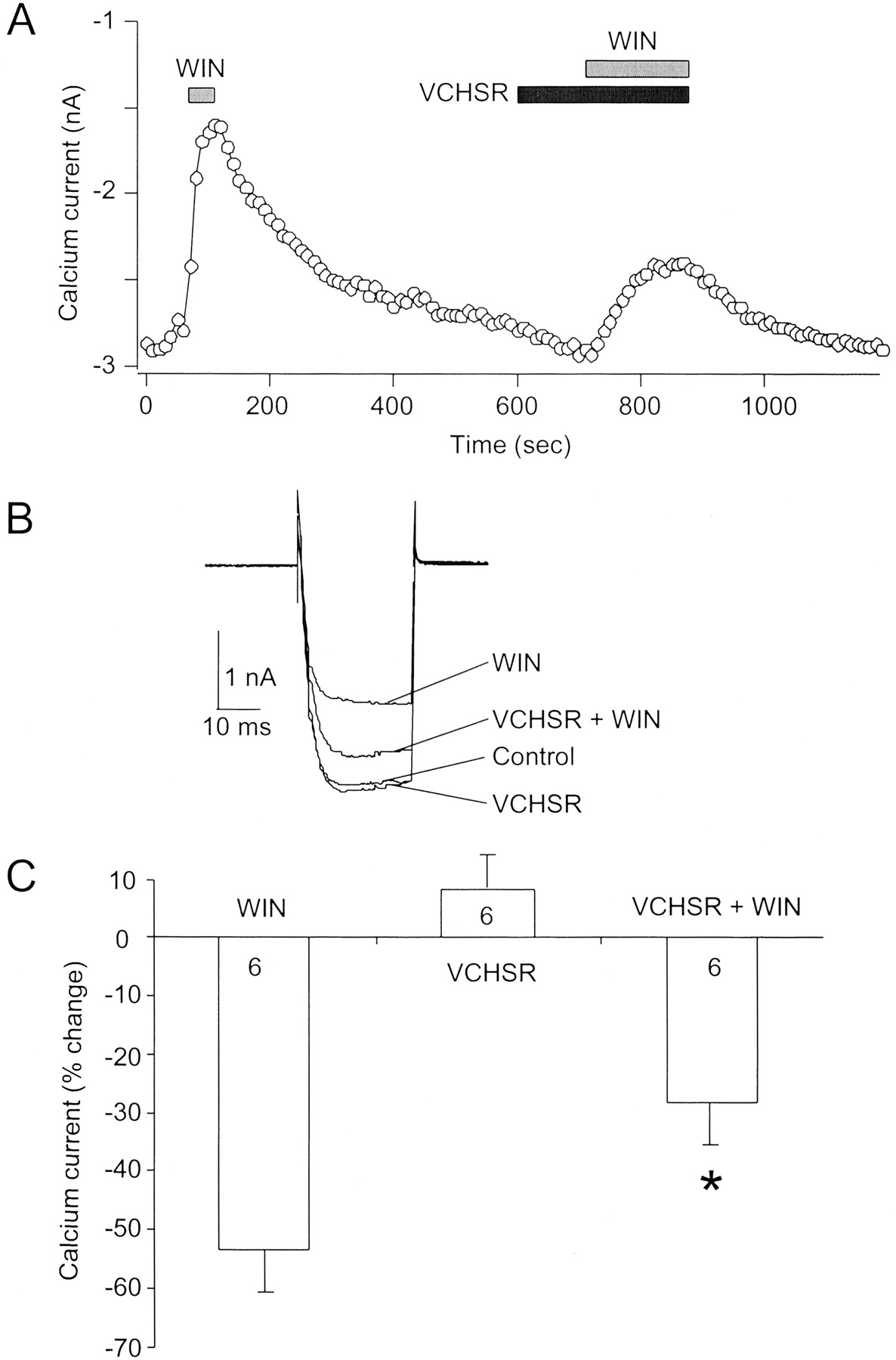
VCHSR attenuated the inhibition of the Ca2+ current by the cannabinoid agonist WIN55,212-2. A, Ca2+ current amplitude recorded over time during a patch-clamp experiment from a superior cervical ganglion neuron expressing the human CB1 cannabinoid receptor. Application of 1 μM WIN rapidly inhibited the Ca2+ current. The current slowly recovered during washout of WIN55,212-2. Application of 1 μM VCHSR slightly increased the Ca2+ current and attenuated the effect of a subsequent application of WIN55,212–2. B, superimposed Ca2+ current traces for the same neuron shown in A in the absence (control) and presence of WIN, VCHSR, and VCHSR + WIN. C, graph of the percentage of change in Ca2+ current amplitude in the presence of WIN, VCHSR, and VCHSR + WIN. VCHSR significantly attenuated the effect of WIN55,212-2 (p < 0.05). However, VCHSR alone produced a change in the Ca2+ current that was not significantly different (p = 0.259) from the control current amplitude. The number of neurons tested is indicated.
These results show that VCHSR behaved as a neutral antagonist because 1 μM VCHSR significantly (n = 6, p < 0.05) attenuated the WIN-induced inhibition of Ca2+ current, but alone VCHSR produced a change in the Ca2+ current that was not significantly different (p = 0.259) from the control current amplitude. Experiments performed with 10 μM VCHSR (data not shown) were consistent with results pictured in Fig. 7, indicating that VCHSR behaves as a neutral antagonist in SCG neurons expressing the human CB1 receptor.
Discussion
The extended ternary complex model for GPCR activation invokes the existence of two receptor conformational states, a ground or inactive R state and an active R* state, which are in equilibrium with each other (Leff, 1995). An agonist has higher affinity for R* and agonist binding is thought to shift the equilibrium toward R*, resulting in G protein activation with an increase in GDP/GTP exchange. An inverse agonist has higher affinity for R and its binding shifts the equilibrium toward R, resulting in a decrease in the activation of the signaling pathway. The binding of a neutral/null antagonist is thought not to alter the equilibrium between R and R* because the neutral antagonist has equal affinity for both states.
The central hypothesis tested in this article is that the interaction between K3.28(192) and SR141716A in WT CB1, an interaction that is possible only in the R state, causes SR141716A to have higher affinity for the R state and consequently behave as an inverse agonist. Pan et al. (1998) reported that SR141716A antagonized the Ca2+ current inhibition induced by the cannabinoid agonist WIN55212-2 in neurons heterologously expressing either rat or human CB1 receptors (Fig. 1). Furthermore, when applied alone, SR141716A increased the Ca2+ current, indicating that SR141716A can act as an inverse agonist by reversal of tonic CB1 receptor activity.
These investigators reported that the Ca2+current was also tonically inhibited in neurons expressing the mutant CB1 K3.28(192)A receptor. Surprisingly, SR141716A had no effect on the Ca2+ current in these neurons, but SR141716A could still antagonize the effect of WIN55212-2 (Fig. 1). There are two possible explanations for these results.
First, it is possible that the K3.28(192)A mutation results in a receptor population 100% in the inactive state (R ← R*). In this case, SR141716A would have no effect when applied alone because there is no R* population to convert back into the inactive state. However,Pan et al. (1998) found that the facilitation ratios for neurons expressing WT CB1 (1.41 ± 0.09, n = 4) and neurons expressing mutant K3.28(192)A receptors (1.42 ± 0.06,n = 8) were both significantly different (p < 0.001) from the Ca2+current facilitation ratio in uninjected neurons (1.17 ± 0.01,n = 11). The finding that the K3.28(192)A mutant has the same facilitation ratio as WT CB1 receptor suggests that both the mutant and WT CB1 can exist in a spontaneously active, G protein-coupled R* state.
The second possible explanation (and the one offered by Pan et al., 1998) is that the K3.28(192) site is critical for the inverse agonist activity of SR141716A, and therefore SR141716A becomes a neutral antagonist at the K3.28(192)A mutant receptor. The modeling, mutation and Ca2+ current assay results reported herein are clearly consistent with this latter interpretation of the results depicted in Fig. 1 for WT CB1 versus the K3.28(192)A mutant.
Central to the hypothesis tested herein is the determination of the interactions available to SR141716A in R and R*. Modeling results reported herein suggest that the overall extent of aromatic stacking (both direct and indirect) created by SR141716A binding is equivalent in the R and R* states. However, SR141716A has an additional interaction, a hydrogen bond with K3.28(192), only in the R state. Thus, the collection of aromatic and hydrogen bonding interactions in the R state suggest that SR141716A will have higher affinity for the R (over the R*) state of CB1, rendering it an inverse agonist.
It is important to emphasize that although SR141716A displayed inverse agonism in the Ca2+ assay, it also competitively antagonized the effect produced by WIN55,212-2 in this assay (Pan et al., 1998). This effect is consistent with modeling studies reported herein, which show that SR141716A will have affinity for the R* state due to aromatic stacking interactions (Fig. 5A). SR141716A should therefore be able to compete with an agonist for binding to the R* state.
Modeling studies reported herein also show that because VCHSR has no hydrogen bonding capability in its C3 substituent, it can engage only in aromatic stacking interactions in both the R and R* states of WT CB1. Because the overall extent of aromatic stacking (both direct and indirect) created by VCHSR binding is nearly equivalent in the R and R* states, VCHSR will have equal affinity for both states and would therefore be expected to behave as a neutral antagonist at WT CB1. In fact, this is what was seen herein in Ca2+current assay results for VCHSR (Fig. 7). In SCG neurons, expressing the human CB1 receptor, VCHSR behaved as a neutral antagonist because 1 μM VCHSR significantly (n = 6, p < 0.05) attenuated CB agonist (WIN) induced inhibition of Ca2+ current, but alone produced a change in the Ca2+ current that was not significantly different from the control current amplitude.
Consistent with the modeling results reported herein, the mutant thermodynamic cycle results show that K3.28 is a direct interaction site with SR141716A. It is important to note, however, that although the modeling results suggest that it is the carboxamide oxygen of SR141716A that interacts with K3.28(192), the mutant cycle calculations indicate only that there is a direct interaction between K3.28(192) and the C3 substituent of SR141716A. We are currently engaged in a synthesis effort to develop analogs that may help to identify the specific K3.28(192) hydrogen bonding site within the C3 substituent of SR141716A.
Implications for Models of Inverse Agonism.
The data presented in this article are consistent with the most widely discussed mechanism of inverse agonism in which the inverse agonist preferentially binds to the ground state (R) over the R* state (Samama et al., 1994), thus suppressing agonist-independent (constitutive) activation. An alternative mechanism has been proposed for the μ-opioid receptor in which the inverse agonist binds preferentially to the uncoupled forms of the receptor (R and R*), rather than to the receptor coupled to G protein (R*G), suppressing constitutive activity (Costa et al., 1992). For other GPCRs, inverse agonist preferential binding to a particular conformational state (i.e., R, R*, R*G, etc.) has not been demonstrated. In such cases, it has been suggested that the inverse agonist binds to the receptor and stabilizes the receptor in an inactive state that cannot activate G protein (McLoughlin and Strange, 2000).
We have shown herein that the inverse agonism of SR141716A can be explained by a two-state model in which SR141715A can interact with K3.28(192) only in the CB1 inactive state. A two-state representation for the CB1 receptor in the presence of SR141716A is consistent with models proposed by Bouaboula et al. (1997) and by Vásquez and Lewis (1999). Based upon results which showed that the interaction of SR141716A with the CB1 receptor can sequester Giproteins, preventing the signaling of other Gi-coupled receptors, Bouaboula et al. (1997)proposed a three-state model for GPCR activation in which agonists stabilize the R+ form, inverse agonists stabilize the R− form, and antagonists stabilize the inactive state, R0. The R−state herein is one in which the inverse agonist converts a tonically active hCB1 receptor into an active negative state in which the receptor is coupled to a GDP-bound G protein. Those G proteins trapped by the inverse agonist would be unavailable to couple to other receptors. Results presented by Bouaboula et al. (1997) suggest that the SR141716A·CB1 complex acts as a reversible negative dominant of Gi. Therefore, it can be expected for the CB1/SR141716A system that the R− and R+ states will predominate and the inactive R0 state can be expected to have a very low population. As a result, the system can be expected to behave essentially as a two-state system.
Vásquez and Lewis (1999) have further documented the ability of WT CB1 receptors to sequester G proteins. These investigators proposed that WT CB1 receptors exist predominantly in either of two states, a G protein-coupled inactive R-GGDP state or an active R*-GGTP state. It is possible that the inactive state represented herein in which SR141716A interacts with K3.28(192) and stabilizes the F3.36/W6.48 interaction (Fig. 4A) may correspond to the CB1 conformation recognized by Gi/0 in its GGDP state. Experiments are currently underway to test this hypothesis.
Acknowledgments
We thank Jannie Jones for technical assistance. This work was supported by National Institute on Drug Abuse Grants DA03934 and DA00489 (to P.H.R.), DA10350 (to D.L.L.), and DA11551 (to Z.H.S.).
Footnotes
- Received June 10, 2002.
- Accepted August 23, 2002.
-
RTI International is a trademark of Research Triangle Institute.
Abbreviations
- SR141716A
- N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- WT
- wild-type
- CB
- cannabinoid
- VCHSR
- 5-(4-chlorophenyl)-3-[(E)-2-cyclohexylethenyl]-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole
- WIN
- WIN55212-2 [(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone]
- R
- inactive state
- hCB
- human cannabinoid
- TMH
- transmembrane helix
- Rho
- rhodopsin
- R*
- active state
- CM
- conformational memory
- CG
- conjugate gradient
- SCG
- superior cervical ganglion
- MEP
- molecular electrostatic potential
- GPCR
- G protein-coupled receptor
- MC
- monochlorophenyl
- DC
- dichlorophenyl
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}