


Abstract
We examined the role of G-protein coupled receptor kinase-2 (GRK2) in the homologous desensitization of mGlu4 metabotropic glutamate receptors transiently expressed in human embryonic kidney (HEK) 293 cells. Receptor activation with the agonist l-2-amino-4-phosphonobutanoate (l-AP4) stimulated at least two distinct signaling pathways: inhibition of cAMP formation and activation of the mitogen-activated protein kinase (MAPK) pathway [assessed by Western blot analysis of phosphorylated extracellular signal-regulated kinase (ERK) 1 and 2]. Activation of both pathways was attenuated by pertussis toxin. Overexpression of GRK2 (but not GRK4) largely attenuated the stimulation of the MAPK pathway by l-AP4, whereas it slightly potentiated the inhibition of FSK-stimulated cAMP formation. Transfection with a kinase-dead mutant of GRK2 (GRK2-K220R) or with the C-terminal fragment of GRK2 also reduced the mGlu4-mediated stimulation of MAPK, suggesting that GRK2 binds to the Gβγ subunits to inhibit signal propagation toward the MAPK pathway. This was confirmed by the evidence that GRK2 coimmunoprecipitated with Gβγ subunits in an agonist-dependent manner. Finally, neither GRK2 nor its kinase-dead mutant had any effect on agonist-induced mGlu4 receptor internalization in HEK293 cells transiently transfected with GFP-tagged receptors. Agonist-dependent internalization was instead abolished by a negative-dominant mutant of dynamin, which also reduced the stimulation of MAPK pathway by l-AP4. We speculate that GRK2 acts as a “switch molecule” by inhibiting the mGlu4 receptor-mediated stimulation of MAPK and therefore directing the signal propagation toward the inhibition of adenylyl cyclase.
Metabotropic glutamate (mGlu) receptors, which belong to the third class of the G protein-coupled receptor (GPCR) superfamily, modulate excitatory synaptic transmission and are implicated in different aspects of central nervous system physiology, including motor control, motor coordination, sensory perception and pain transmission, learning and memory processes, and developmental plasticity (Nakanishi, 1994; Conn and Pin, 1997). mGlu receptors form a family of eight subtypes (mGlu1 to 8), which are divided into three groups based on sequence homology, pharmacological profile, and transduction pathways. Group I includes mGlu1 and -5 receptors, which are coupled to Gq proteins, whereas group II (mGlu2 and -3) and group III (mGlu4, -6, -7, and -8) receptors are coupled to Gi proteins in heterologous expression systems. Individual mGlu receptor subtypes are considered as targets for drugs of potential use in a variety of disorders, including anxiety, schizophrenia, stroke, Parkinson's disease, epilepsy, and neuropathic pain (reviewed by Bruno et al., 2001). This outlines the importance of unraveling the molecular mechanisms that regulate mGlu receptor function starting with the early steps of signal propagation (De Blasi et al., 2001).
Homologous desensitization of GCPRs is mediated by a family of enzymes called G-protein coupled receptor kinases (GRKs). This family includes GRK1, which corresponds to rhodopsin kinase, GRK2, and -3, which are ubiquitous and are activated by G-protein βγ subunits, and GRK4, -5, and -6. Phosphorylation of GPCRs by GRKs causes receptor desensitization and internalization through mechanisms that involve additional proteins, such as β-arrestins (Kohout and Lefkowitz, 2003). The role of different GRKs in the homologous desensitization of mGlu1 receptors has been elucidated. Recombinantly expressed mGlu1 receptors are desensitized by GRK4 in an agonist-dependent manner, and knock-down of GRK4 in cultured cerebellar Purkinje cells (which natively express mGlu1 receptors) impairs receptor desensitization (Sallese et al., 2000a). GRK2 is also involved in desensitization and internalization of mGlu1 receptors, although its action does not require an intact kinase activity (Dale et al., 2000; Dhami et al., 2002; Mundell et al., 2003; Iacovelli et al., 2003). mGlu5 receptor signaling is regulated by GRK2, but not GRK4, in heterologous expression systems (Sorensen and Conn, 2003).
Little is known on how GRKs regulate receptor signaling at group II and III mGlu receptor subtypes. We examined the regulation of mGlu4 receptors, which protect neurons against excitotoxic death (Bruno et al., 1995, 1996, 2000; Gasparini et al., 1999; Lafon-Cazal et al., 1999) and are emerging as a promising drug target in the treatment of epilepsy (Dietrich et al., 1999; Klapstein et al., 1999; Lie et al., 2000; Wong et al., 2001). mGlu4 receptors are negatively coupled to adenylyl cyclase, and their activation inhibits glutamate release from nerve terminals (reviewed by Pin and Duvoisin, 1995). Our recent studies on cultured cerebellar granule cells show that activation of mGlu4 receptors stimulates the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI-3-K) pathways through a Gi/Go protein sensitive to pertussis-toxin. Activation of both pathways mediates the protective activity of mGlu4 receptors against apoptosis by trophic deprivation in cultured cerebellar granule cells (Iacovelli et al., 2002). Thus, mGlu4 receptors activate multiple transduction pathways, which may subserve different functions under physiological or pathological conditions. The overall effect of mGlu4 receptor activation will critically depend on how the signal is propagated toward a specific pathway. We now report that GRK2 may determine the fate of the cell response to mGlu4 receptor activation by desensitizing the stimulation of the MAPK pathway without affecting the inhibition of cAMP formation.
Materials and Methods
Materials. Polyclonal anti-mGlu4 receptor and monoclonal anti-GRK2/3 (clone C5/1) were purchased from Upstate Biotechnology (Lake Placid, NY); polyclonal anti-ERK1/2, polyclonal anti-GRK4, and polyclonal anti-Gβ were from Santa Cruz Biotechnology (Santa Cruz, CA); monoclonal anti-phospho-ERK1/2 was from Cell Signaling Technology (Beverly, MA); monoclonal anti-pan Ras was from Oncogene (San Diego, CA). PP1, PP2, and pertussis toxin (PTX) were purchased from Calbiochem (San Diego, CA). l-2-Amino-4-phosphonobutanoate (l-AP4) was purchased from Tocris Cookson (Bristol, UK); all other drugs were purchased from Sigma-Aldrich (Milan, Italy). The N-terminal domain of GRK2 (Ala2-Thr187)(GRK2-Nter) and the full-length GRK4 cDNA were prepared as described previously (Sallese et al., 2000b). The plasmids encoding for the kinase-dead mutant of GRK2 (GRK2-K220R) and the C-terminal domain of GRK2 (Gly495-Leu689) (GRK2-Cter) were kindly provided by C. Scorer (GlaxoSmithKline, Uxbridge, Middlesex, UK); mGlu4 receptor and GFP-mGlu4 receptor cDNA were kindly provided by J. P. Pin (Montpellier, France); the kinase dead mutant of Ras (RasN17) was kindly provided by J. de Gunzburg (Paris, France); and DynK44A cDNA was kindly provided by J. Benovic.⇓
Quantification of l-AP4—induced mGlu4 receptor internalization
Receptor internalization in HEK293 cells transfected with GFP-mGlu4 receptor alone (control) or co-transfected with GRK2, GRK2-K220R, and Dyn-K44A. Data (means ± S.E.) are the ratio of cytosolic MFP/membrane MFP. L-AP4 = 100 μM. n indicates number of cells.
Cell Culture and Transfection. Human embryonic kidney (HEK) 293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and antibiotics (100 U/ml penicillin, 100 μg/ml streptomycin). Cells were transfected in 10-mm Falcon dishes using 8 μl of LipofectAMINE2000 (Invitrogen, Carlsbad, CA) in OptiMEM medium, and 10 μg of cDNA for 4 h. The cells used for determination of cAMP were cotransfected with 2.5 μg/dish of adenylyl cyclase type V cDNA (Aramori et al., 1997). One day later, cells were split into six-well dishes previously coated with poly(l-lysine) (0.01%), and the experiments were performed 72 h after transfection.
Immunoblotting. At the end of the final incubation, cells were rapidly rinsed in ice-cold PBS and solubilized in Triton X-lysis buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 10% glycerol, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 mM sodium orthovanadate, 50 mM sodium fluoride, and 10 mM β-glycerophosphate) as described previously (Iacovelli et al., 2002). Protein cell lysates (80 μg) were separated by SDS-PAGE electrophoresis, blotted onto nitrocellulose, and probed using specific antibodies. The antibodies were used at the following dilution: anti-phospho-ERK1/2, 1:1000; anti-ERK1/2, 1:2000; anti-mGlu4 receptor, 1:1000; anti-GRK2/3 (clone C5/1), 1:7000; anti-GRK4, 1:2000; and anti-Ras, 1:5000. The immunoreactive bands were visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) using horseradish peroxidase-conjugated secondary antibodies.
Coimmunoprecipitation. HEK293 cells were cotransfected with cDNA encoding for mGlu4 receptor, Gβ1 and Gγ2 subunits (6 μg for each cDNA in 10-mm cell culture dishes). After 72 h, the cells were stimulated with l-AP4 (100 μM, for 10 min), rapidly rinsed with ice-cold PBS, and solubilized in Triton X lysis buffer for 15 min. The lysates were clarified by centrifugation (10,000g for 10 min). Cell lysates (800 μg total) were immunoprecipitated with 5 μg of anti-Gβ antibody for 4 h at 4°C followed by the addition of 50 μl of di Protein-A Sepharose pre-equilibrated in buffer A (20 mM HEPES, 150 mM NaCl, 0.1% Triton X-100, and 5% glycerol), for a further 2 h at 4°C. Immunoprecipitates were washed four times in buffer A, and the pellets were boiled in Laemmli's buffer for 5 min before electrophoresis. Immunoprecipitates and starting materials were subjected to 10% SDS-polyacrylamide gels under reducing condition. After electrophoresis, proteins were transferred to polyvinylidene difluoride membranes and immunoblotted using anti-GRK2 antibody (Upstate Biotechnology).
cAMP Assay. Cultures were incubated in Hanks' balanced salt solution buffer, pH 7.4, containing 0.5 mM 3-isobutyl-1-methylxanthine. l-AP4 (or vehicle) was added 10 min before forskolin (FSK) stimulation (10 mM). After 20 min, the reaction was stopped by substituting the buffer with ice-cold ethanol. Extraction and measurement of cAMP was carried out as described previously (Iacovelli et al., 1996) by RIA (RPA 509; Amersham Biosciences).
Confocal Microscopy. HEK293 cells were transfected with GFP-mGlu4 receptor and the EAAC1 glutamate transporter and cotransfected with empty vector, GRK2 wild type, GRK2-K220R, the GRK2 kinase-dead mutant, or DynK44A, the dynamin dominant-negative mutant cDNA. Twenty-four hours after transfection, the cells were plated onto 35-mm glass-bottomed culture dishes at a density of 2 × 105 and starved for 18 h. The cells expressing GFP-mGlu4 receptor were observed under confocal microscopy (laser scanning microscope; PerkinElmer Life and Analytical Sciences, Boston, MA) using a Nikon 60× numerical aperture 1.4 oil immersion lens. The cells were kept in culture medium at 37°C during analysis. GFP-mGlu4 receptor fluorescent signals were collected sequentially using the “time series” function of the UltraVIEW software (PerkinElmer Life and Analytical Sciences) with single line excitation (488 nm). l-AP4 (100 μM) was applied to the cells during the scanning of GFP-mGlu4 receptor-transfected cells. The internalization of the mGlu4 receptor was quantified as described previously (Sallese et al., 2000a; Iacovelli et al., 2003). For each cell, we traced four lines crossing the cell side by side outside the nucleus. Each line was used to determine the fluorescence at different times during treatment, usually at time 0 (basal) and after 5- and 10-min exposure to l-AP4; for each line, we measured the green fluorescence corresponding to the pixels of the line (UltraVIEW software). The fluorescence corresponding to plasma membrane was that of the 5 to 10 pixels at each side, whereas the fluorescence of the cytosol was that of the 70 to 90 pixels in between. We determined the mean fluorescence per pixel (MFP) of the plasma membrane and the MFP of the cytosol, and we calculated the ratio of cytosol MFP to membrane MFP and multiplied by 100. When this ratio is enhanced, such as after 5-min exposure to l-AP4, it reflects both the increase of the cytosolic fluorescence and the reduction of membrane fluorescence. In a typical experiment for one line under basal conditions, the cytosol MFP was 458 and the membrane MFP was 911 (the ratio = 50%); after 5 min exposure to l-AP4 the cytosol MFP was 703 and the membrane MFP was 422 (the ratio = 167%).
Results
mGlu4 Signaling in Transfected HEK293 Cells. HEK293 cells transfected with mGlu4 receptor cDNA expressed detectable amounts of the receptor protein, as assessed by Western blotting and confocal microscopy (Fig. 1 and Fig. 5). At least three signaling pathways have been associated with mGlu4 receptors: inhibition of adenylyl cyclase activity, stimulation of the MAPK pathway, and stimulation of the PI-3-K pathway (Tanabe et al., 1993; Thomsen et al., 1997; Iacovelli et al., 2002). We examined these pathways in mGlu4 receptor-expressing HEK293 cells challenged with the selective agonist l-AP4. Activation of mGlu4 receptors stimulated the MAPK pathway and inhibited adenylyl cyclase activity, as assessed by Western blot analysis of phosphorylated ERK1/2 (Fig. 2B), and measurements of FSK-stimulated cAMP formation (Fig. 2A), respectively. Both effects were observed at l-AP4 concentrations ranging from 1 to 100 μM (Fig. 2). We could not assess the mGlu4 receptor-mediated stimulation of the PI-3-K pathway in HEK293 cells because, even when mGlu4 transfected cells were serum-starved, the basal levels of phosphorylated Akt were very high, and any further stimulation of the pathway by l-AP4 could not be detected (data not shown).

Immunoblots of mGlu4 receptors, the Ras dominant-negative mutant, RasN17; GRK2; the GRK2 kinase-dead mutant, GRK2-K220R; and GRK4 in HEK293 cells transfected with the respective cDNAs. Mock indicates HEK293 transfected with empty vectors. The two bands of mGlu4 receptors represent receptor monomers (100 kDa) and dimers (200 kDa). Each lane was loaded with 80 μg of proteins from individual culture dishes. Note that control cells express wild-type Ras and GRK2 but not mGlu4 receptor or GRK4 (the latter band at 66 kDa).
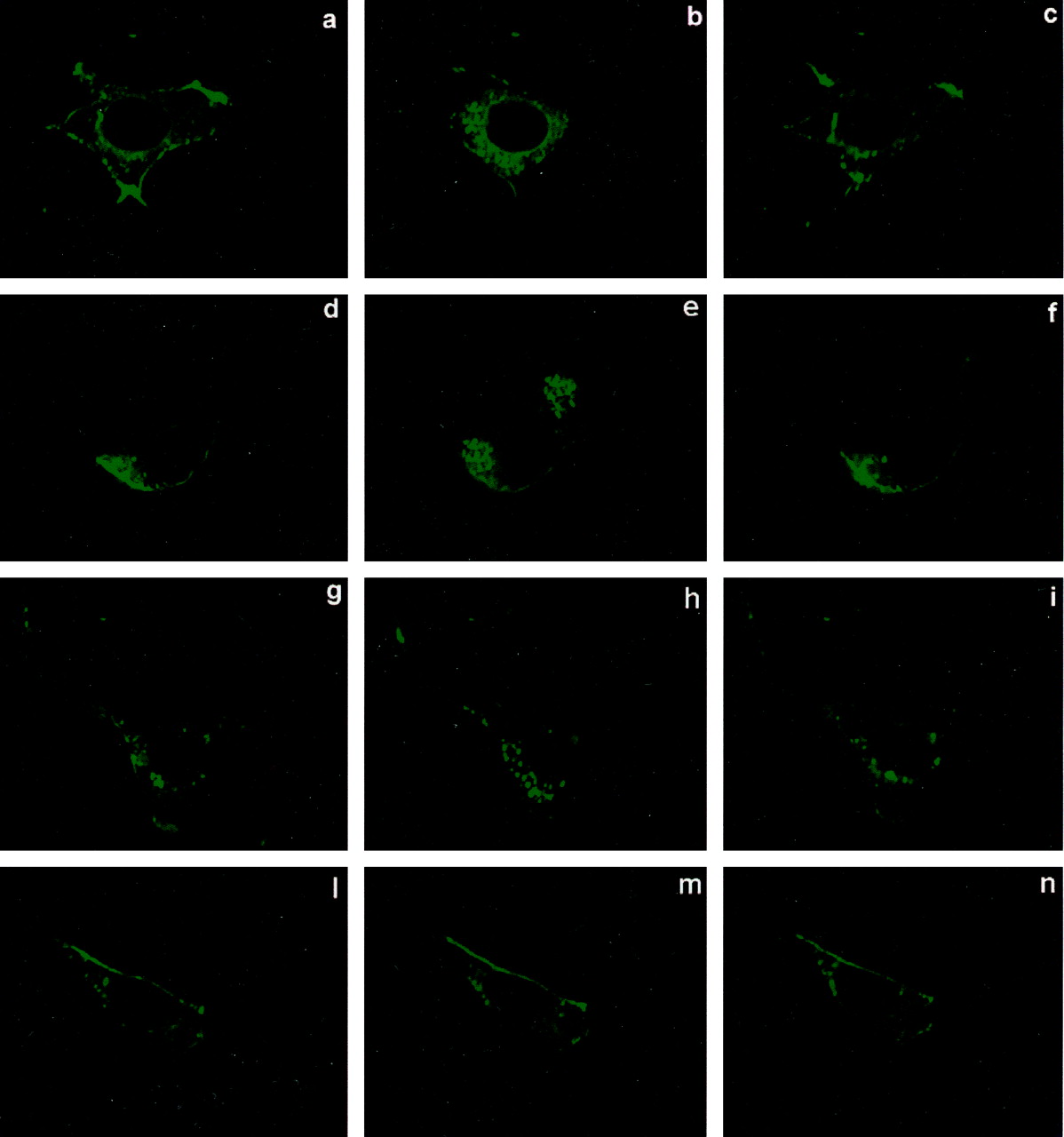
Agonist-dependent internalization of mGlu4 receptors in HEK293 cells. Cells were transfected to express the receptor alone (a-c) or with GRK2 (d-f), GRK2-K220R (g-i) or DynK44A (l-n). The images, obtained by real-time confocal microscopy, show the distribution of mGlu4 receptors tagged with GFP under basal conditions (a, d, g, and l) and after a 5- (b, e, h, and m) or 10-min (c, f, i, and n) exposure to 100 μM l-AP4. Note that the receptor is mainly localized in the plasma membrane but is also present in intracellular compartments under basal conditions. Receptor internalization is clearly shown after 5 min of exposure to l-AP4. This process was reversible and the distribution returned to basal conditions at 10 min. Neither overexpression of GRK2 nor expression of GRK2-K220R affected agonist-induced mGlu4 receptor internalization; in contrast, expression of DynK44A prevented receptor internalization. Images are representative of 10 to 20 cells per condition from three independent experiments.
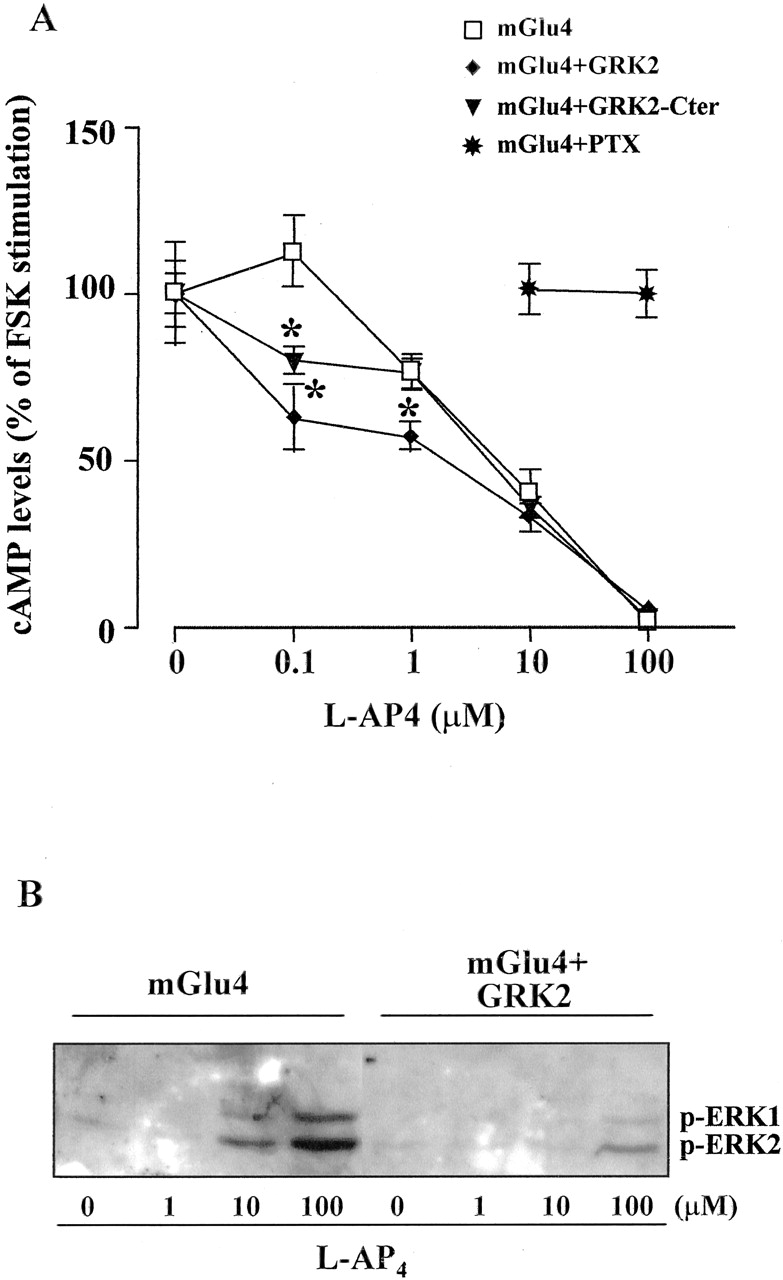
A, concentration-dependent inhibition of FSK-stimulated cAMP formation by l-AP4 in HEK293 cells expressing mGlu4 receptor alone or coexpressed with GRK2 or GRK2-Cter. Cells were treated with different concentrations of l-AP4 and then stimulated with 10 mM FSK. Data obtained with cells pretreated with PTX (1 μg/ml for 12 h) are also shown. Values are means ± S.E.M. from six determinations (two independent experiments with three culture dishes per group). *, p < 0.05 versus mGlu4 receptor alone (one-way analysis of variance + Fisher's least significant difference). B, concentration-dependent stimulation of MAPK by l-AP4 in cells expressing the mGlu4 receptor alone or coexpressed with GRK2. The experiment was performed one additional time with identical results.
mGlu4-Dependent MAPK Activation Involves Ras and Src. The finding that in transfected HEK293 cells, MAPK are stimulated by the mGlu4 receptor confirms our previous results in cultured cerebellar granule cells that natively express mGlu4 receptors (Iacovelli et al., 2002). However, the proteins involved in this mechanism of signal transduction are not defined. To address this point, HEK293 cells were transfected with the mGlu4 receptor and pretreated with PTX (1 μg/ml) for 12 h. The stimulation of the MAPK by l-AP4 was reduced by 60 to 70%, indicating that this pathway is mediated by a PTX-sensitive heterotrimeric G protein (Fig. 3C). The PTX treatment also blocked the mGlu4 receptor-dependent inhibition of adenylyl cyclase activity (Fig. 2A), suggesting that the receptor is interacting with the same G protein to stimulate both pathways. The l-AP4-induced ERK1/2 phosphorylation was reduced by 80 to 90% in cells treated with the Src inhibitors PP1 and PP2 (10 μM), indicating that mGlu4 receptor-dependent MAPK stimulation requires Src activation (Fig. 3C). To test the possible involvement of the monomeric G protein Ras in MAPK activation, we transfected HEK293 cells with a Ras dominant-negative mutant, RasN17, which blocks the activity of endogenous Ras (Iacovelli et al., 2001). The stimulation of MAPK by l-AP4 was reduced by ∼65% in cells expressing RasN17 (Fig. 3, A and B; expression shown in Fig. 1).

Time-dependent stimulation of MAPK by l-AP4 (100 μM) in HEK293 cells expressing mGlu4 receptors. A, representative immunoblot of phosphorylated ERK1/2 (p-ERK1/2) in cells transfected with mGlu4 receptor alone or with RasN17. Unphosphorylated ERK1/2 are also shown. Densitometric analysis is shown in B, where data (means ± S.E.M.) were calculated from three independent experiments. *, p < 0.05 (Student's t test) versus control cells. C, agonist-dependent MAPK activation in mGlu4-expressing cells treated with the Src inhibitors PP1 and PP2 (10 μM) or with PTX (1 μg/ml). PP1 and PP2 were preincubated for 30 min and PTX was preincubated for 12 h before the application of l-AP4 (100 μM, 10 min). Neither PP1 and -2 nor PTX had any effect on basal phospho-ERK1/2 levels (data not shown). The experiment was repeated one additional time with similar results.
GRK2 Selectively Regulates MAPK Signaling by Interacting with Gβγ To study the regulation of mGlu4 receptor signaling by receptor kinases we transfected HEK293 cells with the cDNA encoding for GRK2 (expression shown in Fig. 1). Overexpression of GRK2 slightly increased the ability of l-AP4 in inhibiting forskolin-stimulated cAMP formation at low agonist concentrations (0.1-1 μM), whereas the dose-response curve was unaffected at agonist concentrations >10 μM (Fig. 2A). By contrast, the mGlu4 receptor-stimulated ERK1/2 phosphorylation was substantially blunted at all the l-AP4 concentrations tested (1-100 μM) (Fig. 2B).
GPCR phosphorylation by GRKs usually results in the desensitization of all the pathways that are mediated by the receptor. By contrast, evidence that the overexpression of GRK2 desensitizes the effects of the mGlu4 receptor on MAPK but not on adenylyl cyclase pathways suggests that GRK2 could be acting at a postreceptor level, possibly by a phosphorylation-independent mechanism, as has been shown for other GPCRs (Sallese et al., 2000b). To test this hypothesis, we transfected HEK293 cells with the cDNA encoding a GRK2 mutant GRK2-K220R in which the kinase activity was disrupted by mutagenesis (expression shown in Fig. 1). l-AP4-induced MAPK activation was inhibited to a similar extent by the wild-type GRK2 and by the kinase-dead mutant GRK2-K220R, indicating that GRK2 was acting by a phosphorylation-independent mechanism. We also examined the stimulation of MAPK by l-AP4 in cells transfected with the cDNA encoding the C-terminal domain (GRK2-Cter), or the N-terminal domain (GRK2-Nter) of GRK2, because both these domains contain sites of interaction with Gβγ (Ferguson, 2001; Eichmann et al., 2003). Both of these functional domains mimicked the action of GRK2 in inhibiting the stimulation of MAPK (Fig. 4, A, B, and D), suggesting that interaction of GRK2 with Gβγ subunits is critical for the regulation of mGlu4 receptor-induced MAPK activation. Accordingly, expression of GRK4, which does not interact with Gβγ subunits (Ferguson, 2001) had no effect on l-AP4-stimulated MAPK (Fig. 4C). To document the interaction between GRK2 and Gβγ in intact cells, we performed coimmunoprecipitation experiments. HEK293 cells were cotransfected with mGlu4 receptor cDNA plus the cDNA encoding for Gβ1 and Gγ2 subunits, and after 72 h they were stimulated with l-AP4. We found that GRK2 was coimmunoprecipitated with Gβγ in an agonist-dependent manner (an estimated ∼3-5% of the starting material) thus demonstrating the receptor-induced direct interaction between these proteins (Fig. 4E).

Regulation of mGlu4-receptor-mediated MAPK activation in HEK293 cells. Representative immunoblots of phosphorylated ERK1/2 from HEK293 cells expressing mGlu4 alone or coexpressed with GRK2, GRK2-K220R, GRK2-Cter, GRK2-Nter or with GRK4 are shown (A-C). Densitometric analysis of phosphorylated ERK1/2 in cultures exposed to l-AP4 (100 μM) for 5 min is shown in D Values are expressed as percentage of unstimulated cultures (basal) and were calculated from four to six individual cultures. *, p < 0.05 (one-way analysis of variance + Fisher's least significant difference) versus cells expressing the mGlu4 receptor alone. E, coimmunoprecipitation of GRK2 with the Gβ subunit in cells expressing the mGlu4 receptor and treated with l-AP4 (100 μM, 10 min). HEK293 were cotransfected with mGlu4 receptor cDNA plus cDNA encoding for Gβ1 and Gγ2 subunits. Total cell lysates (800 μg) from untreated and l-AP4-treated cells were immunoprecipitated with anti-Gβ antibody, and GRK2 was detected by immunoblot. An aliquot of starting material (15%) is included for comparison. IP, immunoprecipitating antibody; SM, starting material.
mGlu4 Receptor Internalization and MAPK Activation are Dynamin-Dependent. Using confocal microscopy analysis, we investigated mGlu4 receptor internalization in HEK293 cells transfected with the cDNA encoding for GFP-mGlu4 receptor fusion protein. The mGlu4 receptor was mainly localized in the plasma membrane of unstimulated HEK293 cells (Fig. 5a), although some intracellular vesicles were present under basal conditions, similar to what was reported for mGlu1 and mGlu5 receptors (Dale et al., 2001; Fourgeaud et al., 2003). Exposure to l-AP4 induced the internalization of the receptor in intracellular vesicles with maximal effect at 5 min of agonist treatment, whereas after 10 min, the receptor localization was similar to that of untreated cells (Fig. 5, b and c). The coexpression of GRK2 and GRK2-K220R did not affect this pattern of agonist-promoted internalization of mGlu4 receptor (Fig. 5d-i). By contrast the expression of the dynamin dominant negative mutant dynK44A completely prevented agonist-dependent receptor internalization, indicating that the mGlu4 receptor is internalized by a dynamin-dependent mechanism (Fig. 5, l-n). To test whether mGlu4 receptor internalization is required for the agonist-dependent MAPK activation, as has been shown for several other GPCRs (Lefkowitz et al., 2002), we measured l-AP4-stimulated ERK1/2 phosphorylation in HEK293 cells transfected with dynK44A. In the presence of the dynamin dominant-negative mutant, the activation of MAPK by l-AP4 was almost completely blunted, suggesting that mGlu4 receptor internalization is involved in this signaling pathway (Fig. 6).
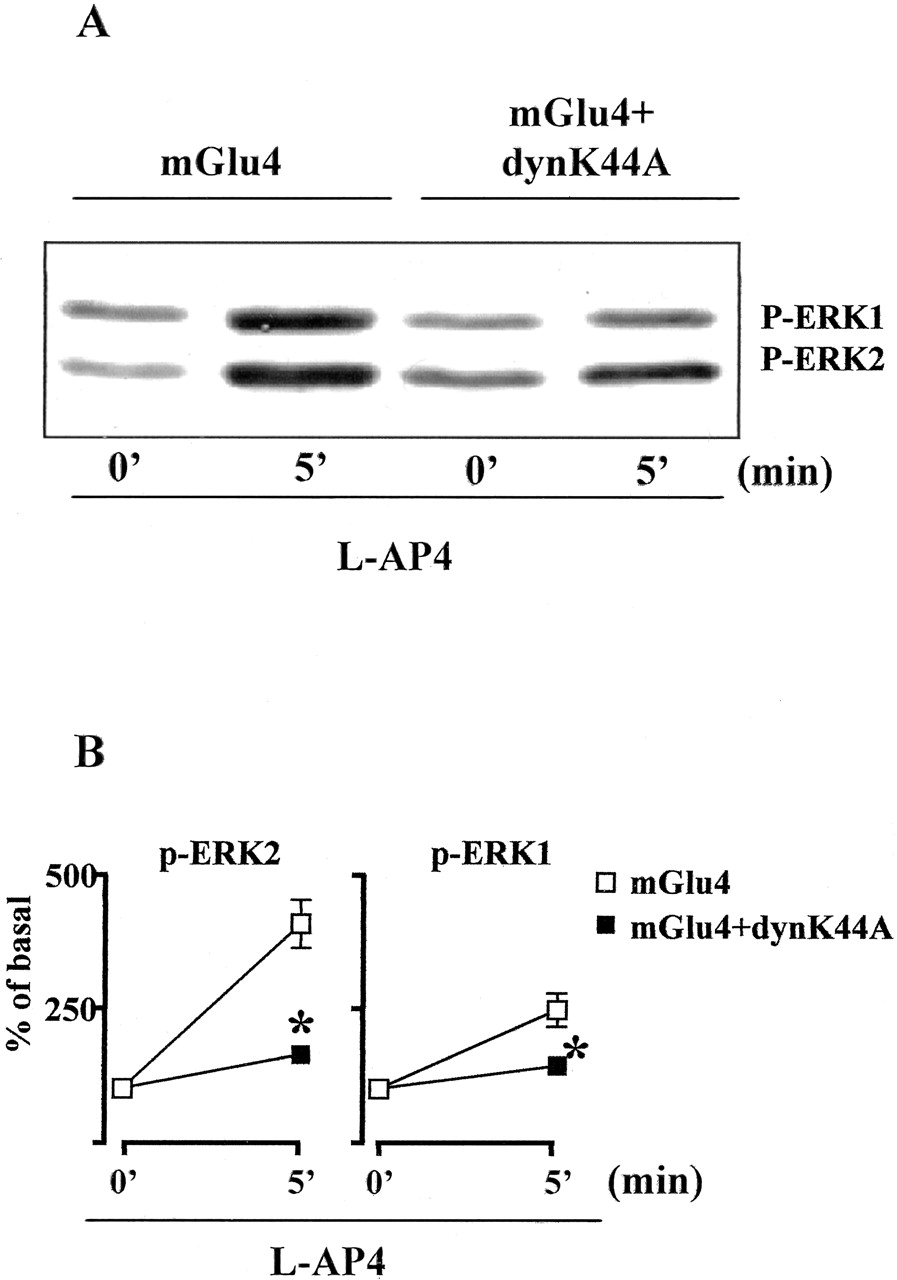
mGlu4-receptor mediated ERK1/2 phosphorylation in cells expressing DynK44A. A representative immunoblot is shown in A. Densitometric analysis calculated from three independent experiments is shown in B. *, p < 0.05 (Student's t test) versus control cells. l-AP4, 100 μM.
Discussion
The stimulation of the mGlu4 receptor transiently expressed in HEK293 cells by l-AP4 induces the inhibition of adenylyl cyclase activity and the activation of MAPK. This is consistent with our data obtained in cultured cerebellar granule cells (Iacovelli et al., 2002), indicating that the coupling of the mGlu4 receptor in transfected HEK293 cells is similar to that occurring in cells natively expressing the receptor. Both inhibition of adenylyl cyclase activity and MAPK activation were blunted by PTX treatment, suggesting that the receptor is interacting with the same PTX-sensitive G protein (presumably Gi). It is likely that the primary event is Gi activation by the mGlu4 receptor, after which the pathway bifurcates and Giα inhibits adenylyl cyclase whereas Gβγ subunits activate MAPK. Accordingly, the expression of GRK2-Cter, which is widely used to inhibit the Gβγ-dependent signaling, drastically decreased the l-AP4-stimulated MAPK activation. The use of specific inhibitors of Src and the expression of the Ras dominant-negative RasN17 suggested that the mGlu4 receptor activates MAPK through an action of the Gβγ subunits on the Src/Ras pathway, as shown for other GPCRs (reviewed by Lefkowitz et al., 2002).
Overexpression of individual GRKs is widely used as a model for the study of how these protein kinases regulate signal transduction of GPCRs (reviewed by Ferguson, 2001). We have used this strategy to examine the regulation of mGlu4 receptor signaling by GRK2 and GRK4, which were formerly shown to mediate homologous desensitization and internalization of mGlu1 and -5 receptors (Dale et al., 2000; Dhami et al., 2002; Iacovelli et al., 2003; Mundell et al., 2003; Sorensen and Conn, 2003). GRK2 is known to mediate homologous desensitization of a variety of receptors coupled to Gi, including α2-adrenergic, CCR5, μ-opiate, A1 adenosine, and lysophosphatidic acid receptors (Jewell-Motz and Liggett, 1996; Aramori et al., 1997; Zhang et al., 1998; Iacovelli et al., 1999; Iacovelli et al., 2002). Unexpectedly, overexpression of GRK2 did not reduce, but slightly potentiated, the inhibition of FSK-stimulated cAMP formation by the agonist l-AP4 in mGlu4 receptor-expressing HEK293 cells. This suggests that the classic Giα-coupled signaling pathway activated by mGlu4 receptors (i.e., the inhibition of adenylyl cyclase) is not desensitized by GRK2. This pathway might be regulated by other protein kinases, such as protein kinases C and A, which are known to phosphorylate group III mGlu receptors and impair their ability to inhibit synaptic transmission in the hippocampus (Macek et al., 1998; 1999; Cai et al., 2001). In contrast, overexpressed GRK2, but not GRK4, inhibited the stimulation of the MAPK pathway induced by l-AP4, suggesting that GRK2 may drive receptor signaling toward the inhibition of adenylyl cyclase rather than the activation of MAPK. The increased potency of l-AP4 in inhibiting adenylyl cyclase activity in cells overexpressing GRK2 is consistent with this hypothesis. These results are different from those obtained in HEK293 cells expressing the mGlu1 receptor or in PC12 cells, where GRK2 enhances the stimulation of MAPK by quisqualate and NGF, respectively (Iacovelli et al., 2003; Rakhit et al., 2001). This excludes that GRK2 has a nonspecific effect on the MAPK pathway and suggests that the enzyme affects the early steps of mGlu4 receptor signaling. The following data indicate that GRK2 desensitizes the mGlu4-stimulated MAPK pathway by interacting with the Gβγ subunit: 1) GRK2 coimmunoprecipitated with Gβ in an agonist-dependent manner; 2) the action of GRK2 was mimicked by the kinase-dead mutant GRK2-K220R, which lacks any catalytic activity but can still interact with Gβγ subunits (Sallese et al., 2000b); 3) the C-terminal domain of GRK2 (which interacts with the Gβγ subunits) also shared the action of GRK2; and 4) no effect was seen with GRK4, which does not contain a PH domain and is therefore unable to interact with Gβγ subunits (Ferguson, 2001).
We also examined the role of GRK2 in mGlu4 receptor internalization. For these experiments, we used confocal microscopy, and receptor internalization was quantified by analysis of the GFP-mGlu4 fluorescence redistribution in real-time experiments. We could not analyze receptor internalization by flow cytometry or by enzyme-linked immunosorbent assay, which provide an accurate quantitative estimate of changes in cell surface receptors, because the N-tagged mGlu4 receptor needed for such experiments was not available in our laboratory. Neither GRK2 nor its kinase-dead mutant had any detectable effect on agonist-induced mGlu4 receptor internalization, whereas a dominant-negative mutant of dynamin nearly abolished internalization. This indicates that internalization is not mediated by a classic GRK/β-arrestin pathway but involves a dynamin-dependent pathway (Claing et al., 2002; Perry and Lefkowitz, 2002). We suggest that mGlu4 receptor stimulation activates MAPK by a mechanism that requires receptor internalization and involves Gβγ signaling to downstream effectors, such as Src and Ras. GRK2 regulates this pathway acting on Gβγ subunits without affecting receptor internalization.
Although inhibition of adenylyl cyclase is related to classic functions of mGlu4 receptors, such as the modulation of glutamate release, the role of MAPK activation in mGlu4 receptor function is a topic for future investigation. Studies carried out in cultured cerebellar granule cells show that activation of the MAPK and PI-3-K pathways mediates the protective action of mGlu4 receptors against apoptosis by trophic deprivation (Iacovelli et al., 2002). An attractive hypothesis is that the extent of GRK2 expression determines the fate of the cell response to mGlu4 receptor activation by regulating the balance between “trophic” effects mediated by the MAPK pathway and classic “synaptic” effects mediated by the inhibition of adenylyl cyclase. It will be interesting to examine whether this balance can be disrupted by molecules that buffer GRK2 (such as excessive amounts of Gβγ subunits) or by an “overactivation” of other GPCRs regulated by GRK2.
Footnotes
-
This work was supported by Telethon-Italy grant 1238.
-
ABBREVIATIONS: MGlu, metabotropic glutamate; GPCR, G-protein coupled receptor; GRK, G protein-coupled receptor kinase; MAPK, mitogen-activated protein kinases; PI-3-K, phosphatidylinositol-3-kinase; PTX, pertussis toxin; PP1, 4-amino-1-tert-butyl-3-(1′-naphthyl)pyrazolo[3,4-d]pyrimidine; PP2, AG 1879: 4-amino-5-(4-chlorophenyl)-7-(tert-butyl)pyrazolo[3,4-d]pyrimidine; l-AP4, l-2-amino-4-phosphonobutanoate; HEK, human embryonic kidney; FSK, forskolin; GFP, green fluorescent protein; MFP, mean fluorescence per pixel; ERK, extracellular signal-regulated kinase.
- Received August 4, 2003.
- Accepted January 29, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}