


Abstract
Dopamine and endogenous cannabinoids display complex interactions in the basal ganglia. One possible level of interaction is between CB1 cannabinoid and D2 dopamine receptors. Here, we demonstrate that a regulated association of CB1 and D2 receptors profoundly alters CB1 signaling. This provides the first evidence that CB1/D2 receptor complexes exist, are dynamic, and are agonist-regulated with highest complex levels detected when both receptors are stimulated with subsaturating concentrations of agonist. The consequence of this interaction is a differential preference for signaling through a “nonpreferred” G protein. In this case, D2 receptor activation, simultaneously with CB1 receptor stimulation, results in the receptor complex coupling to Gαs protein in preference to the expected Gαi/o proteins. The result of this interaction is an increase in the second messenger cAMP, reversing an initial synergistic inhibition of adenylyl cyclase activity seen at subthreshold concentrations of cannabinoid agonist. Additionally, a pertussis toxin insensitive component in the activation of extracellular signal-regulated kinase (ERK) 1/2 kinases by the cannabinoid agonist CP 55,940 [(1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol] is revealed in cells stably expressing both CB1 and D2 receptors. Thus, concurrent receptor stimulation promotes a heterooligomeric receptor complex and an apparent shift of CB1 signaling from a pertussis toxin-sensitive inhibition to a partly pertussis toxin-insensitive stimulation of adenylyl cyclase and ERK 1/2 phosphorylation.
It is well established that dopamine and the endogenous cannabinoids display complex interactions in their control of basal ganglia circuitry. For example, studies have shown an increase in dopamine turnover and release after cannabinoid CB1 receptor stimulation of the striatum (Romero et al., 1995; Szabo et al., 1999) and stimulation of rat striatal D2 receptors in vivo triggers the release of the endocannabinoid anandamide. In turn, the released anandamide inhibits the facilitatory role on movement derived from dopamine D2 receptor stimulation (Giuffrida et al., 1999; Beltramo et al., 2000). Cannabinoid CB1 receptors are densely localized in the basal ganglia (Herkenham et al., 1991; Glass et al., 1997). Specifically, CB1 receptors are located presynaptically on striatal GABAergic projections to the substantia nigra, globus pallidus, and entopeduncular nucleus (Herkenham et al., 1991). Additionally, CB1 receptors are found on corticostriatal terminals and medium spiny neuron dendrites (Gerdeman and Lovinger, 2001; Rodriguez et al., 2001). Thus, CB1 receptors and dopamine D2 receptors are expressed on the same neuronal elements. Individually, both CB1 and D2 receptor agonists inhibit forskolin-stimulated adenylyl cyclase activity in a convergent signal transduction pathway (Meschler and Howlett, 2001). However, these two receptors were found to display a complex signaling interaction in primary striatal neuron cultures (Glass and Felder, 1997). Although individual activation of CB1 and D2 receptors clearly leads to inhibition of adenylyl cyclase, costimulation of these receptors results in a substantial increase in cAMP accumulation, indicating the initiation of alternate signaling pathways when both receptors are activated by agonist (Glass and Felder, 1997). To date, however, no studies have examined the molecular mechanism underlying the interactions between these two receptors.
The richness of GPCR signaling is not solely a consequence of a single heptahelical receptor engaging in multiple specific interactions with different G proteins, but it is also the result of the receptors themselves functioning as a complex. Two or more GPCRs may form a signaling complex with like receptors or form hybrid complexes between members of different GPCR families. Heteromultimers of GPCRs have been shown to possess unique characteristics distinct from the component receptors. Although higher order receptor associations likely exist, for simplicity complexes resulting from GPCR interaction are commonly referred to as “dimers” and the phenomenon as “dimerization”.
A clear example of physical interactions between GPCRs comes from studies of receptor trafficking. Functional GABAB receptors are believed to be heterodimeric combinations of two GPCRs, GABABR1 and GABABR2 (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998), with neither receptor functional alone (Kaupmann et al., 1997; Margeta-Mitrovic et al., 2000; Pagano et al., 2001). The impact of dimerization on ligand binding and signaling is not clearly understood, yet distinct examples for unique properties of receptor dimers have been identified (George et al., 2002). Again, the GABAB receptor, a class C GPCR, serves as a model system; in the heterodimer, GABABR1 is thought to bind the ligand (Galvez et al., 1999), whereas GABABR2 is thought to be the primary G protein contact site (Margeta-Mitrovic et al., 2001; Robbins et al., 2001). Similarly, when two functional class A opioid receptor subtypes, κ and δ, are coexpressed they exhibit markedly different ligand recognition compared with individually expressed receptors (Jordan and Devi, 1999), suggesting that κ-/δ-opioid receptor heterodimerization results in a change in the binding properties of opioid ligands. Several recent studies have also suggested that dimerization may play a role in signaling (George et al., 2002). Heterodimerization between two fully functional receptors has been shown to lead to a synergistic increase in signaling in response to a combination of ligands (Jordan and Devi, 1999; Gomes et al., 2000). Coexpression of μ- and δ-opioid receptors leads to a novel pertussis toxin-insensitive inhibition of adenylyl cyclase at high agonist concentrations (George et al., 2000). Conversely, the β2/β3-adrenergic receptor heterodimer exhibits a pertussis toxin-insensitive activation of adenylyl cyclase and ERK 1/2 mitogen-activated protein kinase (Breit et al., 2004).
Immunohistochemical data suggests that cannabinoid CB1 receptors form homodimers in vivo (Wager-Miller et al., 2002), and biochemical studies indicate that D2 dopamine receptor homodimers may form a new binding site that is capable of binding selective ligands (Ng et al., 1996; Zawarynski et al., 1998). Although functional interactions between CB1 receptors and OX1 orexin receptors (Hilairet et al., 2003), or serotonin (5-hydroxytryptamine) receptors (Devlin and Christopoulos, 2002) have been suggested, data regarding CB1 receptor heterodimer formation is limited. Here, we provide evidence for a physical interaction between two class A GPCRs, the CB1 cannabinoid, and D2 dopamine receptors. Additionally, coactivation of the receptors results in modulation of the relative amount of interacting receptors and initiates a signaling cascade distinct from the component receptors.
Materials and Methods
Reagents were of 99% purity or higher and obtained from commercial sources. CP 55,940 and SR 141716A were provided by the National Institute on Drug Abuse Reagents and Resource Service or purchased from Tocris Cookson Inc. (supplied by Australian Laboratory Services NZ Ltd., Auckland, New Zealand). The N-terminal tagged FLAG-D2L construct was generously provided by Dr. L. Devi (New York University, New York, NY). A nucleotide sequence for the HA epitope (YPYDVPDYA) was flanked by KpnI and BamHI restrictions sites and inserted on the 5′ end of the CB1 receptor coding sequence in the pEF-4a plasmid (Invitrogen, Carlsbad, CA) using standard molecular techniques.
Cell Lines. HEK 293 cells were grown in DMEM with 10% fetal bovine serum and penicillin/streptomycin. Transfections were carried out in 35-mm dishes with 2 μg of DNA and LipofectAMINE (Invitrogen) following the manufacturer's protocol. To establish stable cell lines, cells were selected with G418 (Geneticin) (for the FLAG-D2 construct) or Zeocin (for the HA-CB1 construct). Resistant colonies were evaluated for surface expression of the construct by immunostaining using antibodies against the HA epitope (HA.11; Covance, Berkeley, CA) or the FLAG epitope (M2; Sigma-Aldrich, St. Louis, MO) and a Cy3- or fluorescein isothiocyanate-conjugated secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). The CB1/D2 cell lines were generated by transfecting a stably expressing HEK FLAG-D2L cell line with the HA-CB1 construct followed by selection and evaluation as described above.
Radioligand Binding. Cells were harvested in ice-cold lysis buffer (10 mM MOPS, 1 mM EGTA, and 100 μM AEBSF, pH 7.4) and homogenized in a glass Dounce homogenizer. P2 membranes were prepared by initial centrifugation at 4000g. The supernatant was then centrifuged at 20,000g, and the pellet resuspended in TME buffer (50 mM Tris-HCl, pH 7.5, 3 mM MgCl2, and 1 mM EDTA) containing saturating sucrose and stored at –80°C until used. Several single and coexpressing clones from both individually expressing and coexpressing cell lines were screened by receptor saturation binding assay using 1 to 36 nM [3H]SR 141716A (CB1) or 0.3 to 20 nM [3H]raclopride (D2). Binding reactions were performed for 90 min at 30°C in TME buffer with 5 mg/ml fraction V bovine serum albumin in a final reaction volume of 100 μl containing 20 μg of membrane protein. The incubation was terminated by addition of 200 μl of ice-cold TME buffer, and samples were filtered though a printed filtermat A (GF-C) filter (PerkinElmer Life and Analytical Sciences, Boston, MA) and washed twice with 300 μl of TME buffer on a cell harvester (Inotech Biosystems, Rockville, MD). The filter was then dried before the addition of Multilex (melt-on scintillant; PerkinElmer Life and Analytical Sciences) and was counted for 5 min in a Microbeta Trilux. Displacement binding assays were performed as described above at either 5 nM [3H]SR 141716A or 10 nM [3H]raclopride with 10–11 to 5 × 10–5 M CP 55,940 or quinpirole, respectively. To test for changes in affinity due to dimerization, displacement of [3H]SR 141716A by CP 55,940 was performed in the presence and absence of 100 nM quinpirole, whereas [3H]raclopride displacement by quinpirole was performed in the presence and absence of 100 nM CP 55,940.
Measurement of cAMP. Cells were plated in poly-l-lysine-coated 96-well plates at 40,000 cells/well and allowed to grow overnight in full media. Cells were then incubated at 37°C with serum-free DMEM containing 5 mg/ml bovine serum albumin and 500 μM 3-isobutyl-1-methyl xanthine (Sigma-Aldrich) for 20 to 30 min before drugs being added at 2× concentration in the same media. cAMP accumulation was terminated after 15 min by removal of the media and addition of 50 μl of ice-cold absolute ethanol and incubation at –20°C for at least 10 min. The ethanol was then evaporated off, and the cell lysate was resuspended in 50 μl of cAMP assay buffer (20 mM HEPES and 5 mM EDTA, pH 7.5). Twenty-five microliters of this buffer was incubated with 25 μl [3H]cAMP (∼1 nM final concentration) and 50 μl of 0.02% (w/v) cAMP-dependent protein kinase (Sigma-Aldrich) (in 1 mM sodium citrate and 2 mM dithiothreitol, pH 6.5) overnight at 4°C. Unbound cAMP was absorbed by the addition of 50 μl of charcoal [5% (w/v) charcoal, 0.2% (w/v) bovine serum albumin in cAMP assay buffer], and centrifugation at 3000g for 5 min. The supernatant was mixed with Starscint (PerkinElmer Life and Analytical Sciences) and counted for 5 min/well in a Microbeta Trilux. When required, cells were treated with 10 ng/ml pertussis toxin for 16 h before cAMP assay.
Immunoprecipitation and Western Immunoblotting. Cells at 80% confluence in poly-d-lysine-coated six-well tissue culture plates were washed and equilibrated in HEPES-buffered saline (130 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, and 10 mM HEPES, pH 7.5) containing 1.0 mg/ml bovine serum albumin. Drug treatments were added from a 1000× concentrated stock in dimethyl sulfoxide and incubated at 37°C for 7 min. The plates were placed on ice, washed with ice-cold phosphate-buffered saline, and then scraped into IP buffer (150 mM NaCl, 1 mM EDTA, 1 mM EGTA, and 20 mM Tris, pH 7.5, containing 0.5% CHAPS) and incubated for 10 min. The soluble supernatant, obtained after centrifugation at 14,000g for 10 min., was added to M2-agarose resin (Sigma-Aldrich) and incubated overnight at 4°C. The resin was washed twice with IP buffer, and the bound proteins were eluted with 100 mM glycine, pH 2.5. Eluted proteins were resolved by SDS-PAGE in 7.5% Tris/glycine gels and transferred to nitrocellulose. After a 1-h block in 5% nonfat dry milk in Tris-buffered saline with 0.05% Tween 20, the blots were incubated with primary antibody in 2.5% nonfat dry milk/Tris-buffered saline with 0.05% Tween 20 overnight. The immunocomplex was visualized with enhanced chemiluminescence reagent after extensive washing and incubation with horseradish peroxidase-conjugated secondary. Exposure of the blot to radiographic film provided a permanent record. In a select set of experiments, the supernatant from the 14,000g centrifugation was subjected to additional centrifugation at 100,000g for 1 h at 4°C. This supernatant was immunoprecipitated as described previously. The FLAG and HA immunoreactivities were visualized concurrently after SDS-PAGE and transfer to nitrocellulose with a dual wavelength detection system (Odyssey Imager; Li-Cor, Lincoln, NE) following the manufacturer's protocols.
Immunocytochemistry Staining and Fluorescent Imaging. Immunocytochemistry of the cells was conducted using procedures well established in the laboratory (Hsieh et al., 1999). In brief, cells were grown on poly-d-lysine-coated glass coverslips. Drug treatments were performed on cells washed with HEPES-buffered saline containing 0.2 mg/ml bovine serum albumin and applied in the same buffer. The cells were washed with ice-cold phosphate-buffered saline and fixed with 4% formaldehyde solution for 20 min. After blocking for 1 h in Tris-buffered saline with 5% donkey serum and 0.1% saponin, the cells were incubated with primary antibody overnight at 4°C in Tris-buffered saline with 2.5% donkey serum. Immunostaining for D2 receptors was conducted with an affinity-purified polyclonal antibody isolated from rabbits immunized with a fusion protein containing amino acids 216 to 311 of the human D2 long isoform expressed from the pET30c plasmid (Novagen, Madison, WI). CB1 receptors were detected with the HA.11 monoclonal antibody. After extensive washes, immunoreactivity was visualized with a fluorescently labeled secondary antibody, incubated for 1 h at room temperature, washed again, dried, mounted, and visualized on a Leica SP1/MP confocal microscope at the W. M. Keck Center for Advanced Studies on Neuronal Signaling at the University of Washington.
Measurement of ERK 1/2 Phosphorylation. Cells in 12-well tissue culture plates were grown to 80% confluence and then incubated overnight in DMEM containing 1% fetal bovine serum. Drug treatments were the same as described for the immunoprecipitations. After a 7-min incubation, the cells were placed on ice, the buffer was removed, and SDS-PAGE loading buffer was added directly to the well. Proteins were resolved as described above in 10% Tris/glycine gels. After the blocking step, the blots were incubated with a phospho-ERK 1/2-specific antibody (Cell Signaling Technology Inc., Beverly, MA) overnight. Again, the immunocomplex was visualized by enhanced chemiluminescence and exposure to radiographic film.
Results
Coexpression, Immunoprecipitation, and Western Blotting of CB1 and D2 Receptors. Fluorescent immunostaining of FLAG-D2 (Fig. 1A) and HA-CB1 (Fig. 1B) receptors indicates that these proteins are both expressed on the cell membrane in HEK cells. After treatment with 10 nM CP 55,940 and 100 nM quinpirole for 7 min at 37°C, D2 receptors remain at the cell surface together with the majority of the CB1 receptors (data not shown). Immunoprecipitation reactions were carried out on HEK 293 cells stably expressing FLAG-tagged D2 receptors, HA-tagged CB1 receptors, or both, using M2 anti-FLAG antibody resin. After separation of the proteins by SDS-PAGE, Western immunoblotting was performed using a rabbit anti-FLAG antibody (Sigma-Aldrich) or the HA.11 monoclonal antibody (Covance). Successful immunoprecipitation of D2 receptors is evident in Fig. 1C, lane 1 by three immunoreactive bands. In this gel system, CB1 receptors migrate with an apparent molecular mass of ∼66 kDa, corresponding to the major immunoreactive band, as indicated by the arrow in Fig. 1D, lane 1. Often, higher HA.11-immunoreactive bands were seen. These likely represent nondissociated oligomers of CB1 receptors with D2 receptors and possibly other proteins. Blocking the interaction of the M2 resin with the FLAG-D2 receptor by coincubation with excess FLAG peptide prevents the immunoprecipitation of FLAG-D2 receptors (Fig. 1C, lane 2) as well as HA-CB1 receptors (Fig. 1D, lane 2). No association was seen when cells individually transfected with HA-CB1 or FLAG-D2 were extracted, and then the extracts were mixed and immunoprecipitation and Western immunoblotting were performed (data not shown).

CB1 receptors immunoprecipitate with D2 receptors. Immunostaining of membrane localized FLAG-D2 (A) and HA-CB1 (B) receptors coexpressed in HEK cells. Fixed cells were incubated with anti-D2 and anti-HA antibodies after permeabilization and blocking. The primary antibodies were visualized after incubation with fluorescently conjugated secondary antibodies by confocal microscopy. Scale bar, 25 μm. Soluble lysates from HEK cells stably expressing HA-CB1 and FLAG-D2L receptors were immunoprecipitated with M2 anti-FLAG resin. Precipitated proteins were resolved on a 7.5% acrylamide SDS-PAGE and detected by Western immunoblot with a rabbit anti-FLAG (C) or the HA.11 monoclonal antibody (D). Immunoprecipitated FLAG-D2 receptors are detected in C, lane 1. In D, lane 1, the coprecipitated HA-CB1 receptor immunoreactivity is visible at ∼66 kDa. Addition of 7.5 μg of FLAG peptide to the lysate before immunoprecipitation prevents the immunoprecipitation of both receptors (C and D, lane 2).
Agonist Modulation of Dimer Formation. To assess the influence of agonist stimulation on receptor dimerization, we investigated the consequence of treatment with either the D2 receptor agonist quinpirole, the CB1 receptor agonist CP 55,940 or both, on coimmunoprecipitation of the tagged CB1/D2 receptor complex. Figure 2A depicts a representative increase in HA-immunoreactive proteins from CB1/D2-expressing HEK cells treated with quinpirole and an increasing concentration of CP 55,940 and immunoprecipitated with M2 resin. To ensure that the observed increases in HA-CB1 immunoreactivity are specific to a solubilized receptor complex with FLAG-D2 receptors, immunoprecipitations with M2 resin were conducted on supernatants of cell lysates after 14,000g and 100,000g centrifugation. In both supernatants, HA-CB1 receptors are coimmunoprecipitated with FLAG-D2 receptors (Fig. 2B, lane 1 and 5, top), and treatment with 10 nM CP 55,940 and 100 nM quinpirole resulted in an increase in HA immunoreactivity in the immunoprecipitation product (Fig. 2B, lanes 3 and 7, top). Conversely, FLAG-D2 immunoreactivity was not significantly different between basal and drug-treated conditions (Fig. 2B, lanes 1 and 3 and lanes 5 and 7, bottom). Furthermore, when compared across drug treatment and supernatant preparation, the FLAG-D2 immunoreactivity varied by less than 10%. For each condition evaluated, inclusion of the FLAG peptide blocked immunoprecipitation of both FLAG-D2 and HA-CB1 (Fig. 2B, even-numbered lanes). When compared across several experiments, we find that stimulation with either quinpirole or CP 55,940, respectively, did not significantly increase the level of dimer compared with basal (Fig. 2, C and D). In contrast, if the cells were treated with the most efficacious concentration of quinpirole (100 nM) and increasing concentrations of CP 55,940, a concentration-dependent increase in dimer was observed (Fig. 2, A and C). A similar increase was observed with 10 nM CP 55,940 and increasing concentrations of quinpirole (Fig. 2D). The CB1 receptor inverse agonist SR 141716A inhibited the 20% increase in dimer produced by 100 nM quinpirole in the absence of CB1 receptor agonist in a concentration-dependent manner, indicating that dimer formation required the CB1 receptor to be in an active conformation (Fig. 2E).

Receptor activation regulates CB1/D2 receptor dimers. HEK cells stably expressing CB1 and D2 receptors were treated with agonist(s) as indicated for 7 min at 37°C before preparation of IP lysates. A, soluble fraction was immunoprecipitated with M2 resin, and the product was resolved by SDS-PAGE. Coimmunoprecipitation is detected under basal conditions (lane 1), with little increase after incubation with quinpirole (lane 2). However, increased HA-CB1 receptors are detected by Western immunoblot with the HA.11 monoclonal antibody in samples treated with both CP 55,940 and quinpirole (lanes 3–6). B, CB1 and D2 receptors coimmunoprecipitate in supernatants subjected to 14,000g (lane 1) or 100,000g (lane 5) centrifugation, indicating a solubilized receptor complex. Treatment with 10 nM CP 55,940 and 100 nM quinpirole before cell lysis resulted in increased HA-CB1 in the immunoprecipitation product from supernatants prepared under either condition (lanes 3 and 7). The amount of FLAG-D2 immunoreactivity is not statistically different among the samples. However, addition of the FLAG peptide to the supernatant blocks the immunoprecipitation of both FLAG D2 and HA-CB1 receptors (even-numbered lanes). The relative amount of CB1 receptor in the IP product from several experiments was determined by densitometry of HA immunoreactivity on the radiographic film is plotted in C. D, relative HA-CB1 receptor immunoreactivity in the IP product after treatment with a constant concentration of CP 55,940 and increasing concentrations of quinpirole. E, samples were pretreated for 5 min with SR 141716 and then treated with 100 nM quinpirole for 7 min at 37°C before preparation of IP lysates. The relative HA-CB1 immunoreactivity from the samples is plotted as percentage of difference from basal HA immunoreactivity. Data are mean values ± S.E.M. with n = 4 to 9 replicates. *, p < 0.05 compared with no drug control; †, p < 0.05 compared with 100 nM quinpirole.
Signaling and Binding Interactions. CB1 and D2 receptor interactions were examined for their ability to modulate levels of cAMP and activation of ERK 1/2. As shown in Table 1, both receptors inhibited adenylyl cyclase when expressed individually in HEK 293 cells. Additionally, in stably cotransfected HEK cells, application of a single agonist resulted in inhibition of forskolin stimulated cAMP accumulation with equivalent efficacy and potency to singly expressing cells (Table 1). Despite similar receptor levels (D2, 537 ± 51 fmol/mg; CB1, 460 ± 71 fmol/mg), the D2 receptor was significantly more efficacious in producing inhibition (D2, 66 ± 6% inhibition versus CB1, 44 ± 4% inhibition; p = 0.004).
Characterization of stably transfected HEK cell lines
Receptor number (Bmax) and affinity (Kd) was determined by [3H]SR 141716A binding (CB1) or [3H]raclopride binding (D2) as described under Materials and Methods. Log IC50 values for the displacement of [3H]SR 141716A by CP 55,940 performed in the presence and absence of 100 nM quinpirole, and [3H]raclopride displacement by quinpirole performed in the presence and absence of 100 nM CP 55,940 are presented. The ability of the receptor activation to alter forskolin (fsk)-stimulated cAMP accumulation was determined as described under Materials and Methods. Emax values for each concentration-response curve are presented as a percentage of the total forskolin-mediated stimulation. After overnight treatment with pertussis toxin, D2-mediated inhibition was blocked in both singly and dually expressing cells, whereas a small, but significant concentration-dependent increase in cAMP accumulation was observed for both CB1-containing cell lines. Data are mean ± S.E.M. for three experiments performed in quadruplicate for cAMP assays or duplicate for binding assays.
Next, concentration-response relationships for adenylyl cyclase inhibition by each receptor were repeated in the presence of maximally efficacious agonist concentrations for the alternate receptor (100 nM). As can be seen in Fig. 3, this resulted in a biphasic interaction. For CB1 receptor stimulation in the presence of 100 nM quinpirole, an increase in the extent of inhibition was observed at low CP 55,940 concentrations, whereas at higher concentrations the combination resulted in an increase in the level of cAMP (Fig. 3A). For quinpirole dose-response curve at 100 nM CP 55,940, a U-shaped dose-response curve was also observed, with high concentrations of both drugs resulting in an increase in cAMP accumulation (Fig. 3B).
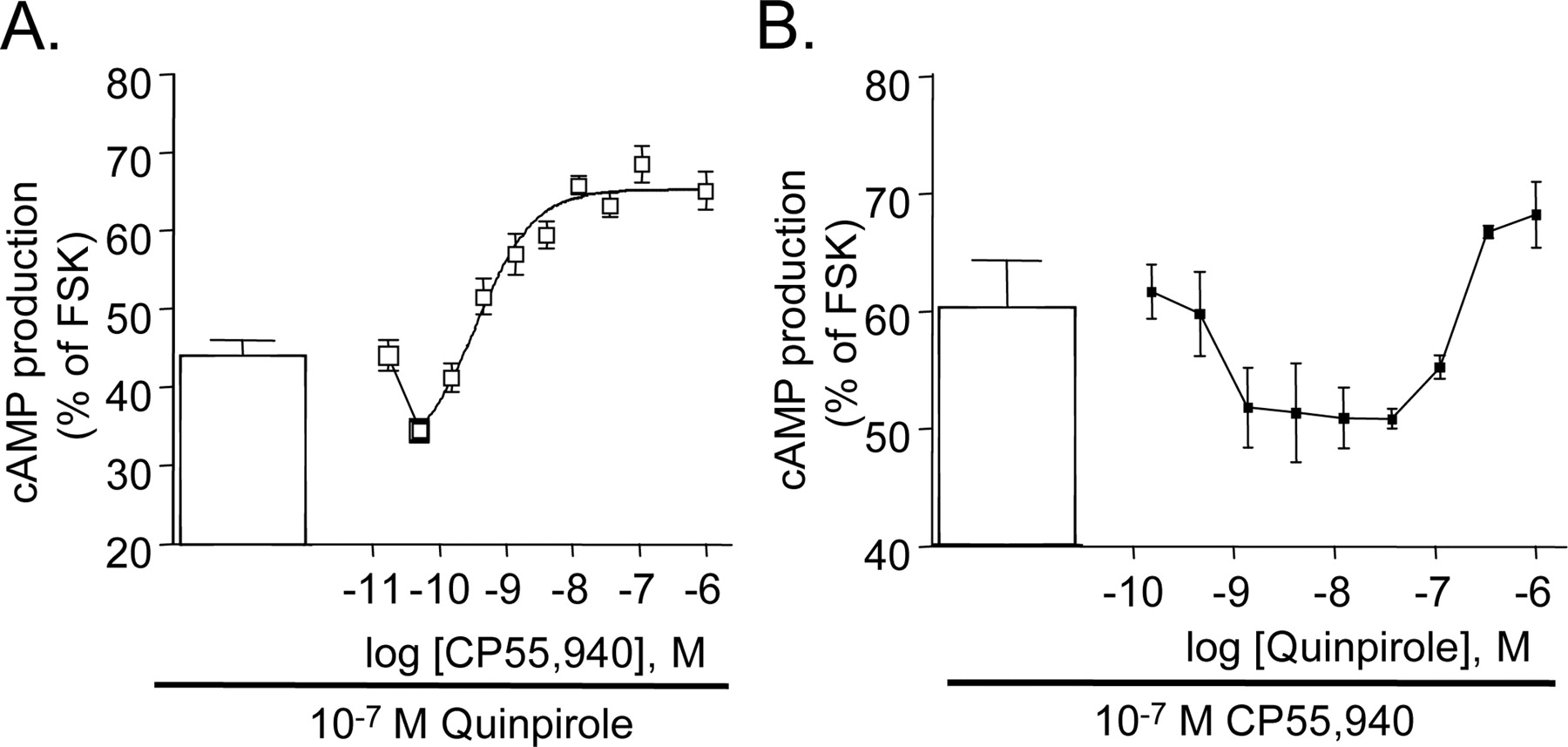
Accumulation of cAMP in HEK 293 cells stably expressing both CB1 and D2 receptors when stimulated with both agonists. A, CP 55,940 concentration-response curve in the presence of 100 nM quinpirole. B, quinpirole concentration-response curve in the presence of 100 nM CP 55,940. Data shown are mean ± S.E.M. for a representative experiment of three or more experiments performed in triplicate.
To determine whether these changes represented a shift in agonist binding capacity, the binding affinities of quinpirole or CP 55,940 were assessed by their ability to displace a specific receptor antagonist (raclopride or SR 141716A, respectively). For each receptor, these assays were performed in the presence and absence of maximal occupancy of the second receptor. However, agonist affinities were not significantly altered for either drug (Table 1).
Influence of Pertussis Toxin Treatment on cAMP Production. Overnight treatment with pertussis toxin ablated D2 receptor-mediated inhibition of adenylyl cyclase in both singly and coexpressing HEK 293 cells (Table 1). In contrast, CB1 receptor stimulation resulted in a small but statistically significant increase in cAMP accumulation after pertussis toxin treatment in both singly and CB1/D2-expressing cell lines (Table 1) consistent with previous observations in primary striatal cultures (Glass and Felder, 1997).
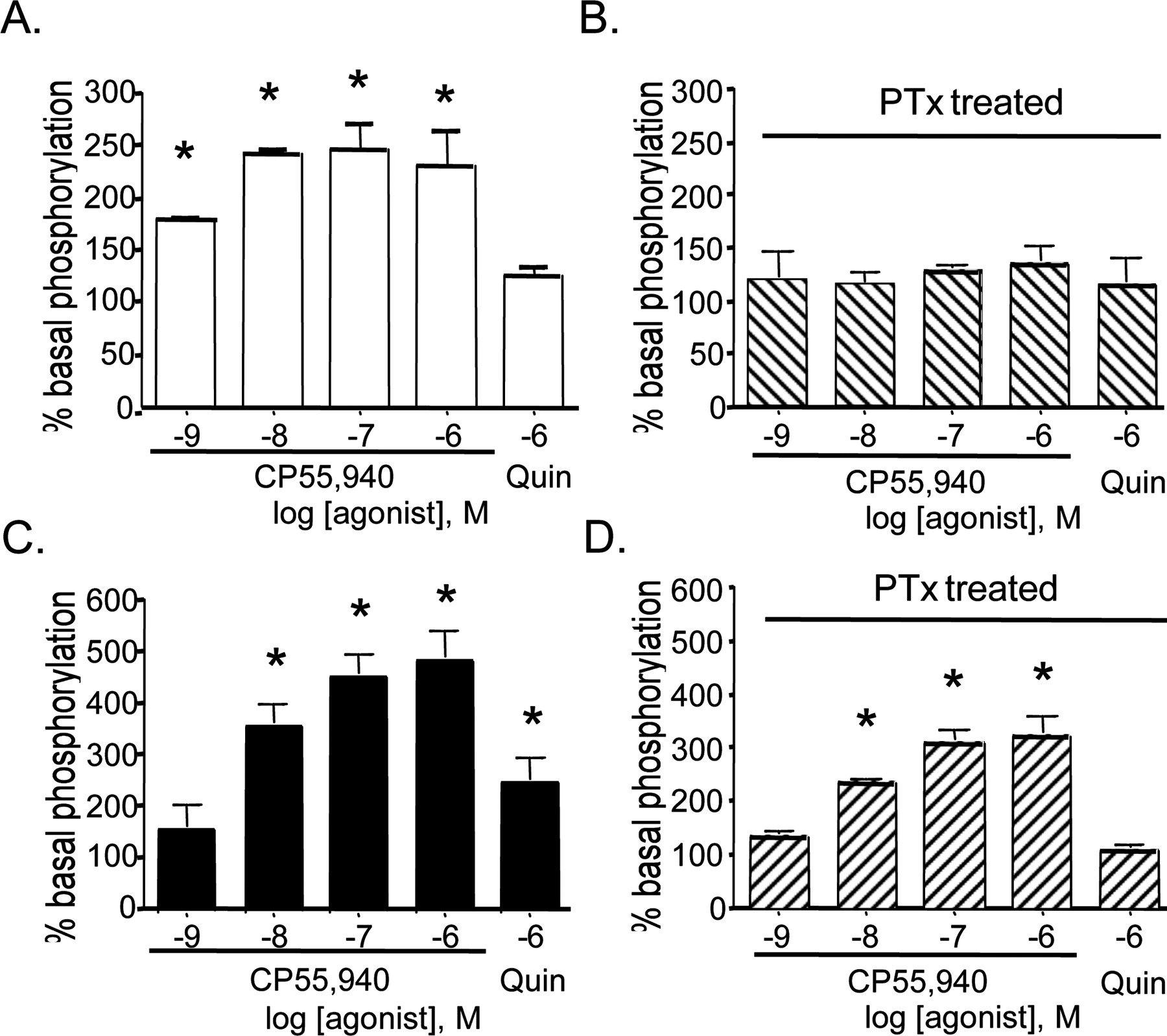
Activation of ERK 1/2. Stimulation of HEK 293 cells expressing CB1 receptors (Fig. 4A) with CP 55,940 for 7 min increased phosphorylation of ERK 1/2, reaching a maximum of 246 ± 26% of basal. Quinpirole stimulation of D2 receptors expressed in HEK cells resulted in a 186 ± 24% increase of ERK 1/2 phosphorylation over basal (data not shown). In HEK cells expressing the receptors individually, overnight incubation with 100 ng/ml pertussis toxin resulted in the blockade of CB1 (Fig. 4B) or D2 (data not shown) receptor-mediated ERK 1/2 phosphorylation. Stimulation of HEK 293 cells expressing both CB1 and D2 receptors with either CP 55,940 or quinpirole increased phosphorylation of ERK 1/2 (Fig. 4C) as described for singly expressing cells. In comparison with the cAMP assay, CB1 receptor activation in HEK cells expressing CB1 and D2 receptors resulted in a more efficacious stimulation of ERK 1/2 phosphorylation (487 ± 49% of basal), relative to D2 receptor stimulation (248 ± 49% of basal phosphorylation) (Fig. 4C). Furthermore, coexpression of CB1 and D2 receptors caused a statistically significant increase in maximal CB1 receptor-mediated ERK 1/2 phosphorylation, whereas D2 receptor activation of ERK 1/2 was not significantly changed. In contrast to HEK cells expressing either CB1 or D2 receptors, an overnight treatment of HEK cells expressing both receptors with pertussis toxin resulted in only a partial attenuation of the CP 55,940-induced ERK 1/2 phosphorylation, whereas it abolished the quinpirole-mediated activation (Fig. 4D).

Activation of ERK 1/2 in HEK HA-CB1 and HEK HA-CB1/FLAG-D2 cells. Cells were treated with agonist for 7 min at 37°C and then lysed directly into SDS-PAGE loading buffer. After Western immunoblot, phospho-ERK 1/2 immunoreactivity was determined by densitometry. Agonist-induced activation of ERK 1/2 in HEK HA-CB1 cells (A) and after overnight pretreatment with 100 ng/ml pertussis toxin (B). Activation of ERK 1/2 in HA-CB1- and FLAG-D2 receptor-expressing HEK cells (C) and after overnight pretreatment with 100 ng/ml pertussis toxin (D). Data are mean ± S.E.M. with n = 4 to 6 determinations. *, p < 0.05 compared with no drug control by one-way analysis of variance.
Discussion
The extensive colocalization of CB1 and D2 receptors within the basal ganglia suggests the potential for interplay between the signal cascades of these two receptors. Striatal neurons expressing both receptor types display distinct signaling pathways with receptor coactivation, including the putative coupling of CB1 receptors to Gαs proteins (Glass and Felder, 1997). In these studies, we demonstrate that stable expression of CB1 and D2 receptors in HEK 293 cells results in a pertussis toxin-insensitive component to CB1 receptor activation of ERK 1/2 and a stimulation of adenylyl cyclase activity after simultaneous activation of both receptors. Additionally, we have identified that the CB1 and D2 receptors form a basal level of associated receptor complex that is regulated by receptor activation and therefore may mediate the associated signaling pathways.
Although CB1 and D2 receptor dimers were found under basal conditions, there is no necessity for coexpression of both receptors for proper surface expression. In contrast to GABAB receptors, generation of cell lines expressing only a single receptor type with the appropriate signal transduction and membrane localization was readily possible. Evaluation of the cotransfected cell clones using nonpermeabilized, live cell immunostaining as well as confocal microscopy of permeabilized, fixed immunostained cells (Fig. 1, A and B) ensured that both the CB1 and D2 receptors are expressed at the cell surface, an important feature since it has been demonstrated that the CB1 receptor can be poorly trafficked to the cell surface in some heterologous expression systems (Andersson et al., 2003; our unpublished observations.) The specificity of the receptor interaction requires that both receptors be expressed in the same cells since neither coculture of singly transfected cells nor mixing of the detergent extracts from singly transfected cells resulted in detectable coimmunoprecipitation. Additionally, CB1 receptors display selectivity toward interacting partners since stable coexpression of CB1 and GABAB receptors did not result in detectable dimerization as determined by immunoprecipitation (data not shown). Remarkably, CB1/D2 receptor complexes are stable and not disrupted by detergent solubilization of the cells with CHAPS (Fig. 1D) or Triton X-100 (data not shown). However, the denaturing conditions during electrophoresis are sufficient to disrupt the complex because the majority of the CB1 immunoreactivity is found at a mobility appropriate for the monomeric receptor. Consistent with other reports (Zawarynski et al., 1998), a portion of the D2 immunoreactivity is found at higher molecular masses and may represent a more stable D2 homodimer (Fig. 1C).
Treatment of the CB1/D2-expressing cells with either receptor agonist in isolation has little effect on relative dimer levels. However, when both receptors are simultaneously stimulated by agonist, there is a marked increase in the amount of CB1 receptor coimmunoprecipitated, suggesting that the active conformation of the CB1 and D2 receptor enhances their interactions. The similar affinities for radio-labeled antagonists for their respective receptor in both dually and singly expressing cell lines, along with the inability of activation of one receptor to alter agonist affinity for the second receptor, suggest that the binding site for these ligands is not altered by dimer formation. Similarly, the β2/ β3-adrenergic heterodimer displays the same ligand affinities as its component receptors, yet displays markedly distinct signal transduction (Breit et al., 2004). As yet, signaling efficacy of the CB1/D2 dimer and the proportion of the receptor in the signaling conformation remain open issues. However, a dynamic pool of receptor complexes consisting of CB1 homodimers, D2 homodimers, and CB1/D2 heterodimers likely exists in these cells and is likely to occur in other cells such as medium spiny neurons.
Dynamic regulation of receptor dimers has been studied in a number of homo- and heterodimer receptor systems, with varying results from coimmunoprecipitation and energy transfer techniques. Many proposed homo- and heterodimers are insensitive to agonist stimulation (Jordan and Devi, 1999; Zeng and Wess, 1999; AbdAlla et al., 2000). In response to agonist stimulation, the relative proportion of receptor dimers has been suggested to increase for β2-adrenergic (Hebert et al., 1996; Angers et al., 2000), SST5 (Rocheville et al., 2000), chemokine CCR5 (Vila-Coro et al., 2000), and thyrotropin-releasing hormone (Kroeger et al., 2001) receptors and decrease for δ-opioid homodimerization (Cvejic and Devi, 1997) and dopamine D1/adenosine A1 receptor heterodimerization (Gines et al., 2000). Nevertheless, these studies need cautious interpretation. It is possible that ligand binding may stabilize constitutive heterodimers, thereby making them more readily identifiable (Ramsay et al., 2002; Percherancier et al., 2005). To try to assess the stability of our heterodimer, we have used two solubilization methods, CHAPS and Triton X-100, and found identical amounts of HA-CB1 receptor immunoreactivity after FLAG immunoprecipitation under both conditions, suggesting the dimer is stable under these conditions. Alternatively, ligand binding to both receptors simultaneously may alter the conformation of the D2 receptor such that the FLAG epitope is more available on the heterodimers than on the homodimers, and therefore a higher percentage of the receptors precipitated seem to be heterodimers. No differences in the total amount of FLAG-D2 immunoprecipitated were observed under any conditions tested (Fig. 2B), and we have established that agonist binding to CB1 receptors does not seem to alter the binding site of D2 receptors significantly (as measured by ligand affinity; Table 1). In our study, conformational changes in the receptors affecting dimer stability but not ligand binding can not be ruled out; however, altered access to the epitope seems an unlikely explanation given their N-terminal location.
Distinct signal transduction interactions were elicited by concurrent agonist stimulation in the cells expressing both CB1 and D2 receptors. At a maximally active concentration of quinpirole, low concentrations of a CB1 receptor agonist result in a potentiation of adenylyl cyclase inhibition. As the concentration of CP 55,940 was increased, however, no further reduction in cAMP was observed; rather, increasing concentrations of CP 55,940 result in an increase of the second messenger. This result was in agreement with that previously observed in primary cultures of striatal neurons (Glass and Felder, 1997) in which a CB1 receptor agonist dose-dependent increase in cAMP was observed after maximal D2 receptor agonist inhibition. In contrast, a recent report suggested that simply coexpressing the CB1 and D2 receptors was sufficient to result in stimulation of cAMP in response to CB1 receptor activation (Jarrahian et al., 2004). However, this study used transiently transfected CB1 receptors that results in different expression levels, receptor localization, and widely varying ratios of CB1-to-D2 receptor from cell to cell. These results from transiently expressing cells could not be replicated in our model. Since signal transduction is dependent on GPCR location (Luttrell et al., 1999) and transient expression results in a portion of intracellularly localized CB1 receptors, aberrant signaling pathways of the CB1 receptor after transient transfection is a likely explanation.
The addition of increasing concentrations of quinpirole to cells already inhibited with a maximally effective concentration of CP 55,940 resulted in a U-shaped dose-response curve in cAMP assays. Low concentrations of quinpirole increased the inhibition observed with CP 55,940 alone, whereas higher concentrations resulted in increasing cAMP levels. Previous studies strongly suggest that this stimulation is mediated by coupling of CB1 to Gαs (Glass and Felder, 1997). However, how activation of D2 leads to this switch in G protein affinity requires further study. It is possible that D2 receptor activation may enhance the coupling of CB1 receptors to Gαs by altering the ratio of Gαi- versus Gαs-proteins available to the receptor. However, the relatively weak coupling of CB1 receptors to Gαs in these cells after pertussis toxin treatment suggests that lack of availability of Gαi may be insufficient on its own to efficiently switch signaling.
Coexpression of CB1 and D2 receptors also strongly modified ERK 1/2 signaling. CB1 receptor activation in cells expressing CB1 and D2 receptors caused nearly a 2-fold greater phosphorylation of ERK 1/2 compared with cells expressing the CB1 receptor alone. This increase in maximal stimulation of ERK 1/2 was observed only for CB1 receptor activation because the Emax for D2 receptor stimulation was not affected in a significant manner by coexpression. Consistent with the expected Gαi/o-mediated pathway and previously published results, pertussis toxin was able to completely inhibit ERK 1/2 activation in cells expressing either the CB1 (Bouaboula et al., 1995) or D2 receptor (Luo et al., 1998) individually. However, in cells expressing CB1 and D2 receptors, inactivation of Gαi/o proteins by pertussis toxin resulted in only a 35% reduction in CP 55,940-stimulated phosphorylation but ablated activation through D2 receptors. Interestingly, the EC50 for the pertussis toxin-insensitive component of ERK 1/2 activation is less than 10 nM and is similar to the half-maximal concentrations of CP 55,940 for adenylyl cyclase stimulation as well as agonist stimulated dimer formation.
Taking the data as a whole in cells expressing both CB1 and D2 receptors, we see CP 55,940 as weakly inhibiting to adenylyl cyclase despite a significant G protein activation occurring as indicated by ERK 1/2 phosphorylation. In contrast, the D2 receptor-mediated inhibition of adenylyl cyclase was significantly more robust than its ERK 1/2 phosphorylation, suggesting that the signaling pathways for these two effectors diverge early. In cells expressing CB1 and D2 receptors, as likely occurs in some neurons, CP 55,940 stimulates ERK 1/2 phosphorylation in part through a pertussis toxin-insensitive pathway. Additionally, in these cells, once adenylyl cyclase activity is maximally inhibited by D2 receptor activation, CB1 receptor stimulation partially reverses the inhibition. Similar concentrations of agonists also increase CB1/D2 receptor dimer formation. Thus, it is reasonable to propose that CP 55,940 signaling through the CB1/D2 dimer activates Gαs protein, and that CB1 receptor agonist-induced increases in dimer levels partially reverse D2 receptor-mediated inhibition of adenylyl cyclase.
Coexpression of CB1 and D2 G protein-coupled receptors in HEK 293 cells models the colocalization of these receptors on striatal GABAergic neurons in vivo. Here, we have shown an interaction between CB1 and D2 receptors that is dynamic, regulatable, and dependent on the active conformation of the receptors. In addition, costimulation of the receptors has been shown to modulate receptor signaling of two distinct intracellular pathways. Our data show that the proportion of CB1 and D2 receptors existing as heterodimers may play a role in determining the responses of medium spiny neurons, and other neurons expressing both receptors, to neurotransmitter. Furthermore, depletion of either receptor pool in striatal pathologies such as Huntington's disease (Glass et al., 2000) may preclude adequate agonist-regulation of dimer number and disrupt normal signaling.
Footnotes
-
This work was supported by an Auckland Medical Research Foundation grant (to M.G.) and by National Institutes of Health grants DA11322 (to K.M.), DA00286 (to K.M.), DA07278 (to C.S.K.), and DA013410 (to K.M. and C.S.K.).
-
K.M. and M.G. contributed equally to this work.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.006882.
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; ERK, extracellular signal-regulated kinase; CP 55,940, (1R,3R,4R)-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-4-(3-hydroxypropyl)cyclohexan-1-ol; SR 141716A, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride; HA, hemagglutinin; TME, Tris/MgCl2/EDTA; HEK, human embryonic kidney; DMEM, Dulbecco's modified Eagle's medium; MOPS, 4-(N-morpholino)propanesulfonic acid; AEBSF, 4-(2-aminoethyl)benzenesulfonyl fluoride; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate; PAGE, polyacrylamide gel electrophoresis; IP, immunoprecipitation.
- Received September 2, 2004.
- Accepted February 14, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}