


Abstract
Neuronal nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels found throughout the central and peripheral nervous systems. They are crucial to normal physiology and have been clearly implicated in nicotine addiction. In addition, they are possible therapeutic targets in a wide range of pathological conditions, including cognitive disorders, Parkinson's disease, and neuropathic pain. Nicotinic ligands are usually classified as agonists (or partial agonists), competitive antagonists, or noncompetitive antagonists. Sazetidine-A is a new nicotinic ligand that shows a different pharmacological profile from any of these known classes of ligands. Sazetidine-A competes with very high binding affinity (Ki ≈ 0.5 nM) and selectivity for the α4β2 nAChR subtype (Ki ratio α3β4/α4β2 ∼ 24,000). Despite its high affinity, sazetidine-A neither activates nAChR channel function nor prevents channel activation when it is applied simultaneously with nicotine. However, when it is pre-incubated for 10 min with the receptors, it potently blocks nicotine-stimulated α4β2 nAChR function (IC50 ≈ 30 nM). The action of sazetidine-A may be explained by its very low affinity for the resting conformation of the α4β2 nAChRs, and its very high affinity for the desensitized state of the receptor. We propose that sazetidine-A is a “silent desensitizer” of nAChRs, meaning that it desensitizes the receptor without first activating it. Furthermore, comparison of the effects of sazetidine-A and nicotine at α4β2 nAChRs suggests that the predominant effects of nicotine and other nicotinic agonists are related to desensitization of the receptors and that sazetidine-A potently mimics these effects.
nAChRs are members of the Cys-loop family of neurotransmitter-gated ion channels (Karlin, 2002; Le Novere et al., 2002; Lindstrom, 2002). High-resolution structural models have greatly advanced our understanding of these receptors (Brejc et al., 2001; Unwin, 2005). They are composed of five subunits assembled around a central ion channel. A large extracellular domain, composed of N-terminal portions of all five subunits, contains two to five ligand binding sites formed between an α subunit and an adjacent subunit. Each subunit has four transmembrane α-helices, M1-M4. The M2 helices from the five subunits form a long narrow ion-conducting pore. The binding of the endogenous agonist acetylcholine or a drug such as nicotine initiates a series of conformational changes, which are translated to the pore through a pathway in the α subunits formed between three residues of the extracellular portion and two residues that link the M2 and M3 transmembrane helices (Lee and Sine, 2005).
In addition to acetylcholine and nicotine, many other natural products and synthetic drugs act on nAChRs (Daly, 2005; Jensen et al., 2005). These nicotinic compounds have been classified into three groups: agonists (or partial agonists), which act on acetylcholine binding sites and lead to the opening of nAChR channels; competitive antagonists, which bind to the agonist sites without stimulating the opening of nAChR channels and so block access by agonists; and noncompetitive antagonists, which act on sites of nAChRs other than acetylcholine binding sites (e.g., within the ion channel) to block functions of agonists. In addition, some drugs have been proposed to be allosteric potentiating ligands, which may enhance the effects of agonists by acting at a site close to but distinct from the acetylcholine binding sites to increase the probability of channel opening in the presence of an agonist (Pereira et al., 2002).
nAChRs have at least four discrete conformations: a resting state, an open state, and two or more desensitized states (Katz and Thesleff, 1957; Karlin, 1967; Changeux and Edelstein, 1998). Agonists have dual effects at nAChRs. They initially open the channel, allowing conduction of Na+,K+, and Ca2+ ions, which can lead to membrane depolarization and altered cell function, but agonists then drive the receptor into a desensitized state, temporarily rendering it unresponsive to further agonist stimulation. The desensitized state may be brief, as is usually the case with the endogenous agonist acetylcholine, or it may be prolonged, as seems to occur when nicotine is the agonist, especially with repeated exposure (Quick and Lester, 2002). In addition to these two immediate effects, long-term exposure to nicotine or other agonists increases the density of nAChRs in brain (Marks et al., 1983; Schwartz and Kellar, 1983).

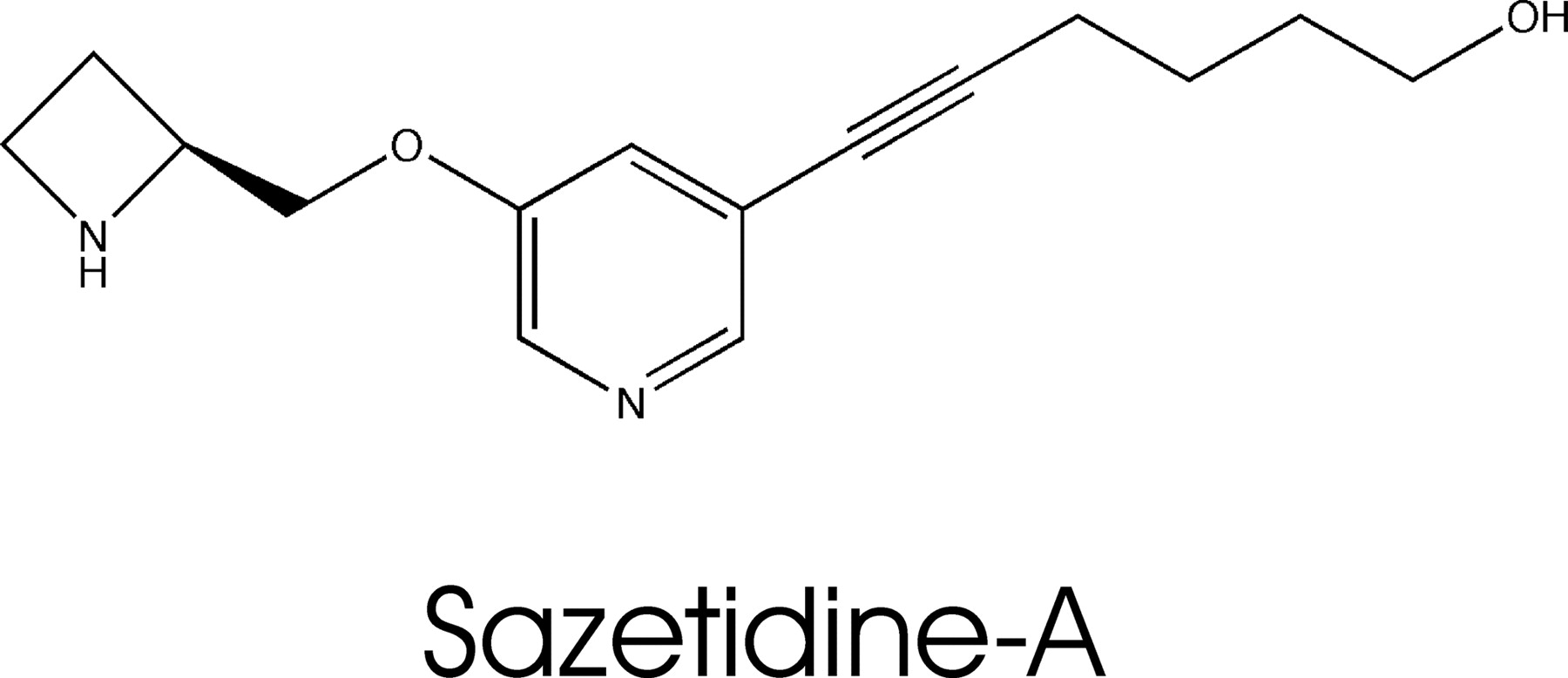
Structure of sazetidine-A, 6-(5-(((S)-azetidin-2-yl)methoxy)pyridine-3-yl)hex-5-yn-1-ol.
In this report, we have characterized the pharmacological actions of sazetidine-A (Fig. 1), a new nicotinic ligand with very high affinity and selectivity for nAChRs containing β2 subunits. Sazetidine-A is an analog of A-85380, which itself has high affinity and selectivity for nAChRs containing β2 subunits. Because the 5-iodo analog of A-85380 retains high affinity and displays even higher selectivity for α4β2 nAChRs (Mukhin et al., 2000; Xiao and Kellar, 2004), we examined other substituents at this position to determine the possibility of creating high-affinity nAChR ligands that could be used for affinity isolation and/or fluorescent labeling, among other purposes. In the course of those studies, we synthesized sazetidine-A. As described here, we found that it retains high affinity for α4β2 nAChRs and is more selective.
It is noteworthy that despite its high affinity for α4β2 receptors, sazetidine-A did not activate these receptors, and it did not inhibit activation when added simultaneously with nicotine. But after a 10 min preincubation, sazetidine-A very potently inhibited nicotine-stimulated α4β2 nAChR activation. These results suggest that although sazetidine-A does not have measurable agonist activity, it potently drives the receptors into a desensitized state. Taken together, these data imply that sazetidine-A may represent a new class of nicotinic cholinergic drugs with a novel mechanism of action. These drugs may have important potential for treatment of nicotine addiction, as well as other conditions involving nAChRs.
Materials and Methods
Materials and Chemicals
[3H](±)Epibatidine ([3H]EB) and [86Rb]rubidium chloride (86RbCl) were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). All other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO), unless otherwise stated. Varenicline was synthesized using methods described previously (O'Donnell et al., 2004; Coe et al., 2005).
Synthesis of Sazetidine-A
Sazetidine-A was synthesized from a precursor, (s)-5-bromo-3-{[1-(tert-butoxycarbonyl)-2-azetinyl]-methoxy}pyridine(Musachio et al., 1999). This precursor was coupled with 5-hexyn-1-ol via palladium-catalyzed Sonogashira reaction at 80°C, in the presence of P(t-Bu)3, to afford (s)-5-(5-hexyn-1-ol)-3-{[1-(tert-butoxycarbonyl)-2-azetidinyl]-methoxy}pyridine. The coupling product was then de-protected with 1 M HCl in diethyl ether to give 6-(5-(((S)-azetidin-2-yl)methoxy)pyridin-3-yl)hex-5-yn-l-ol (sazetidine-A).
(s)-5-(5-Hexyn-1-ol)-3-{[1-(tert-butoxycarbonyl)-2-azetidinyl]-methoxy}pyridine. To a v-vial containing of (s)-5-bromo-3-{[1-(tert-butoxycarbonyl)-2-azetinyl]-methoxy}pyridine (60 mg, 0.17 mmol) was added a solution of 5-hexyn-1-ol (0.022 ml, 0.2 mmol) in isopropylamine (1 ml), followed by charging with copper(I) iodide (3.3 mg, 0.017 mmol), (PPh3)2PdCl2 (3.7 mg, 0.0085 mmol), and tri-tert-butly phosphine (10% in hexane, 22.5 mg, 0.011 mmol). The mixture was heated at 80°C for 36 h. The solution was then cooled, diluted with EtOAc (5 ml), filtered through a small pad of silica gel, and concentrated. The residue was purified by flush chromatography using ethyl acetate/dichloromethane (2:3) to give 60 mg (81%) of desired product. 1H NMR (CDCl3, δ) 1.42 (s, 9H), 1.68-1.78 (m, 4H), 2.22-2.40 (m, 2H), 2.47 (t, J = 6 Hz, 2H), 3.70 (t, J = 5.6 Hz, 2H), 3.88 (t, J = 7.2 Hz, 2H), 4.11 (dd, J1 = 10, J2 = 2.8 Hz, 2H), 4.41 (m, 1H), 7.48 (m, 1H), 7.62 (m, 2H). FAM-HRMS: calculated for C20H28N2O4: 360.4473; found: 361.2132 (M+H)+.
(s)-5-(5-Hexyn-1-ol)-3-(2-azetidinylmethoxy)pyridine. To an ethanolic solution (0.2 ml) of (s)-5-(5-Hexyn-1-ol)-3-{[1-(tert-butoxycarbonyl)-2-azetidinyl]-methoxy}pyridine (20 mg, 56 μmol), was added 1 ml of 1 M HCl in anhydrous diethyl ether. The reaction was allowed to proceed for 12 h before collection of the resulting white precipitate via suction filtration and washing with ice-cold diethyl ether. Desired product (15 mg, 78%) was obtained. 1H NMR (CD3OD, δ) 1.71 (m, 4H), 2.56 (m, 2H), 2.69 (t, J = 8.0 Hz, 2H), 3.60 (t, J = 2.8 Hz, 2H), 4.10 (m, 2H), 4.55 (m, 2H), 4.82 (m, 1H), 8.21 (m, 1H), 8.59 (m, 2H). FAM-HRMS: calculated for C15H20N2O2: 260.1525; found: 261.1613 (M+H)+.
Cell Lines and Cell Culture
Tissue culture medium and antibiotics were obtained from Invitrogen Corporation (Carlsbad, CA), unless otherwise stated. Fetal bovine serum and horse serum were provided by Gemini Bio-Products (Woodland, CA). The cell lines KXα3β4R2, expressing rat α3β4 nAChRs, and KXα4β2, expressing rat α4β2 nAChRs, were established previously by stably transfecting human embryonic kidney 293 cells with combinations of rat nAChR subunit genes (Xiao et al., 1998; Xiao and Kellar, 2004). These cell lines were maintained in minimum essential medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin G, 100 mg/ml streptomycin, and 0.7 mg/ml G418 (Geneticin) at 37°C with 5% CO2 in a humidified incubator. The cell line SH-EP1-pcDNA-hα4β2, expressing human α4β2 nAChRs, was described previously (Eaton et al., 2003) and was generously provided by Dr. Ronald J. Lukas (Barrow Neurological Institute, Phoenix, AZ). The SH-EP1-pcDNA-hα4β2 cells were grown in Dulbecco's modified Eagle's medium supplemented with 5% fetal bovine serum, 10% horse serum, 100 units/ml penicillin G, 100 μg/ml streptomycin, 320 μg/ml hygromycin B (Calbiochem, La Jolla, CA), 400 units/ml Zeocin at 37°C with 5% CO2 in a humidified incubator.
Radioligand Binding Assay. Binding of [3H]EB to nAChRs was measured as described previously (Xiao et al., 1998). In brief, cultured cells at >80% confluence were removed from their flasks (80 cm2) with a disposable cell scraper and placed in 10 ml of 50 mM Tris-HCl buffer, pH 7.4, 4°C. The cell suspension was centrifuged at 10,000g for 5 min and the pellet was collected. For cells undergoing prolonged treatment with nicotinic agonists during culturing, the pellet was washed two more times by centrifugation in fresh buffer to remove residual drug. The cell pellet was then homogenized in 10 ml buffer with a Polytron homogenizer (12-mm aggregate, 26,000 rpm, 20 s; model PT2100; Kinematica, Basel, Switzerland) and centrifuged at 36,000g for 10 min at 4°C. The membrane pellet was resuspended in fresh buffer, and aliquots of the membrane preparation equivalent to 30 to 200 μg of protein were used for binding assays. The concentration of [3H]EB used was ∼100 pM for competition binding assays and ∼2.4 nM, which is a saturating concentration for the α4β2 receptor subtype, for measuring receptor density. Nonspecific binding was assessed in parallel incubations in the presence of 300 μM nicotine. Bound and free ligands were separated by vacuum filtration through Whatman GF/C filters treated with 0.5% polyethylenimine. The filter-retained radioactivity was measured by liquid scintillation counting. Specific binding was defined as the difference between total binding and nonspecific binding. Data from saturation and competition binding assays were analyzed using Prism 4 (GraphPad Software, San Diego, CA). Additional statistical analyses are indicated in the figure legends.

Competition by sazetidine-A for nAChR binding sites labeled by [3H]epibatidine. Competition binding assays were carried out in SH-EP1 cells expressing human α4β2 nAChRs (hα4β2), or HEK cells expressing rat α4β2 nAChRs (rα4β2) or rat α3β4 nAChRs (rα3β4), as described under Materials and Methods. The concentration of [3H]EB used was 100 pM. Data shown are representative of three to five independent experiments. See Table 1 for a summary of the Ki values for sazetidine-A binding at these receptors.
86Rb+ Efflux Assay. Functional properties of compounds at nAChRs expressed in the transfected cells were measured using 86Rb+ efflux assays as described previously (Xiao et al., 1998). In brief, cells were plated into 24-well plates coated with poly-d-lysine. The plated cells were grown at 37°C for 18 to 24 h to reach 85 to 95% confluence. The cells were then incubated in growth medium (0.5 ml/well) containing 86Rb+ (2 μCi/ml) for 4 h at 37°C. The loading mixture was then aspirated, and the cells were washed four times with 1 ml of buffer (15 mM HEPES, 140 mM NaCl, 2 mM KCl, 1 mM MgSO4, 1.8 mM CaCl2, and 11 mM glucose, pH 7.4). One milliliter of buffer with or without compounds to be tested was then added to each well. After incubation for 2 min, the assay buffer was collected for measurements of 86Rb+ released from the cells. Cells were then lysed by adding 1 ml of 100 mM NaOH to each well, and the lysate was collected for determination of the amount of 86Rb+ that was in the cells at the end of the efflux assay. Radioactivity of assay samples and lysates was measured by liquid scintillation counting. Total loading (cpm) was calculated as the sum of the assay sample and the lysate of each well. The amount of 86Rb+ efflux was expressed as a percentage of 86Rb+ loaded. Stimulated 86Rb+ efflux was defined as the difference between efflux in the presence and absence of nicotine. For obtaining an IC50 value, inhibition curves were constructed in which different concentrations of an antagonist were included in the assay to inhibit efflux stimulated by 100 μM nicotine. IC50 values were determined by nonlinear least-squares regression analyses (GraphPad Software).
Results
Binding Affinities of Sazetidine-A at nAChR Subtypes. The structure of sazetidine-A is shown in Fig. 1. All ligand binding assays were carried out using rat or human nAChRs heterologously expressed in human cells or in native rat brain tissues. As shown in Fig. 2 and summarized in Table 1, sazetidine-A competes with very high affinity for rat and human α4β2 nAChRs heterologously expressed in cells or from rat brain; moreover, its binding affinity for the heterologously expressed rat α4β2 receptors is 24,000 times higher than for rat α3β4 receptors. As shown in Table 1, the selectivity of sazetidine-A for α4β2 receptors over α3β4 receptors (Ki ratio) was much greater than that of nicotine or dihydro-β-erythroidine (DHβE), both of which are relatively selective ligands for α4β2 nAChRs in binding assays (Xiao and Kellar, 2004). The affinity of sazetidine-A for heterologously expressed rat and human α4β2 subtype is comparable with its affinity for native nAChRs from rat forebrain, which expresses predominantly the α4β2 subtype (Whiting and Lindstrom, 1987; Flores et al., 1992).
Comparison of binding affinities of Sazetidine-A, nicotine, and DHβE at nAChR subtypes
Binding was measured using 100 pM [3H]EB as described under Materials and Methods. Values are the mean ± S.E.M. of three to five independent experiments. The Ki values of nicotine and DHβE at rat nAChRs were published previously (Xiao and Kellar, 2004) and are shown here for comparison. Approximately 90% of the nAChRs in rat forebrain are the α4β2 subtype.
Effects of Sazetidine-A on nAChR Function. The functional effects of sazetidine-A on nAChRs were assessed by measuring agonist-stimulated 86Rb+ efflux from stably transfected cells. Consistent with previous reports, nicotine stimulates 86Rb+ release mediated by both α4β2 (Eaton et al., 2003) and α3β4 (Xiao et al., 1998) nAChR subtypes in a concentration dependent manner (Fig. 3). In contrast, sazetidine-A showed no agonist activity at either nAChR subtype over a wide concentration range (Fig. 3). This indicates that sazetidine-A does not activate nAChR channel function, or that its action is so weak, and/or that the duration of its action is so transient that no measurable channel function can be detected by the 86Rb+ efflux method.
The antagonist activity of sazetidine-A was evaluated by measuring its blockade of nicotine-stimulated 86Rb+ efflux. It is interesting that when sazetidine-A was added simultaneously with nicotine no antagonist activity in cells expressing either α4β2or α3β4 nAChRs was detected (Fig. 4 and Table 2). However, when preincubated with the cells for 10 min, sazetidine-A potently inhibited nicotine activation of the α4β2 receptors with an IC50 value of ∼30 nM, and it nearly completely inhibited activation at ∼200 nM. In contrast, even with the 10 min preincubation, sazetidine-A was still ineffective in blocking α3β4 nAChRs (Fig. 4 and Table 2).
Inhibition of nAChR function by sazetidine-A, DHβE, varenicline, and nicotine
nAChR function in cells expressing human α4β2 nAChRs and rat α3β4 nAChR was measured in the nicotine-stimulated 86Rb+ efflux assays, as described under Materials and Methods. The IC50 values were derived from inhibition curves as shown in Fig. 4 and are the mean ± S.E.M. of three to seven independent experiments.

Comparison of agonist activity of sazetidine-A and nicotine. The agonist activity of sazetidine-A and nicotine at nAChRs was compared by measuring the stimulation of 86Rb+ efflux by each compound in cells expressing human α4β2 and rat α3β4 nAChRs, as described under Materials and Methods. Data shown are representative of four to seven independent experiments. The EC50 values (mean ± S.E.M.) for nicotine activation of channel function were 10 ± 1 μM at the human α4β2 receptors and 34 ± 5 μM at the rat α3β4 receptors. EC50 values for sazetidine-A could not be calculated.

Comparison of inhibitory effects of sazetidine-A on nAChR channel function with or without a 10 min pretreatment. The 86Rb+ efflux assays in cells expressing human α4β2 and rat α3β4 nAChRs were carried out as described under Materials and Methods. Cells were either preincubated with buffer alone and then exposed simultaneously to 100 μM nicotine and the indicated concentration of sazetidine-A (0 min, no pretreatment group), or the cells were preincubated for 10 min with the indicated concentration of sazetidine-A alone and then exposed to 100 μM nicotine plus sazetidine-A (10 min, pretreatment group). Concentration-effect curves are representative of three to seven independent experiments. See Table 2 for a summary of IC50 values for sazetidine-A from all experiments.
For comparison, we also examined three other nicotinic ligands in the same manner. DHβE, a relatively potent and selective competitive antagonist of α4β2 nAChRs, blocked nicotine activation of the α4β2 subtype with an IC50 value of ∼1.4 μM (Table 2). In contrast to sazetidine-A, however, the potency of DHβE was similar with or without the 10 min preincubation. DHβE also blocked nicotine activation of the α3β4 subtype but with ∼100-fold lower potency than it did the α4β2 subtype (Table 2). Varenicline, a new drug recently approved for smoking cessation, like sazetidine-A, has high affinity and selectivity for α4β2 nAChRs (Coe et al., 2005). However, whereas varenicline was found to be a partial agonist at α4β2 nAChRs expressed in frog oocytes or at receptors mediating nicotine effects on dopamine turnover and release in rat brain (Coe et al., 2005), it potently and completely inhibited the α4β2 receptor-mediated channel response when it was preincubated with the cells for 10 min (Table 2). Under these conditions, the inhibitory potency of varenicline was comparable with that of sazetidine-A. Nicotine itself also potently inhibited channel function of the α4β2 receptors after the 10-min pretreatment (Table 2); in fact, in these studies, nicotine was ∼50 times more potent at inhibiting α4β2 nAChRs than at activating them (compare Table 2 and Fig. 3).
Recovery of Function after Sazetidine-A. We next examined recovery of channel function after the α4β2 nAChRs were pretreated with 1 μM sazetidine-A or 10 μM nicotine for 10 min. When nicotine stimulation was measured immediately after removal of buffer containing sazetidine-A (the no recovery group), ∼10% of channel function was observed (Fig. 5). In cells pretreated with sazetidine-A and then washed and incubated in fresh medium for 1 h, channel function recovered to ∼35%; after incubation in fresh medium for 20 h, recovery of function was nearly complete (Fig. 5). For comparison, after a 10-min pretreatment with nicotine, channel function was ∼15% of control when no recovery period was included, but it was nearly fully recovered within 1 h after incubation in fresh medium (Fig. 5).

Recovery of α4β2 nAChR channel function after desensitization by a 10-min treatment with sazetidine-A or nicotine. The 86Rb+ efflux assays in cells expressing human α4β2 nAChRs were carried out as described under Materials and Methods. Cells were incubated with buffer containing 1 μM sazetidine-A or 10 μM nicotine for 10 min. The buffer containing the drug was then removed, and 86Rb+ efflux stimulated by 100 μM nicotine was either measured immediately (no recovery group), or the cells were washed thoroughly and incubated in fresh medium for the recovery period indicated (1- or 20-h recovery groups) before their nAChR channel function was measured. Data are the mean ± S.E.M. of three to five independent experiments. Nicotine-stimulated 86Rb+ efflux in both recovery groups (1- and 20 h) are significantly higher than in their corresponding no recovery groups (analysis of variance, Dunnett's multiple comparison test; *, P < 0.05).

Increase in α4β2 nAChR binding sites induced by Sazetidine-A and nicotine. Cells expressing human α4β2 nAChRs were grown in the presence of 1 μM sazetidine-A or 10 μM nicotine for 5 days before membrane homogenates were prepared and nAChR binding sites were measured with 2.4 nM [3H]EB, a concentration that saturates these receptors and thus reflects the binding site density. Values are the mean ± S.E.M. of three independent experiments. Binding values in both treated groups are significantly higher than in the control group (analysis of variance, Dunnett's multiple comparison test; *, P < 0.01).
Up-Regulation of α4β2 nAChR Binding Sites by Sazetidine-A. Long-term administration of nicotine increases α4β2 nAChR density in mouse and rat brain (Marks et al., 1983; Schwartz and Kellar, 1983; Flores et al., 1992). This effect is also seen in cells in culture and with other nicotinic agonists (Peng et al., 1994; Gopalakrishnan et al., 1997). To determine whether sazetidine-A can up-regulate α4β2 nAChRs, cells were grown in the presence of 1 μM sazetidine-A for 5 days before the binding sites in membrane homogenates were measured with a saturating concentration of [3H]EB, which closely approximates the binding site density. As shown in Fig. 6, the binding site density was ∼2 times higher in membrane homogenates from cells exposed to sazetidine-A than in control cells. As a comparison, in cells grown in the presence of 10 μM nicotine, binding site density was increased ∼5-fold (Fig. 6). The concentrations of sazetidine-A and nicotine that were used in these experiments are ∼1500 and ∼1200 times their respective Ki values.
Discussion
The present study demonstrates that the main pharmacological action of sazetidine-A at α4β2 nAChRs is different from that of other known nicotinic ligands; in fact, it may represent a new type of drug action at these receptors. Two lines of evidence lead to this conclusion. First, sazetidine-A has very high affinity (Ki value less than 1 nM) for α4β2 nAChRs in equilibrium binding assays, in which it presumably interacts with the desensitized form of the receptors (Fig. 2, Table 1); despite its high affinity for the desensitized receptor, however, sazetidine-A does not activate the channel function of the receptors in their resting state when it is applied alone (Fig. 3), and it does not potentiate or block channel function when it is applied simultaneously with nicotine (Fig. 4). Thus, it is neither an agonist nor a classic competitive antagonist. Second, it does, however, potently block α4β2 nAChR function when it is preincubated for 10 min with the cells expressing these receptors; in fact, under these conditions, sazetidine-A is one of the most potent inhibitors of α4β2 nAChR function ever reported, 50 times more potent than DHβE (Fig. 4, Table 2).
We term this inhibitory action “silent desensitization”—i.e., desensitization of the receptors without first activating them. The explanation for this action may derive from the fact that nAChRs can exist in a resting state and two or more desensitized states, which represent distinct but interconvertible conformations of the receptor (Katz and Thesleff, 1957; Karlin, 1967; Changeux and Edelstein, 1998). The agonist-induced desensitization of nAChRs has been studied extensively (Grady et al., 1994; Marks et al., 1994, 1996; Fenster et al., 1997). We propose that sazetidine-A has very low affinity for the resting state of the α4β2 nAChRs, but very high affinity for one or more of the desensitized states of the receptor. Thus, when added to cells with nAChRs in the resting state, sazetidine-A, because of its low affinity, would act neither as an agonist to open the channel nor as an antagonist to block nicotine's effects. But preincubation would allow sazetidine-A to bind to receptors that spontaneously convert to a desensitized state, and because of its high affinity for those receptors, sazetidine-A would maintain them in that high-affinity, desensitized state. Over the 10-min preincubation time period used here, as more receptors are trapped in the desensitized state, channel function would be progressively inhibited. This action would thus allow sazetidine-A to bind to these receptors in their desensitized state without first activating them. Sazetidine-A is therefore different from nicotine and other nicotinic agonists, which can desensitize nAChRs but only after first activating them to some measurable extent (Hulihan-Giblin et al., 1990; Marks et al., 1994; Fenster et al., 1997). We propose that sazetidine-A and other nicotinic ligands with similar properties be classified as “silent desensitizers” of nAChRs.
Compared with nicotine, sazetidine-A has at least 10 times higher binding affinity for α4β2 nAChRs and is ∼500 times more selective for the α4β2 subtype over the α3β4 subtype (Table 1). Sazetidine-A doesn't activate nAChRs, but it is ∼6 times more potent than nicotine in inhibiting α4β2 nAChR function after a 10-min pretreatment (Table 2), presumably by the desensitizing action described above. The desensitizing effects of both sazetidine-A and nicotine are reversible. Thus, after removing sazetidine-A, receptor function recovered ∼35% in 1 h and nearly 100% in 20 h, whereas in the case of nicotine, function recovered fully in 1 h (Fig. 5). Long-term exposure to nicotine increases α4β2 nAChRs (Flores et al., 1992; Peng et al., 1994; Gopalakrishnan et al., 1997). Likewise, incubation of cells with sazetidine-A for 5 days significantly increased α4β2 nAChR density (Fig. 6). Although the increase was less than that seen after incubation with nicotine, the difference could have been due to the lower concentration of sazetidine-A used. The concentrations used here for both sazetidine-A and nicotine were ∼1200 to 1500 times their affinity (Ki) at the agonist binding site, but other factors related to the mechanisms underlying up-regulation, such as penetration into the cell, may be involved. Therefore, of the three actions of nicotine at α4β2 nAChRs—activation of the receptor channel, desensitization of the receptor and up-regulation of the receptor density—sazetidine-A displays only two: desensitization and up-regulation. This property may make sazetidine-A a very useful research tool with which to study the relationships between receptor activation, desensitization, and up-regulation.
Nicotine, the classic agonist that defines nAChRs, is probably the most widely used addictive drug. Moreover, with more than a billion cigarette smokers worldwide, smoking is estimated to be responsible for ∼5 million premature deaths each year (Ezzati and Lopez, 2003). Several lines of evidence suggest that the α4β2 nAChR is the major brain nAChR subtype that mediates nicotine addiction (Flores et al., 1992; Picciotto et al., 1998; Marubio et al., 2003; Tapper et al., 2004; Maskos et al., 2005). In addition, this receptor may also mediate some of the cognitive enhancing effects of nicotine (Picciotto et al., 1995; Maskos et al., 2005).
The actions of nicotine to both stimulate and block responses of autonomic ganglia were demonstrated more than 100 years ago (Langley and Dickinson, 1889); and the concept of desensitization of nAChRs in muscle was proposed almost 50 years ago (Katz and Thesleff, 1957). Thus, considering nicotine's dual actions to activate and then desensitize nAChRs, an important question is how each of these two essentially diametrically opposed actions contributes to the pharmacological effects of nicotine in the CNS. Most investigations of nicotine's pharmacological actions have focused on its activity as an agonist and the stimulatory effects it produces; consequently, there is a rich literature on the agonist actions of nicotine in both ganglia and the CNS. However, in almost all studies of nicotine and nAChR function, the desensitizing actions of nicotine are recognized (Quick and Lester, 2002; Giniatullin et al., 2005). In rats, for example, a single injection of nicotine initially stimulates prolactin release, but it then blocks subsequent nicotine-stimulated prolactin release for several hours or longer (Sharp and Beyer, 1986; Hulihan-Giblin et al., 1990). To explain this observation, nicotine was proposed to act as a “time-averaged antagonist” of nAChRs (Hulihan-Giblin et al., 1990), meaning that after a brief initial stimulation of brain nAChRs, it causes a long-lasting desensitization of the receptors that prevents their function for an extended period of several hours or longer. Our present results suggest that the primary functional effect of sazetidine-A is to desensitize α4β2 nAChRs without any detectable activation of the receptor population.
nAChRs are allosteric proteins, and their behavior at the molecular level is complex (Monod et al., 1965; Karlin, 1967; Changeux and Edelstein, 1998; Karlin, 2002). The action of sazetidine-A might be explained by an allosteric model of ligand-gated channel function of nAChRs. In fact, many of the important pharmacological effects of nicotine on the CNS might be due to its ability to desensitize the α4β2 nAChR. Thus, as long as its concentration is high enough to occupy the desensitized receptors, it acts essentially as an antagonist. After a 10-min pretreatment, nicotine potently inhibited α4β2 receptor function with an IC50 value 180 nM (Table 2). Similar inhibitory effects of nicotine and other agonists were also observed with a 3-min pretreatment of both human and rat α4β2 nAChRs (Fitch et al., 2003). Moreover, a recent study found that after a 6-h pretreatment, nicotine blocked α4β2 receptor function with an IC50 of 6 nM (Kuryatov et al., 2005). The nicotine concentration in arterial blood typically exceeds 200 nM during cigarette smoking and is maintained above 100 nM for more than 10 min after smoking (Henningfield et al., 1993; Gourlay and Benowitz, 1997). The concentration of nicotine in brain is thought to be even higher (Ghosheh et al., 2001). A recent PET imaging study in human smokers found that after subjects smoked one cigarette, more than 80% of their brain α4β2 nAChRs appeared to be occupied by nicotine for several hours (Brody et al., 2006).
Taken together, these findings suggest that nicotine acts primarily as an antagonist in vivo as long as its concentration is high enough to maintain receptor desensitization. Furthermore, the data suggest that the main reason people smoke might be to keep brain α4β2 nAChRs desensitized. A corollary to this hypothesis is that the withdrawal symptoms reported by smokers during abstinence may be due to increased central nervous system and autonomic stimulation mediated by increased activity of nAChRs as they recover from desensitization. This may especially involve β4-containing nAChRs (Salas et al., 2004).
It will be interesting to compare behavioral effects of sazetidine-A with those of nicotine in animal models, such as drug discrimination, self-administration, and drug with-drawal. Moreover, receptor desensitization might also mediate some of nicotine's potentially beneficial effects in such conditions as cognitive disorders, neuropathic pain, and Tourette's syndrome. If so, then drugs like sazetidine-A may prove to be especially useful therapeutic agents.
Acknowledgments
We thank Dr. Ronald J. Lukas of Barrow Neurological Institute (Phoenix, AZ) for generously providing cell line SH-EP1-pcDNA-hα4β2.
Footnotes
-
This work was supported by National Institutes of Health grants DA017980, DA12976, and DA13199.
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; A-85380, 3-(2(S)-azetidinylmethoxy) pyridine; EB, (±)epibatidine; DHβE, dihydro-β-erythroidine.
-
↵1 Current affiliation: National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland.
-
↵2 Current affiliation: Renovis, Inc., South San Francisco, California.
- Received May 31, 2006.
- Accepted July 14, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}