


Abstract
mRNAs for the neuronal nicotinic acetylcholine receptor (nAChR) α6 and β3 subunits are abundantly expressed and colocalized in dopaminergic cells of the substantia nigra and ventral tegmental area. Studies using subunit-null mutant mice have shown that α6- or β3-dependent nAChRs bind α-conotoxin MII (α-CtxMII) with high affinity and modulate striatal dopamine release. This study explores the effects of β3 subunit-null mutation on striatal and midbrain nAChR expression, composition, and pharmacology. Ligand binding and immunoprecipitation experiments using subunit-specific antibodies indicated that β3-null mutation selectively reduced striatal α6* nAChR expression by 76% versus β3+/+ control. Parallel experiments showed a smaller reduction in both midbrain α3* and α6* nAChRs (34 and 42% versus β3+/+ control, respectively). Sedimentation coefficient determinations indicated that residual α6* nAChRs in β3–/– striatum were pentameric, like their wild-type counterparts. Immunoprecipitation experiments on immunopurified β3* nAChRs demonstrated that almost all wild-type striatal β3* nAChRs also contain α4, α6, and β2 subunits, although a small population of non-β3 α6* nAChRs is also expressed. β3 subunit incorporation seemed to increase α4 participation in α6β2* complexes. 125I-Epibatidine competition binding studies showed that the α-CtxMII affinity of α6* nAChRs from the striata of β3–/– mice was similar to those isolated from β3+/+ animals. Together, the results of these experiments show that the β3 subunit is important for the correct assembly, stability and/or transport of α6* nAChRs in dopaminergic neurons and influences their subunit composition. However, β3 subunit expression is not essential for the expression of α6*, high-affinity α-CtxMII binding nAChRs.
Neuronal nAChRs are a widely distributed, heterogeneous class of cationic channels. Their opening is controlled by the endogenous neurotransmitter acetylcholine or exogenous agonists such as nicotine. They are composed of pentameric assemblies of homologous subunits (Corringer et al., 2000; Lindstrom, 2000). So far, 12 neuronal subunit genes have been identified in vertebrates (α2–α10, β2–β4) (Le Novère and Changeux, 1995; Gotti and Clementi, 2004).
Distinct subunit composition defines the many neuronal nAChR subtypes, which exhibit diverse functional and pharmacological properties. The subtypes may be divided into two subfamilies. The first comprises heteropentameric nAChRs combining ligand binding subunits (α2, α3, α4, and α6) with structural (β2 and β4), and sometimes auxiliary α5 and β3), subunits. The second encompasses nAChRs that bind α-bungarotoxin (αBgtx) and are generally thought to exclusively contain ligand binding subunits (α7, α8, α9, or α10), although other subunits have been implicated by Yu and Role (1998). The second family of nAChRs may be either homopentameric or heteropentameric.
By acting on mesostriatal dopaminergic system nAChRs, nicotine plays an important role in mediating several behavioral effects such as the modulation of locomotor activity, reinforcement, and habit learning (Di Chiara 2000). These effects are thought to be mediated by increased dopamine release in the mesostriatal dopamine system (Picciotto et al., 1998).
Striatal 6-hydroxydopamine-lesioning studies in rats (Zoli et al., 2002) and knockout mice (Champtiaux et al., 2003) indicate that there are two major nAChR subtype populations in the striatum: α6β2* and α4(non-α6)β2*. Both of these populations are heterogeneous, differently expressed by the dopaminergic and nondopaminergic neurons, and involved in mediating the release of dopamine from striatal synaptosomes (Champtiaux et al., 2003; Quik et al., 2003; Salminen et al., 2004). Ligand binding and dopamine release studies have indicated that the two major striatal nAChR populations can be distinguished by their differential interaction with α-conotoxin MII (α-CtxMII), which selectively binds to and blocks the α6β2* population with high affinity (Zoli et al., 2002; Champtiaux et al., 2003). This α6β2* nAChR population may be intimately associated with the β3 subunit. In situ hybridization studies have identified the selective colocalization of α6 and β3 mRNAs in dopaminergic neurons (Le Novère et al., 1996; Azam et al., 2002). Furthermore, β3 subunit-null mice exhibit alterations in behaviors that are controlled by nigrostriatal and mesolimbic dopaminergic activity, and lose much of the α-CtxMII-sensitive portion of striatal dopamine release (Cui et al., 2003).
This study used a combination of ligand binding, immunoprecipitation, and immunopurification techniques to test whether the β3 and α6 subunits are indeed extensively associated with each other. It also examined the consequences of β3-null mutation on the expression, properties, and composition of striatal and midbrain nAChRs. β3 subunit deletion markedly and selectively reduced α6* nAChR expression in both striatum and midbrain, without altering the residual α6β2* nAChRs' α-CtxMII affinity. The results also indicated that almost all β3 subunits are present in α6* nAChRs, where they seem to promote the formation of a complex α4α6β2β3 nAChR subtype. However, some wild-type α6* nAChRs are expressed that do not contain β3. Furthermore, the dopamine cell body and -terminal nAChR populations differ. In particular, the midbrain contains a novel (in mammalian brain) α3β3* nAChR subtype. It seems that β3 expression is not necessary for the expression of all mesostriatal α6β2* nAChRs, but it is critical for the correct assembly and/or transport of a major subset.
Materials and Methods
Antibody Production and Characterization
The polyclonal antibodies (Abs) used were subunit-specific, produced in rabbit against peptides derived from the C-terminal (COOH) or intracytoplasmic loop (CYT) regions of the rat (R), human (H), or mouse (M) subunit sequences, and affinity purified as described previously (Zoli et al., 2002). Most of the Abs were the same as those described previously (Zoli et al., 2002, Champtiaux et al., 2003; Moretti et al., 2004). Given the central role of β3 in this investigation, we generated an antiserum specifically directed against a mouse β3 subunit cytoplasmic peptide (M-CYT), DGTESKGTVRGKFPGKKKQTPTSD, to replace the rat-directed antiserum used previously. These experiments critically depend on Ab specificity and immunoprecipitation efficacy, both of which were carefully checked here or previously (Zoli et al., 2002; Champtiaux et al., 2003; Moretti et al., 2004) in control experiments using tissue obtained from relevant nAChR subunit null mutant animals and/or heterologously expressed nAChRs. Most importantly, the CYT- and COOH-directed β3 Abs failed to immunoprecipitate significant amounts (less than 1%) of [3H]Epi labeled receptors from β3–/– mouse superior colliculus, confirming their specificity. In β3+/+ superior colliculus, however, both Abs were effective, although the CYT-directed Ab was slightly more so than the COOH-directed Ab (28.0 ± 0.9% of sites immunoprecipitated versus 21.2 ± 1.2%, respectively; n = 4). For this reason, we used the anti-β3 CYT Ab exclusively where possible. It is important to note that although the Abs used here have been tested and shown to have high efficacy, few Ab preparations achieve absolutely complete recovery of their targets. Therefore, all immunoprecipitation values should be treated as close, but potentially slightly low, determinations of the proportion of target subunits in the nAChR populations investigated.
Animals
Mice modified to contain a null mutation in the β3 nAChR subunit gene (Cui et al., 2003) were bred at the Institute for Behavioral Genetics (University of Colorado, Boulder, CO). All mice used in this study were maintained on the original mixed C57BL/6J/129SvEv/Tac background. Mice were housed five per cage, and the vivarium was maintained on a 12-h light/dark cycle (lights on from 7:00 AM to 7:00 PM). Mice were given free access to food and water. Mice were genotyped by Polymerase chain reaction, using DNA extracted from tail clippings obtained at approximately 40 days of age. All procedures used in this study were approved by the Animal Care and Utilization Committee of the University of Colorado, Boulder. All mice used in this study were between 60 and 120 days of age.
Preparation of Membranes and 2% Triton X-100 Extracts from Striatum and Midbrain
The tissues were dissected, immediately frozen on dry ice, and stored at –80°C for later use. In every experiment, the tissues from striatum (0.15–0.25 g) or midbrain (0.15–0.25 g) were homogenized in 10 ml of 50 mM sodium phosphate, pH 7.4, 1 M NaCl, 2 mM EDTA, 2 mM EGTA, and 2 mM phenylmethylsulfonyl fluoride (PMSF) with a Potter homogenizer. The homogenates were then diluted further in the same buffer and centrifuged for 1.5 h at 60,000g.
The procedures of homogenization, dilution, and centrifugation of the total membranes were performed twice, after which the pellets were collected, rapidly rinsed with 50 mM Tris-HCl, pH 7, 120 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, and 2 mM PMSF, and then resuspended in the same buffer containing a mixture of 20 μg/ml of each of the following protease inhibitors: leupeptin, bestatin, pepstatin A, and aprotinin. Triton X-100 to a final concentration of 2% was added to the washed membranes, which were extracted for 2 h at 4°C.
The extracts were then centrifuged for 1.5 h at 60,000g, recovered, and an aliquot of the resultant supernatants was collected for protein measurement using the bicinchoninic acid protein assay (Pierce Chemical, Rockford, IL) with bovine serum albumin as the standard.
Binding Assay and Pharmacological Experiments
(±)-[3H]Epibatine (Epi; specific activity, 56–60 Ci/mmol) and 125I-Epi (specific activity, 2200 Ci/mmol) were purchased from PerkinElmer Life and Analytical Sciences (Boston MA); 125I-αBgtx (specific activity, 214 Ci/mmol) was purchased from Amersham Biosciences Inc. (Piscataway, NJ); nonradioactive Epi was from RBI/Sigma (Natick, MA); α-CtxMII was synthesized as described by Cartier et al. (1996). All other compounds were sourced from Sigma-Aldrich (St. Louis, MO).
Membranes. [3H]Epibatidine Binding. β2* and β4* nAChRs bind [3H]Epi with picomolar affinity, and α7 receptors bind it with nanomolar affinity (Gerzanich et al., 1995). To ensure that the α7 subtype did not contribute to [3H]Epi binding, tissue extract and immunoprecipitation epibatidine binding experiments were performed in the presence of 2 μM αBgtx, which specifically binds to the α7 subtype and prevents Epi from binding to the subtypes containing this subunit. Binding to membrane homogenates obtained from striatal and midbrain membranes was performed overnight by incubating aliquots of the membrane with [3H]Epi concentrations ranging from 0.005 to 2.5 nM at 4°C. Nonspecific binding (averaging 5–10% of total binding) was determined in parallel by means of incubation in the presence of 100 nM unlabeled Epi. At the end of the incubation, the samples were filtered on a GFC filter soaked in 0.5% polyethylenimine and washed with 15 ml of 10 mM sodium phosphate, pH 7.4, plus 50 mM NaCl, and the filters were counted in a liquid scintillation counter.
125I-α-Bungarotoxin binding. Saturation experiments were performed by incubating midbrain and striatal membranes overnight with 0.01 to 10 nM 125I-αBgtx at 20°C. For 125I-αBgtx, 2 mg/ml bovine serum albumin was added to the suspension buffer. Specific radioligand binding was defined as total binding minus nonspecific binding determined in the presence of 1 μM nonradioactive αBgtx.
[3H]Epibatidine binding to solubilized receptor. Triton X-100 extracts were preincubated with 2 μM αBgtx for 3 h and then labeled with 2 nM [3H]Epi. Tissue extract binding was performed using DE52 ion-exchange resin (Whatman, Maidstone, UK) as described previously (Vailati et al., 2000).
Immunoprecipitation of [3H]Epibatidine-Labeled Receptors by Anti-subunit–specific Antibodies
Membrane preparations were extracted with 2% Triton X-100 (1 h; 22°C). Extracts were preincubated with 2 μM αBgtx, labeled with 2 nM [3H]Epi and then incubated overnight with a saturating concentration of affinity purified Abs (20–30 μg). The immunoprecipitate was recovered by incubating the samples with beads containing bound anti-rabbit goat IgG (Technogenetics, Milan, Italy). The level of Ab immunoprecipitation was expressed as the percentage of [3H]Epi-labeled receptors immunoprecipitated by the antibodies (taking the amount present in the Triton X-100 extract solution before immunoprecipitation as 100%) or as femtomoles of immunoprecipitated receptors per milligram of protein.
Striatal β3* Population Immunopurification and Analysis
For each immunopurification, striatal membranes of 20 to 30 mice (0.4–0.5 g) were prepared as described above. The membranes (12 ml) were then extracted by addition of 2% Triton X-100 as described above and centrifuged. Extracts (14–15 ml) were incubated three times with 5 ml of Sepharose-4B with bound anti-β3 CYT Abs to remove the β3 subunit-containing receptors (β3* population). This β3* population was eluted from column by means of incubation with 100 μM M-CYT peptide, and the flow-through of the β3 column was then analyzed for the subunit content of the remaining nAChRs. Analysis of the purified β3* population's subunit content was performed by immunoprecipitation using subunit-specific Abs, as described above, after labeling with 2 nM [3H]Epi.
Pharmacological Experiments on Immunoimmobilized Subtypes
Affinity-purified anti-α6 or anti-β2 Abs (10 μg/ml in 50 mM phosphate buffer, pH 7.5) were bound to microwells (Maxi-Sorp; Nunc, Naperville, IL) by means of overnight incubation at 4°C. The following day, the wells were washed to remove the excess of unbound Abs and then incubated overnight at 4°C with 200 μl of 2% Triton X-100 striatal membrane extract prepared from the β3+/+ and β3–/– genotypes (containing 10–30 fmol of 125I-Epi binding sites). After incubation, the wells were washed and immobilized receptors quantified using 125I-Epi binding.
Immobilized nAChRs were incubated overnight at 4°C with 200 μl of 125I-Epi at concentrations ranging from 0.005 to 1 nM. All of the incubations were performed in a buffer containing 50 mM Tris-HCl, pH 7, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 2 mg/ml bovine serum albumin, and 0.05% Tween 20. Specifically labeled ligand binding was defined as total binding minus the binding in the presence of 100 nM unlabeled Epi. Epibatidine and α-CtxMII inhibition of 125I-Epi binding to the immobilized nAChRs was measured by preincubating the indicated concentrations of the compounds for 30 min at room temperature, followed by overnight incubation with 0.05 nM 125I-Epi. After incubation, the wells were washed seven times with ice-cold phosphate-buffered saline containing 0.05% Tween 20, and bound radioactivity was recovered by incubation with 200 μl of 2 N NaOH for 2 h. Bound radioactivity was then determined by γ counting.
Sucrose Gradient Centrifugation
Linear 5 to 20% sucrose gradients in phosphate-buffered saline plus 1 mM PMSF and 0.1% Triton X-100 were prepared using a Buckler gradient maker (Fort Lee, NJ) and stored for 4 h at 4°C before use. The volume of each gradient was 12 ml. Then, 500 μl of 2% Triton X-100 extracts obtained from Torpedo californica electric organ (0.5–1 g, labeled with 6 nM 125I-αBgtx) and 2% Triton X-100 extracts of the striatum or midbrain of β3+/+ or β3–/– mice were loaded on the gradients and centrifuged for 14 h at 40,000 rpm in a Beckman SW41. Fractions of 0.5 ml were collected from the top of the gradient and directly counted on a gamma counter (in the case of the T. californica gradients) or added to the affinity-purified anti-α6 or anti-β2 Abs bound to microwells, and processed as described for the pharmacological experiments.
Data Analysis. The experimental data obtained from the saturation binding experiments performed on immunoimmobilized subtypes were analyzed by means of a nonlinear least square procedure using the LIGAND program as described by Munson and Rodbard (1980). The calculated binding parameters were obtained by simultaneously fitting three independent experiments.
The selection of the best fitting (i.e., one- versus two-site model) and evaluation of the statistical significance of the parameters (i.e., comparison of the binding parameters of the two groups) were based on the F-test for the “extra sum of square” principle. A p value of <0.05 was considered statistically significant (Munson and Rodbard, 1980).
The Ki values of α-CtxMII and epibatidine inhibition binding were also determined by means of the LIGAND program using the data obtained from three independent competition experiments and compared by means of the F-test as described above.
Results
nAChR Expression in the Striatum and Midbrain of β3 Genotypes. To determine the effect of β3 nAChR gene deletion on overall nAChR expression in the mesostriatal pathway, we performed ligand binding studies using membranes prepared from β3 wild-type (β3+/+), heterozygote (β3+/–), and knockout (β3–/–) mice.
[3H]Epibatidine Binding nAChRs. [3H]Epi binding was measured in striatum and midbrain membranes obtained from each β3 genotype. The membranes were preincubated with 2 μM αBgtx to block epibatidine binding at αBtx-sensitive sites. Binding was conducted using a saturating concentration of [3H]Epi (2 nM).
The density of striatal [3H]Epi binding nAChRs was 98.7 ± 4.0, 90.2 ± 2.7, and 93.0 ± 7.0 fmol/mg of protein (mean ± S.E.M. of three experiments) in β3+/+, β3+/–, and β3–/– mice, respectively. These values were not statistically different from each other (Fig. 1A). The density of midbrain [3H]Epi binding nAChRs was higher than in the striatum, being 143.9 ± 5.6, 152.1 ± 6.2 and 137.6 ± 7.6 fmol/mg of protein (mean ± S.E.M.; n = 3) in β3+/+, β3+/–, and β3–/– mice, respectively. Again, no statistically significant differences were seen between the genotypes (Fig. 1C). Both the [3H]Epi binding site densities, and the lack of effect of the β3-null mutation on them are very similar to the results reported by Cui et al. (2003).
125I-α-Bungarotoxin Binding nAChRs. The number of 125I-αBgtx binding nAChRs in the striatum (mean values ± S.E.M. of three experiments) of β3+/+, β3+/–, and β3–/– mice was 50.1 ± 10.0, 51.7 ± 5.3, and 49.8 ± 10.2 fmol/mg protein, respectively (Fig. 1B). The level of 125I-αBgtx nAChRs in mouse midbrain (mean values ± S.E.M. of three experiments) was 71.7 ± 9.5, 67.7 ± 13.0, and 66.0 ± 5.5 fmol/mg protein, respectively (Fig. 1D). Neither region exhibited a statistically significant difference between β3 genotypes (Fig. 1, B and D).
Subunit Composition of Striatal [3H]Epibatidine nAChRs. The results mentioned above demonstrate that deletion of the β3 subunit has no measurable effect on the overall amount of nAChR expression. However, as outlined under Introduction, several distinct nAChR subtypes have been reported in the mesostriatal dopamine pathway. To quantify the relative contribution of each nicotinic subunit to [3H]Epi binding in the striatum, we performed quantitative immunoprecipitation experiments using subunit-specific antibodies and [3H]Epi-labeled nAChRs. The results, expressed as femtomoles of immunoprecipitated nAChRs per milligram of protein are the mean values of three or four separate experiments for each subunit, in each genotype (Fig. 2A).
Most striatal nAChRs from β3+/+ mice could be precipitated by the β2 (85 ± 1.1%) and α4 (67 ± 4.3%) antisera. nAChRs precipitable by the α5 (13.7 ± 2.5%), α6 (18.0 ± 0.7%), and β3 antisera (19 ± 0.7%) were also prevalent, whereas fewer than 3% of the nAChRs apparently contained the α2, α3, or β4 subunits. The overall subunit composition of [3H]Epi binding nAChRs from β3+/+ mouse striatum strongly resembled that previously reported for the striatum of rats, and α4+/+ and α6+/+ mice (Zoli et al., 2002; Champtiaux et al., 2003). Although the level of β3* nAChRs (19%) in β3+/+ striatum is very similar to that previously attributed to α-CtxMII-sensitive sites in striatal tissue (Whiteaker et al., 2000), this fraction is higher than that previously reported for α4+/+ and α6+/+ mice (8%; Champtiaux et al., 2003). This discrepancy probably arose because a new Ab, specifically raised against a cytoplasmic peptide of the mouse β3 subunit, was used in the current experiments (rather than the previously targeted, but slightly different, corresponding rat peptide). This new Ab has a higher immunoprecipitation capacity against mouse β3 subunit, and when tested in the striatum of α6+/+ mice, gave results almost identical to those obtained in β3+/+ mice (C. Gotti and N. Champtiaux, unpublished data), resolving the difference between the present and previous studies.

Expression of [3H]Epi (A) and 125I-αBgtx (B) binding nAChRs in striatum membranes from each β3 genotype (β3+/+, β3+/–, and β3–/–). The membrane homogenates (M) or 2% Triton X-100 extracts (E) were prepared as described under Materials and Methods The reported values are expressed as femtomoles of specific labeled [3H]Epi and 125I-αBgtx binding sites per milligram of protein and are the mean values ± S.E.M. of five experiments performed in triplicate for the different genotypes. Expression of [3H]Epi (C) and 125I-αBgtx (D) binding nAChRs in midbrain membranes from each β3 genotype (β3+/+, β3+/–, and β3–/–). The binding experiments were performed as described for A and B.
Interestingly, β3+/– striatum expressed a population of nAChR subtypes that was indistinguishable from β3+/+ striatum. The lack of difference between nAChR populations isolated from β3+/+ and β3+/– mice (seen here, and in midbrain preparations, see below) presumably indicates that even though β3 mRNA expression in the heterozygotes is significantly reduced (Cui et al., 2003), the residual mRNA transcription drives sufficient subunit protein production to allow receptor assembly similar to that of wild-type mice.
Reassuringly, β3* nAChRs were not detected in β3–/– striatum, confirming the specificity of the new antiserum. β3 subunit-null mutation greatly reduced the expression of striatal α6* nAChRs (from 16.6 ± 2.0 fmol/mg of protein in β3+/+ to 4.0 ± 1.2 fmol/mg of protein in β3–/–). In contrast, deletion of the β3 subunit did not significantly affect expression of striatal α4*, α5*, or β2* nAChRs. The main finding in the striatum of β3–/– mice is therefore a dramatic (76%) decrease in α6* nAChRs.
Subunit Composition of [3H]Epibatidine Binding nAChRs in Midbrain. We also studied the subunit composition of the [3H]Epi binding nAChRs in midbrain and expressed the results as femtomoles of immunoprecipitated nAChRs per milligram of protein (mean values of three or four separate experiments for each subunit in each genotype) (Fig. 2B).

A, immunoprecipitation analysis of the subunit content of the [3H]Epi nAChRs expressed in striatum labeled with 2 nM [3H]Epi. Immunoprecipitation was carried out as described under Materials and Methods using saturating concentrations (20–30 μg) of anti-subunit Abs. The amount immunoprecipitated by each antibody was subtracted from the value obtained in control samples containing an identical concentration of normal rabbit IgG, and the results obtained with each Ab are expressed as femtomoles of immunoprecipitated, labeled [3H]Epi nAChR per milligram of protein. Results are the mean values ± S.E.M. of four to five experiments performed in duplicate for each antibody. Statistical analyses were made using Student's paired t test. The significance of the difference from controls was *, p < 0.05; **, p < 0.01; and ***, p < 0.001. B, immunoprecipitation analysis of the subunit content of the [3H]Epi nAChRs expressed in midbrain labeled with 2 nM [3H]Epi. The reported values, (expressed as in A) are the mean values ± S.E.M. of three to four experiments performed in triplicate.
We found that the [3H]Epi binding nAChRs expressed in midbrain of β3+/+ mice are much more heterogeneous than those expressed in the striatum, with almost all possible subunits being expressed at variable levels. The majority of sites could be precipitated by the β2 (75.4 ± 3.5%) and α4 (68.9 ± 4.9%) antisera, whereas the other antisera were much less efficacious: α2 (4.3 ± 0.8%), α3 (10.8 ± 0.8%), α5 (6. 1 ± 0.9%), α6 (9.3 ± 1.0%), β3 (10.2 ± 1.5%), and β4 (7.9 ± 3.0%). Similar to the situation in striatum, β3+/– midbrain provided almost identical results to those obtained from β3+/+ tissue.
However, the composition of nAChRs in β3–/– midbrain was different from that in β3+/+ and β3+/– striatum. Again, the main finding in β3–/– midbrain was a strong reduction in α6* nAChRs (41%; from 11.6 ± 0.9 to 6.8 ± 0.6 fmol/mg of protein) this time accompanied by a smaller, but still significant, decline in α3* nAChRs (34%; from 15.3 ± 1.1 in WT to 10.1 ± 0.7 fmol/mg protein). No significant effects of β3 gene deletion on the expression of nAChRs containing any other subunit were observed.
Immunopurification of β3* nAChR Subtypes from Striatum. To identify the subunits assembled with the β3 subunit, we immunodepleted the striatal extract of β3* nAChRs using an affinity column with bound anti-β3 CYT Abs. Selective immunodepletion was confirmed by the decrease in β3* nAChRs from 18% in the total striatal extract to 1% in the flow-through of the β3 column.
To identify the subunit composition of the captured β3* nAChRs, they were eluted from the affinity column using an excess of the β3 CYT peptide, labeled with [3H]Epi and then immunoprecipitated with subunit specific antisera. As shown in Fig. 3, the anti-α4, α6, β2, and β3 COOH antisera immunoprecipitated 66.6 ± 0.8, 79.3 ± 9.1, 93.9 ± 2.5, and 61.0 ± 6.0% of the purified [3H]Epi-labeled β3* nAChRs, respectively. The anti-α2, α3, α5, and β4 antisera were essentially ineffective, immunoprecipitating 2.0 ± 1.4, 1.1 ± 0.9, 2.7 ± 2.2, and 2.1 ± 2.2% of the purified β3* nAChRs, respectively.
The subunit composition of this purified striatal β3* subtype is somewhat similar to that previously reported for the purified striatal α6* nAChRs (Champtiaux et al., 2003). However, the major differences are a higher proportion of α4* nAChRs, and greater recovery of β3* nAChRs. The higher proportion of α4 in immunocaptured β3* nAChRs, compared with the overall α6* nAChR population, suggests that the α4 subunit may preferentially associate with β3* nAChRs. That α4 is strongly associated with α-CtxMII-sensitive nAChRs reinforces the findings of Champtiaux et al. (2003) and Marubio et al. (2003). However, the preferential association of α4 with β3-containing α6* nAChRs is a novel finding. In fact, the present results strongly indicate that most β3* nAChRs contain all four α4, α6, β2, and β3 subunits and that incorporation of this “auxiliary subunit” (see Introduction) may encourage the formation of these unusually complex neuronal nAChRs. The increased recovery of β3* nAChRs compared with the previous studies reflects the use of a new, more efficacious mouse-specific anti-β3-CYT Ab for the immunocapture procedure. Unfortunately, the same Ab could not be used to probe the β3 content of the recovered nAChRs (excess competing β3-CYT peptide would render the Ab ineffective). The immunoprecipitation efficacy of the remaining, COOH-directed, Ab is somewhat lower (see Materials and Methods), which at least partially explains why the apparent 61.0 ± 6.0% β3 content of the immunopurified nAChRs does not match the theoretical 100%. Factoring in the lower efficacy, this would correspond to 80.6 ± 7.9% recovery by the more efficacious Ab.

Immunoprecipitation analysis of the subunit content of purified β3* nAChRs. The extracts prepared from β3+/+ striatum were incubated on an affinity column with bound anti-β3 M-CYT Abs (see Materials and Methods) to bind the β3* population, which was eluted from the column by means of incubation with the β3M-CYT peptide. Recovered nAChRs were labeled with 2 nM [3H]Epi and then immunoprecipitated by the indicated subunit-specific Abs. Immunoprecipitation was carried out as described for Fig. 2A. The amount immunoprecipitated by each antibody was subtracted from the value obtained in control samples containing an identical concentration of normal rabbit IgG, and the results are expressed as the percentage of total [3H]Epi binding present in the solution before immunoprecipitation. Each data point is the mean value ± S.E.M. of two determinations performed in triplicate using two Abs directed against two separate epitopes of the same subunit (except for β3).
The striatal nAChRs not captured by the β3 affinity column (i.e., those retained in the flow-through buffer) were also analyzed by immunoprecipitation. Almost all flow-through nAChRs were precipitable by α4 (95%) and β2 (91%) Abs, with lower proportions precipitated by the α5 (15%) and α6 Abs (6%). α2, α3, β3, and β4 Abs were ineffective. These results clearly indicate that after β3* nAChR immunodepletion, the remaining major striatal subtypes are the α4β2 and α4α5β2. Interestingly, the small but measurable recovery of α6 receptors in the flow-through fraction (in the absence of significant β3 leakage from the column, see above) indicates that at least a small proportion (approximately one-quarter) α6* nAChRs in wild-type mice do not contain β3 subunits. Unfortunately, the small size of this population precluded further study on its subunit composition.
Sucrose Gradient Analysis of α6* and β2* nAChRs in the Striatum and Midbrain of β3+/+ and β3–/– Mice. Correct assembly of α6* nAChRs in heterologous expression systems is critically dependent on subunit composition (Kuryatov et al., 2000). To ascertain whether α6* subunits were incorporated into correctly assembled pentameric subtypes in β3–/– mice, the size of detergent-solubilized α6* nAChRs retained in β3–/– striatum and midbrain was measured using sucrose density gradient centrifugation. After centrifugation, nAChRs within each fraction were captured using anti-α6 or anti-β2 Ab-coated wells and then quantitated by 125I-epibatidine binding. In both tissues, the α6* nAChRs detergent solubilized from β3–/– mice sedimented as a single species that was slightly larger than T. californica AChR monomers (Fig. 4, A and C), but of the same size as α6* nAChRs present in β3+/+ mice. These results clearly indicate that, although reduced in number, the α6* nAChRs in β3–/– striatum and midbrain have a correct pentameric assembly (Fig. 4, A and C). Parallel analysis of the same fractions for the presence of β2* nAChRs revealed that these, too, have the same pentameric conformation. Moreover, expression levels of β2* nAChRs in striatum and midbrain were unaffected by β3 genotype (Fig. 4, B and D), confirming the immunoprecipitation results described previously (Fig. 2).
Pharmacological Properties of α6* nAChRs in β3+/+ and β3–/– Mouse Striatum. It has already been shown that α6* nAChRs bind α-CtxMII with high affinity (Champtiaux et al., 2002, 2003; Zoli et al., 2002). Both the present and a previous study (Cui et al., 2003) indicate that the β3 subunit is also a component of high-affinity α-CtxMII binding nAChRs. To explore whether β3 subunit-null mutation influences the pharmacology of α6* nAChRs, we immunoimmobilized striatal α6* nAChRs from 2% Triton X-100 extracts of β3+/+ and β3–/– mice, using anti-α6 Abs. The epibatidine and α-CtxMII affinities of the captured α6* nAChRs were then compared between the two β3 genotypes.
Saturation binding analysis revealed no significant differences in the affinity of 125I-Epi binding at α6* nAChRs captured from β3+/+ and β3–/– mouse striatum [apparent Kd value of 41 pM (CV 16%) and 37 pM (CV 14%), respectively]. However, as expected, the Bmax of the α6* nAChRs immunoimmobilized from β3–/– mouse striatum was reduced to approximately one-fourth of that obtained using β3+/+ tissue (Fig. 5A).
Next, α-CtxMII competition binding studies were performed. In agreement with previously reported data (Champtiaux et al., 2003), we found that the α6* nAChRs in β3+/+ have a statistically significant better fit for a two site model with high (Ki of 0.36 nM; CV 59%) and low affinity (Ki of 6 μM; CV 41%) for α-CtxMII (Fig. 5B). It is likely that the high- and low-affinity sites correspond to binding at α6/β2 and α4/β2 interfaces, respectively, considering the complex subunit composition of these nAChRs. α6* nAChRs captured from the striatum of β3–/– mice contained almost identical high- (Ki of 0.37 nM; CV 79%) and low (Ki 5.7 μM; CV 35%)-α-CtxMII affinity binding sites. β3 genotype made no statistical difference in affinity at either site.
Discussion
nAChR β3 subunit deletion substantially reduces 125I-α-CtxMII binding nAChR expression in SN/VTA dopaminergic projections (Cui et al., 2003). This reduction is accompanied by a diminution of α-CtxMII-sensitive striatal synaptosomal [3H]dopamine release (Salminen et al., 2004). The present study applied ligand binding and immunoprecipitation techniques to investigate the consequences of β3 gene deletion on nAChR expression, composition, and pharmacology in this pathway.
This study's most striking result was that nAChR β3 subunit deletion greatly reduces α6* nAChR expression, both in the SN/VTA and their terminal regions. The decrease in α6* nAChR expression in both regions was quantitatively very similar to the decrease in high affinity 125I-αCtxMII binding in the β3–/– mice (Cui et al., 2003). This is in stark contrast to the effects on mRNA expression, because β3 subunit-null deletion had no effect on the expression of non-β3 nAChR subunit mRNAs, including α6 (Cui et al., 2003). In this case, as with the up-regulation of neuronal nAChRs by long-term nicotine treatment (Marks et al., 1992), it seems that alterations in nAChR expression occur at the level of subunit proteins, rather than subunit mRNAs.
Despite the loss of α6* sites in β3–/– mice, neither striatal cell membrane nor Triton X-100 extracts exhibited a significant decrease in overall [3H]Epi binding sites after β3 deletion. Furthermore, the immunoprecipitation studies showed no statistically significant decrease in α4* and β2* nAChRs after β3 gene deletion. Using the mouse-directed anti-β3 Ab, we immunopurified β3* striatal nAChRs. Most β3* nAChRs contained associated α4, α6, and β2 subunits. Thus, it might be expected that loss of α6β3* sites would be accompanied by a loss of the accompanying α4 and β2 subunit expression. Although it is possible that minor changes in α4 and β2 expression may have been obscured within the larger population, these results may instead suggest that α4β2(non-α6)* nAChRs replace the lost α4/α6* nAChRs. This hypothesis is strongly supported by the recent study of Salminen et al. (2004), who demonstrated that loss of α-CtxMII-sensitive striatal [3H]dopamine release in β3–/– mice is accompanied by a compensatory increase in α-CtxMII-resistant function.

Sucrose gradient analysis of α6* (A and C) and β2* (B and D) nAChRs present in striatum or midbrain. Five hundred microliters of 2% Triton X-100 extracts was loaded on to a 5 to 20% (w/v) sucrose gradient in phosphate buffer saline, pH 7.5, 0.1% Triton X-100, and 1 mM PMSF, and centrifuged for 14 h at 40,000 rpm in a Beckman rotor at 4°C. The fractions were collected, added to anti-α6or β2 Abs bound to microwells, left for 24 h, and then assayed for 125I-Epi binding as described under Materials and Methods. As a standard, 125I-αBgtx-labeled T. californica AChRs were subjected to sucrose gradient centrifugation in parallel, the fractions were collected and the radioactivity determined by gamma counting. The arrows indicate in each gradient the position of the T. californica monomer and dimer.
Although most β3 subunits in wild-type mice coassemble with α6, approximately one-quarter of striatal α6* nAChRs do not incorporate the β3 subunit. Intriguingly, the total striatal α6* nAChR population in β3+/+ mice is also approximately 4 times bigger than that of their β3–/– littermates. Thus, the residual α6* population in β3–/– mice may be composed of retained non-β3, α6* nAChRs, rather than representing a new subtype only expressed after β3 subunit deletion. This non-β3, α6* nAChR population has not been identified by previous mouse studies and represents a novel nAChR subtype. The very low expression this residual α6* population in β3–/– mice precluded a detailed immunoprecipitation study. However, circumstantial evidence suggests that many of these nAChRs also contain α4 subunits, like the larger population found in β3+/+ mice. Previous studies on mouse striatal α6* receptors indicate that the [3H]epibatidine binding sites associated with this population may be divided into two populations, one with high α-CtxMII affinity and one with low affinity (Champtiaux et al., 2003). The high α-CtxMII-affinity sites are thought to correspond to α6/β2 subunit interfaces. Those with low α-CtxMII affinity are likely to arise at α4/β2 subunit interfaces, located within α6* nAChR complexes, because low-affinity sites are absent from α6* nAChRs in the striatum of α4–/– mice (Champtiaux et al., 2003). The fact that, in the present study, α6* nAChRs isolated from β3–/– mice contain roughly equal proportions of high- and low-affinity α-CtxMII binding sites would seem to indicate that much of this population also contains α4/β2 subunit interfaces.
Overall, the major effect of β3 gene deletion in both SN/VTA and the striatum was to reduce α6* expression. This further reinforces the concept of a close relationship between the two subunits, as shown by the linkage of α6 and β3 into a gene cluster (Cui et al., 2003), the fact that many α6 nAChRs contain β3 (Champtiaux et al., 2003), and the results described in this article. The specific decrease of α6* nAChRs in β3–/– mice probably indicates that the β3 subunit is important for the formation of the majority of α6β2* or α6α4β2* pentamers. Decreased α6* nAChR expression could be caused by defects in nAChR assembly, degradation, and/or cell trafficking. Our present approach is not capable of distinguishing definitively between these possibilities, but the much greater decline in cell terminal (striatal) versus cell body (SN/VTA) α6* nAChR expression (76% reduction versus 42%) may support a role for β3 in directing nAChRs to the striatal regions of dopamine neurons. This study's finding that striatum contains higher levels of α6β3* than dopamine cell bodies further suggests that this nAChR subtype may be selectively addressed to dopaminergic nerve terminals. A trafficking hypothesis is also supported by previously published data (Champtiaux et al., 2003) showing that αCtxMII inhibits nicotinic responses more effectively on dopaminergic nerve terminals than on cell bodies, and the results of studies on 6-hydroxydopamine- or 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned rodents showing the selective localization of nAChRs containing the α6 and β3 subunits in dopaminergic terminals (Zoli et al., 2002, Champtiaux et al., 2003; Quik et al., 2003). Alternatively, or additionally, nAChR assembly/degradation effects are supported by Kuryatov et al. (2000), who demonstrated that adding the β3 subunit to α6 and β4 subunits greatly improves the yield of functional channels in oocytes.
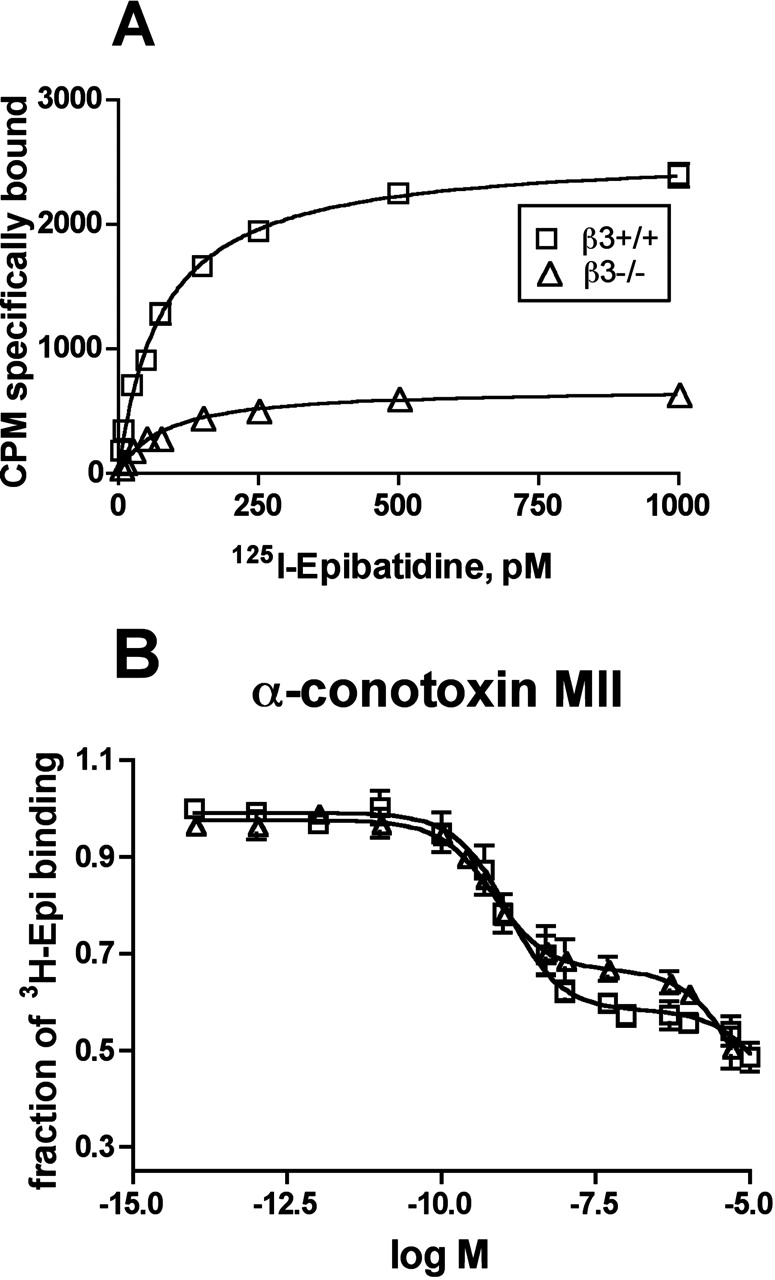
Pharmacological characterization of the α6* nAChRs present in the striatum of the β3+/+ and β3–/– genotypes. A, saturation curve of specific 125I-Epi binding to immunoimmobilized α6* nAChRs. B, inhibition by α-CtxMII of the binding of 125I-Epi to α6 immunoimmobilized nAChRs. α6* nAChRs present in striatum of the β3+/+ and β3–/– genotypes were immunoimmobilized using anti-α6, as described under Materials and Methods. The binding curves were obtained by fitting three separate experiments using the LIGAND program, unless shown, the S.E.M. is in the range of the symbol. In each experiment, each α-CtxMII dilution was tested in triplicate. All of the values are expressed in relation to 125I-Epi specific nAChR binding (considered as 100%).
Interestingly the midbrain, which includes the SN/VTA cell bodies, expressed a slightly different complement of nAChRs than their terminal regions [an additional α3* (10%) and/or β4* (7%) population, whereas the percentage of nAChRs containing the α6 and β3 subunits was lower (8%) than in striatum (18%)]. The midbrain α3* population was reduced by 33% after β3 deletion, which may imply that β3is incorporated into a portion of midbrain α3* nAChRs, together with β2 and/or β4 subunits. This nAChR subtype has previously been described in chick retinal nAChRs (Vailati et al., 2000) and in heterologous systems (Groot-Kormelink et al., 1998), but it has not before been identified in mammals.
The retention of α6* nAChRs in the striatum of β3–/– mice (albeit at reduced density) allowed us to test the β3 subunit's influence on α6* nAChRs' α-CtxMII affinity. Residual α6* nAChRs without the β3 subunit still bound α-CtxMII with high affinity, thus confirming that β3 neither directly nor allosterically affects the high-affinity α-CtxMII binding site. This finding is consistent with the idea that the β3 subunit assembles in a position analogous to that of the muscle type β1. In this scenario, β3 facilitates the formation of properly assembled pentamers, without changing the two acetylcholine binding sites at the α4β2 and/or α6β2 interfaces. One concern was that, as in Xenopus laevis oocytes (Kuryatov et al., 2000), α6 and β2 may assemble to form large aggregates with high α-CtxMII and Epi affinity, but no function. Our sedimentation data indicate that the remaining α6* nAChRs in β3–/– mice form pentameric assemblies. Salminen et al. (2004) have also demonstrated the retention of a small amount of α-CtxMII-sensitive, nAChR-mediated dopamine release in β3–/– mouse striatum, thus indicating that these are indeed functional nAChRs.
In conclusion, our results demonstrate that the nAChR β3 subunit is not necessary for the high-affinity binding of α-CtxMII but it is essential for efficient α6* nAChR assembly and/or selective transport to nerve terminals. It also seems that the β3 subunit encourages formation of unusually complex α4α6β2β3 subtype nAChRs, which constitute the majority of α6* nAChRs in mesostriatal dopamine neurons. It is noteworthy that comparison with the results of Cui et al. (2003) demonstrates that the β3 subunit's effects are mediated by protein/protein interactions, rather than by a reduction of α6 mRNA synthesis/stability after deletion of the β3 nAChR subunit gene. This subunit protein level, rather than gene expression level interaction, is reminiscent of the situation in agonist-mediated up-regulation (Marks et al., 1992) and may indicate that the former has a predominant role in regulating nAChR regulation. There are indications that loss of α6* nAChR expression is compensated for by increased α4β2(non-α6)* nAChR production. In addition, this study has uncovered evidence for two previously uncharacterized mammalian nAChR subtypes [α6(non-β3)β2* and α3β3*].
Footnotes
-
This work was supported in part by grants from the Italian Ministero dell'Istruzione, dell'Università e della Ricerca (MM05152538), European Research Training Network HPRN-CT-2002-00258, Fondo Integrativo Speciale per la Ricerca-Consiglio Nazionale delle Ricerche Neurobiotecnologia 2003, Fondo per gli Investimenti della Ricerca di Base RBNE01NR34, Fondazione Cariplo Grant No. 2002/2010 (to F.C.); Italian Minister of Health RF01.147 and Fondo per gli Investimenti della Ricerca di Base (RBNE01RHZM) 2003 (to C.G.); Colorado Tobacco Research Program 3I-030 (to P.W.); and National Institute on Drug Abuse grant DA12242 (to M.J.M, P.W., and J.M.M.) and National Institute of Mental Health grant MH53631 (to J.M.M.). Production of the β3 subunit-null mutant mice was supported by the National Institute on Drug Abuse animal resources grant DA015663 (to A.C.C.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.011940.
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; αBgtx, α-bungarotoxin; αCntxMII, α-conotoxin MII; Ab, polyclonal antibody; CYT, cytoplasmic peptide; PMSF, phenylmethylsulfonyl fluoride; Epi, epibatidine; WT, wild-type; CV, coefficient of variation; SN/VTA, substantia nigra/ventral tegmental area.
- Received February 9, 2005.
- Accepted March 4, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}