


Abstract
The extracellular part of transmembrane segment V (TM-V) is expected to be involved in the activation process of 7TM receptors, but its role is far from clear. Here, we study the highly constitutively active CXC-chemokine receptor encoded by human herpesvirus 8 (ORF74-HHV8), in which a metal ion site was introduced at the extracellular end of TM-V by substitution of two arginines at positions V:01 and V:05 with histidines [R208H; R212H]. The metal ion site conferred high-potency inverse agonist properties (EC50, 1.7 μM) to Zn(II) in addition to agonist and allosteric enhancing properties at concentrations >10 μM. The chemokine interaction with [R208H;R212H]-ORF74 was altered compared with wild-type ORF74-HHV8 with decreased agonist (CXCL1/GROα) potency (84-fold), affinity (5.8- and 136-fold in competition against agonist and inverse agonist, respectively), and binding capacity (Bmax; 25-fold). Zn(II) in activating concentrations (100 μM) acted as an allosteric enhancer as it increased the Bmax (7.1-fold), the potency (9.9-fold), the affinity (1.7- and 6.1-fold in competition against agonist and inverse agonist, respectively), and the efficacy (2.5-fold) of CXCL1/GROα. The activating properties of Zn(II) were not due to a metal ion site between the ligand and the receptor because CXCL1/GROα analogs in which the putative metal-ion binding residues had been substituted—[H19A] and [H34A]—acted like wild-type CXCL1/GROα. Based on the complex action of Zn(II) and on the chemokine interaction for [R208H;R212H]-ORF74, we conclude that the extracellular end of TM-V is important for the activation of this CXC-chemokine receptor.
The chemokine receptors, which regulate leukocyte movement during homeostasis as well as during inflammation, belong to the superfamily of G protein-coupled Rhodopsin-like seven-transmembrane spanning α-helices (7TM receptors) (Murphy et al., 2000). The endogenous chemokines are divided in two major groups (CC- and CXC-chemokines) based on the presence or absence of a residue between the two first of four conserved cysteines and in two minor groups (XC and CX3C-chemokines). The CXC-chemokines are further divided based on the presence or absence of a Glu-Leu-Arg (ELR) motif just before the first cysteine. It is noteworthy that the chemokine system has been subject to molecular piracy by certain viruses to improve virus survival and spreading. Thus, some pox- and herpesviruses encode chemokines and/or chemokine receptors that presumably originate from the host genome (Murphy, 2001). The ORF74 receptors (located in open reading frame 74 in all sequenced γ2-her-pesviruses) constitute one family of viral chemokine receptors being characterized by a broad-spectrum CXC-chemokine binding and the presence of constitutive activity [ORF74 encoded by human herpesvirus 8 (HHV8), herpesvirus saimiri, and equine herpesvirus], although no constitutive activity has yet been shown for the ORF74 receptor encoded by murine γ-herpesvirus 68 (Arvanitakis et al., 1997; Rosenkilde et al., 1999; Rosenkilde et al., 2004b; Verzijl et al., 2004; Rosenkilde et al., 2005).
The pathways induced constitutively by ORF74-HHV8 include Gq, Gi, and G12/13 and the combined action of these pathways results in cellular transformation and production of angiogenic factors (Bais et al., 1998; Munshi et al., 1999; Rosenkilde et al., 1999). Thus, vascularized tumors develop when ORF74-HHV8 is transplanted into nude mice (Bais et al., 1998), and transgenic expression in mice results in Kaposi's sarcoma-like lesions (Yang et al., 2000). ORF74-HHV8 binds a variety of CXC-chemokines; pro-inflammatory and angiogenic ELR CXC-chemokines act either as agonists (CXCL1-3/GRO-α,β,γ1) or as neutral ligands (e.g., CXCL5/ENA78, CXCL7/NAP2, and CXCL8/IL8), and angiostatic non-ELR CXC-chemokines act as inverse agonists (CXCL10/IP10 and CXCL12/SDF-1α) (Arvanitakis et al., 1997; Rosenkilde et al., 1999). In addition, the HHV8 encoded CC-chemokine vCCL2 acts as an inverse agonist for ORF74-HHV8 (Arvanitakis et al., 1997; Rosenkilde et al., 1999). The interaction of the CXC-chemokines with ORF74-HHV8 and the endogenous receptors (CXCR1-4) involves the N termini of the receptors (mandatory) as well as different parts of the extracellular loops (involved to different extents) (Ahuja et al., 1996; Katancik et al., 2000; Rosenkilde et al., 2000; Cox et al., 2001; Rosenkilde et al., 2004a).
The activation mechanism of rhodopsin-like 7TM receptors involves major movements of the transmembrane helices. Thus, TM-VI, TM-VII, and TM-III in particular have been shown to move apart from each other at their intracellular ends, creating space for the G protein and/or arrestin to interact with the receptor (Farrens et al., 1996). In contrast, the extracellular ends approach each other during receptor activation (constrained either by agonist and/or by interhelical interactions) as shown, for instance, by metal-ion engineering (Schwartz et al., 2006). Thus, active and inactive conformations have been efficiently constrained by building in metal-ion sites in for example the neurokinin (Elling et al., 1995), the opioid (Thirstrup et al., 1996), the adrenergic (Elling et al., 1999, 2006), and the ORF74-HHV8 chemokine receptor (Rosenkilde et al., 1999).
In the present article, we describe the chemokine- and metal ion binding and signaling properties of the [R208H; R212H]-ORF74, in which a metal ion binding site was introduced in the extracellular end of TM-V. It is noteworthy that this mutated receptor was constrained in an agonist-impaired conformation. The action of Zn(II) was rather complex; there was with inverse agonist action with micromolar potency and agonist action for concentrations >10-fold above the EC50 for the inverse agonist activity, and maximal efficacy was reached at 100 μM Zn(II) (Fig. 1B). At this concentration, Zn(II) also acted as an allosteric enhancer of chemokine agonist binding and action, in that it improved the affinity, maximal binding (Bmax), efficacy, and potency of CXCL1/GROα (and of two analogs of CXCL1/GROα in which two putative metal-ion binding sites were substituted with alanines). Thus, by binding to the introduced metal-ion binding site in TM-V (i.e., an allosteric binding site compared with the orthosteric binding site of the CXC-chemokines, described above), Zn(II) acts as an agonist and a positive allosteric modulator (allosteric enhancer) of agonist chemokine binding (and action) in addition to its role as an inverse agonist at low concentrations.
Materials and Methods
Materials. The human chemokines CXCL1/GROα, CXCL6/GCP2, CXCL7/ENA78, CXCL5/NAP2, CXCL12/SDF-1, and CXCL10/IP10 were from Peprotech (Rocky Hill, NJ). Other human chemokines were made in-house by Thomas P. Boesen through Escherichia coli expression, purification, and refolding (CXCL8/IL8) or by COS-7 cell expression, purification, and refolding (vCCL2) (Kledal et al., 1997). ORF74-HHV8 (Genbank accession no. U24275) was cloned from a Kaposi's sarcoma skin lesion biopsy (Rosenkilde et al., 1999). 125I-CXCL8/IL8, [myo-3H]inositol and Bolton-Hunter reagent for iodination of CXCL1/GROα (and CXCL1/GROα analogs) and CXCL10/IP10 were from GE Healthcare (Little Chalfont, Buckinghamshire, UK). AG 1-X8 anion exchange resin (for PI-turnover assay) was from Bio-Rad Laboratories (Hercules, CA).
Cloning and Mutation of CXCL1 His19 to Ala and His34 to Ala. The cDNA encoding CXCL1/GROα was amplified by polymerase chain reaction using the forward primer 5′-GGTGGTCATATGGCGTCCGTGGCCACTGA-3′, containing an NdeI-restriction site, and the reverse primer 5′-GGTGGTGGCTCTTCCGCAGTTGGATTTGTCACTGTT-3′, containing a SapI-restriction site. After digestion with NdeI/SapI, the DNA was purified and introduced to a pTXB1-Vector (IMPACT-CN System; New England Biolabs, Ipswich, MA) digested with the same enzymes to get a C-terminal fusion-protein with an MxeGyrA intein fused to a C-terminal chitin binding domain. Mutations were introduced with the QuikChange site-directed mutagenesis method (Stratagene, La Jolla, CA). To obtain the H19A mutation, the forward primer 5′-CAGACCCTGCAGGGAATTGCCCCCAAGAACATCCAAAGT-3′ and the reverse primer 5′-ACTTTGGATGTTCTTGGGGGCAATTCCCTGCAGGGTCTG-3′ were used. To obtain the H34A mutation, the forward primer 5′-GTGAAGTCCCCCGGACCCGCCTGCGCCCAAACCGAAGTC-3′ and the reverse primer 5′-GACTTCGGTTTGGGCGCAGGCGGGTCCGGGGGACTTCAC-3′ were used. The mutations were confirmed by sequencing of the entire coding region.
Fusion Protein Expression in E. coli. E. coli ER2566 cells transformed with pTXB1 plasmid containing CXCL1/GROα or mutant analogs were grown in Luria Bertani medium containing 100 μg/ml ampicillin. Fusion protein expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside. After overnight expression at 37°C and 220 rpm, cells were harvested by centrifugation at 4600g. Cells were resuspended in buffer A (50 mM Tris-HCl, 1 mM EDTA, and 500 mM NaCl, pH 8) in the presence 20 μM phenylmethylsulfonyl fluoride (PMSF). Lysis was performed using a French pressure cell press (SLM Instruments, Inc., Rochester, NY) with a cell pressure of 15,000 psi followed by a 2-h desoxyribonuclease I treatment using 0.2 U/ml. After centrifugation (18,000g for 30 min), the pellet containing the insoluble fusion protein was solubilized three times with buffer A containing 6 M guanidine-hydrochloride for at least 8h.
Purification and Isolation of CXCL1 Analogs. CXCL1 and analogs were purified as recombinant thioester as described previously (David et al., 2003). The extracts containing the fusion protein were pooled and diluted with buffer A to 3 M guanidine-hydrochloride and loaded manually to the column (Econo Column; Bio-Rad) filled with chitin beads. The flow-through was diluted with buffer A (described above) to 1.5 M guanidine-hydrochloride and again loaded to the column two times. After washing with 10 column-volumes of buffer A, intein cleavage was induced by washing with two column-volumes of the buffer A containing 200 mM dithiothreitol. On-column cleavage proceeded at 4°C for 48 h. Peptide-thioester was eluted at room temperature with buffer A in 3-ml fractions, and the protein content was determined by Bradford (1976) assay. The thioester identity was confirmed by matrix-assisted laser desorption ionization mass spectrometry (Voyager II; Applied Biosystems, Foster City, CA) and the purity was determined by analytical reversed-phase HPLC using an Impaq C18-Column 250 × 4 mm (Bischoff, Leonberg, Germany) with acetonitrile/water (0.1% trifluoroacetic acid). All fractions containing peptide-thioester were pooled, and the pH was adjusted to 9.5. Thioester hydrolysis was performed for 24 h. This solution was then charged with 10 mM tris(2-carboxyethyl)phosphine hydrochloride and the guanidine-hydrochloride concentration was increased to 3 M before filling to a prewelled dialysis tube (molecular mass cutoff, 3500 Da). Refolding was performed as dialysis against buffer A containing 1.5 M guanidine-hydrochloride, 10 mM cysteine, and 1 mM cystine for 8 h, and in the second step against buffer A containing 10 mM cysteine and 1 mM cystine. The refolded product was purified by preparative HPLC, identity was determined by FTICR-mass spectrometry (Bruker Daltonics, Billerica, MA), and purity was confirmed by analytical HPLC.

Introduction of metal-ion binding site in ORF74. Serpentine model of ORF74-HHV8 with an indication of the positions (V:01 and V:05) in which the His residues were introduced in substituted for two Arg residues (A). B-D, PI turnover experiments in transiently transfected COS-7 cells. Dose-response curves of ZnC2 in wt ORF74-HHV8 and [R0208H;R212H]-ORF74 (B). Effect of the chemokines CXCL1/GROα (white columns) and CXCL10/IP10 (black columns) on wt ORF74-HHV8 (left), [R208H;R212H]-ORF74 (middle), and vector-transfected cells (right) (C). Dose-response-curves of CXCL1/GROα (circles) and CXCL10/IP10 (squares) in wt ORF74-HHV8 (filled symbols) and [R208H; R212H]-ORF74 (open symbols) (D). (n = 3-17).
CD Studies of CXCL1 and CXCL1 Analogs. CD spectra of 50 μM CXCL1 and CXCL1 analogs were recorded in 10 mM phosphate buffer, pH 7, in the absence and in the presence of 10 μM ZnCl2 [Zn(II)] and 1 mM ZnCl2 [Zn(II)] using a spectrapolarimeter (J-715; JASCO Labor und Datentechnik GmbH, Groß-Umstadt, Germany). CD was measured from 250 to 190 nm in a nitrogen atmosphere at 20°C using a scanning speed of 20 nm/min with a time constant of 2 s, step resolution of 0.1 nm, a sensitivity range of 20 mdeg, and a band width of 2 nm. The spectra were obtained as average of three recordings in a sample cell of 1-mm pathlength. High-frequency noise was reduced by means of a low-path Fourier transform filter
Iodination of CXCL1/GROα, CXCL1/GROα Analogs and CXCL10/IP10. The Bolton-Hunter reagent was dried by a gentle stream of nitrogen for 30 to 60 min. Chemokine (10 μg) was incubated on ice with 1 mCi of Bolton-Hunter reagent in a total volume of 50 μl of 0.1 mM borate buffer, pH 8.5, for 1 h. The reaction was terminated by addition of 0.25 ml of H2O supplemented with 0.1% (v/v) tri-fluoro-acetic acid. The labeled chemokines were purified by reversed-phase HPLC.
Transfections and Tissue Culture. COS-7 cells were grown at 10% CO2 and 37°C in Dulbecco's modified Eagle's medium with GlutaMAX (Invitrogen, Carlsbad, CA) adjusted with 10% fetal bovine serum, 180 U/ml penicillin, and 45 μg/ml streptomycin. Transfection of COS-7 cells was performed by the calcium phosphate precipitation method (Rosenkilde et al., 1999).
Competition Binding Experiments. COS-7 cells were transferred to 12- or 24-well culture plates 1 day after transfection. The number of cells seeded per well was determined by the apparent expression efficiency of the individual clones and was aimed at obtaining 5 to 10% specific binding of the added radioactive ligand. In the test for specific binding, 3 to 5 × 105 cells/well were used. Two days after transfection, cells were assayed by competition binding for 3 h at 4°C using 12 to 15 pM 125I-CXCL1/GROα (and analogs), 125I-CXCL8/IL8, and 125I-CXCL10/IP10 plus unlabeled ligand in 0.5 ml of a 50 mM HEPES buffer, pH 7.4, supplemented with 1 mM CaCl2, 5 mM MgCl2, and 0.5% (w/v) bovine serum albumin. After incubation, cells were washed two to four times in 4°C binding buffer supplemented with 0.5 M NaCl. Nonspecific binding was determined in the presence of 0.1 μM unlabeled chemokine. Determinations were made in duplicate.
Kinetic Binding Experiments. Association and dissociation reactions were determined in a total volume of 0.4 ml of binding buffer at 4°C using 12 to 15 pM 125I-CXCL1/GROα. After incubation for various periods, the cells were washed quickly four times in 4°C binding buffer supplemented with 0.5 M NaCl. The association and dissociation were measured over a total period of 4 to 7 h. After an incubation period of 180 min with radioligand in the absence or presence of 100 μM ZnCl2 [Zn(II)], 10-7 M CXCL1/GROα was added (in the absence or presence of 100 μM ZnCl2 [Zn(II)]. Determinations were made in duplicate, and the nonspecific binding was determined with 10-7 M CXCL1/GROα (in the absence or presence of 100 μM ZnCl2 [Zn(II)]).
Phosphatidyl Inositol Assay. COS-7 cells were transfected with receptor cDNA. One day after transfection, COS-7 cells (2.5 × 104 cells/well in 24-well plates) were incubated for 24 h with 5 μCi/ml [myo-3H]inositol in 0.3 ml of growth medium per well. Cells were washed twice in 20 mM HEPES, pH 7.4, supplemented with 140 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM CaCl2, 10 mM glucose, and 0.05% (w/v) bovine serum albumin and were subsequently incubated in 0.3 ml of buffer supplemented with 10 mM LiCl at 37°C for 90 min in the presence or absence of ligands. Cells were extracted by addition of 1 ml of 10 mM formic acid per well followed by 30-min incubation on ice. The generated [3H]inositol phosphates were purified on AG 1-X8 anion exchange resin. Determinations were made in duplicate.
Surface Enzyme-Linked Immunosorbent Assay. COS-7 cells were seeded into 96-well plates, 50,000 cells/well, and transfected with 15 ng (per well) of the N-terminal FLAG-tagged variants of ORF74-HHV8, [R208H;R212H]-ORF74, and EBI2 for the ELISA assay. Twenty-four hours after transfection, the cells were treated with the ligands for 5 h at 37°C and were subsequently washed once in TBS (35 mM Tris-HCl and 140 mM NaCl, pH 7.4), fixed in 4% paraformaldehyde for 10 min, and incubated in blocking solution (2% bovine serum albumin in TBS) for 30 min at room temperature. The cells were thereafter kept at room temperature and incubated 2 h with anti-FLAG (M1) antibody (2 μg/ml) in TBS supplemented with 1% bovine serum albumin and 1 mM CaCl2. After three washes in TBS with 1 mM CaCl2 the cells were incubated with goat anti-mouse horseradish peroxidase-conjugated antibody in the same buffer as the anti-FLAG antibody for 1 h. After three washes in TBS with 1 mM CaCl2, the immune reactivity was revealed by the addition of horseradish peroxidase substrate according to manufacture's instruction.
Calculations. IC50 and EC50 values were determined by nonlinear regression and Bmax values calculated using the Prism software (ver. 4; GraphPad Software, San Diego, CA). The calculations of Bmax from our homologous competition binding experiments were done at the assumption that the labeled and unlabeled chemokine had the same affinity for the receptor, and that the presence of Zn(II) affected the labeled and unlabeled chemokine to the same extent.
Results
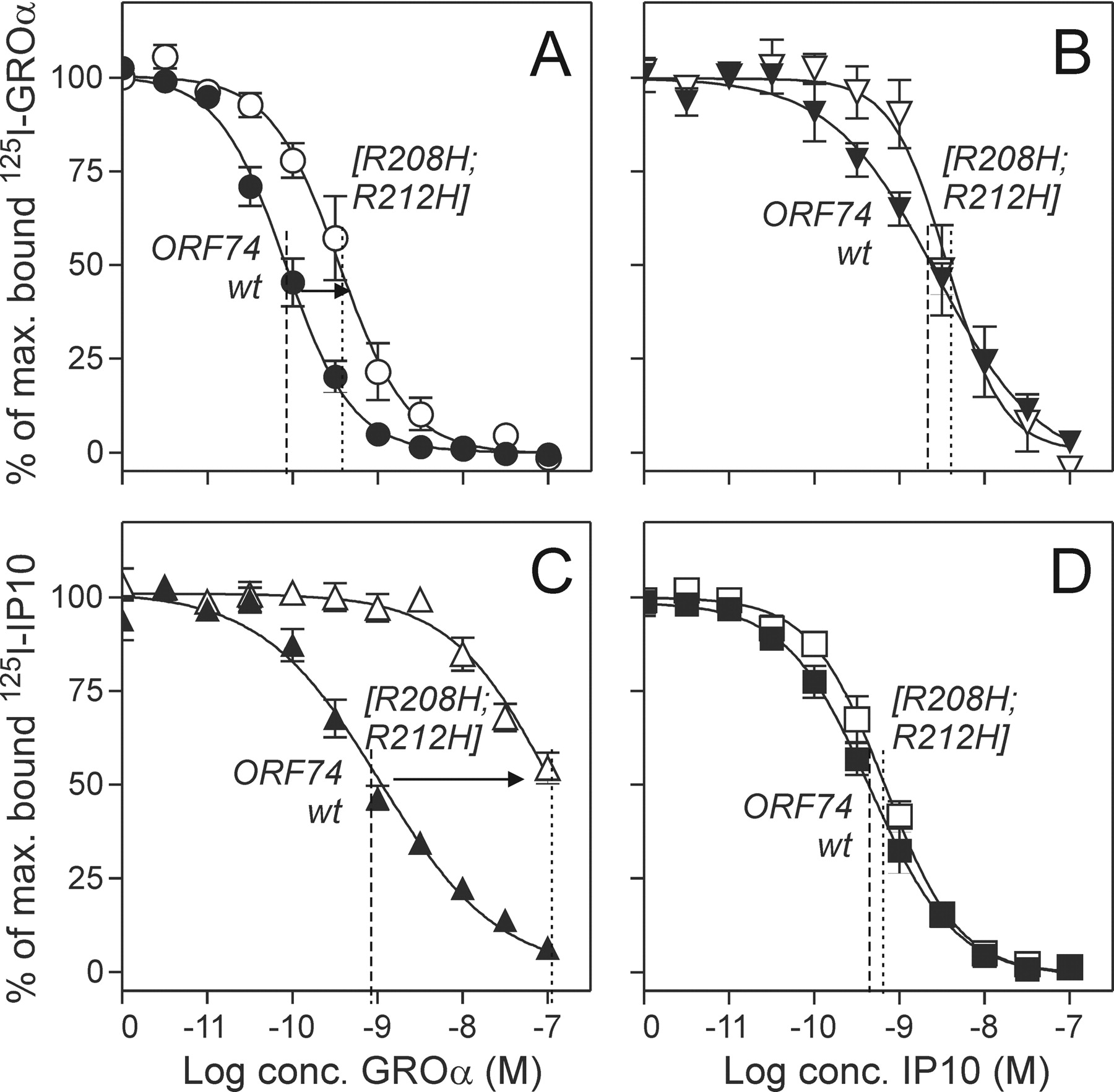
Chemokine Binding and Action in the Mutated [R208H;R212H]-ORF74 Receptor. Three chemokine-based ligand classes (agonists, inverse agonists, and neutral ligands) have previously been shown to bind to wt ORF74-HHV8 with high affinities and similar maximal binding capacities (Bmax) measured by homologous competition binding experiments with 125I-CXCL1/GROα, 125I-CXCL10/IP10, and 125I-CXCL8/IL8 as radioligands (Rosenkilde et al., 1999; Rosenkilde and Schwartz, 2000). We have previously shown that a small molecule [Zn(II)] could inhibit the constitutive activity provided introduction of a metal-ion binding-site in the extracellular end of TM-V (in position V:01 and V:05 by substitution of two arginines with histidines [R208H; R212H]-ORF74) (Rosenkilde et al., 1999) (Fig. 1B). In the present article, we characterize the chemokine-mediated signaling and binding properties of [R208H;R212H]-ORF74. By measuring Gq coupling via phosphatidyl inositol (PI) turn-over and the action—for wt ORF74-HHV8—of the most potent and efficacious agonist (CXCL1/GROα) and inverse agonist (CXCL10/IP10), we found that the agonist potency was decreased >80-fold for [R208H;R212H]-ORF74 compared with wt ORF74-HHV8 (EC50 of ∼105 nM compared with 1.25 nM, Fig. 1, C and D). In contrast, the potency of the inverse agonist was unchanged (EC50 of 3.6 and 3.9 nM, respectively). Thus, [R208H;R212H]-ORF74 was easily inhibited by metal-ion and chemokine inverse agonists, whereas it was highly deficient in chemokine-mediated activation despite surprisingly similar levels of basal activity for the two receptors (Fig. 1C).
A broad spectrum of agonists (CXCL1-3/GROα,-β, and -γ), neutral ligands (CXCL5/ENA78, CXCL7/NAP2, and CXCL8/IL8), and inverse agonists (CXCL10/IP10, CXCL12/SDF-1, and vCCL2) were tested in competition against radiolabeled agonist (125I-CXCL1/GROα) and inverse agonist (125I-CXCL10/IP10), whereas the radiolabeled neutral ligand 125I-CXCL8/IL8 was excluded because of the lack of specific binding to [R208H;R212H]-ORF74 (data not shown). In contrast to the almost equal distribution of agonist and inverse agonist binding conformations for wt ORF74-HHV8, we observed—in the homologous competition binding—a 24.6-fold decrease in the Bmax for the agonist and a 1.3-fold increase in the Bmax for the inverse agonist (Table 1). Furthermore, the affinity for CXCL1/GROα was 5.8-fold decreased (Fig. 2A; Table 1) and the affinities for the two other agonists (CXCL2 and 3/GROβ and -γ) followed the same pattern in competition against 125I-CXCL1/GROα (9.1- and 19-fold decreases, respectively; Table 1). In contrast, the affinity for CXCL10/IP10 was unchanged (measured by homologous competition binding; Fig. 2D) whereas the two other inverse agonists, CXCL12/SDF-1 and vCCL2, showed a slight increase in affinity (2- and 2.5-fold compared with wt ORF74-HHV8 in competition against 125I-CXCL10/IP10; Table 1). We further tested the ability of agonists and inverse agonists to compete with each other. In contrast to the interconvertible agonist and inverse agonist binding states for wt ORF74-HHV8 (Rosenkilde and Schwartz, 2000) we observed—for [R208H; R212H]-ORF74—a lack of competition of the agonists for radiolabeled inverse agonist. Thus, the agonists (CXCL1-3/GROα,-β, and -γ) were all impaired in their competition against 125I-CXCL10/IP10 (136-, 55-, and 84-fold decrease in affinities, respectively; Table 1, Fig. 2C). This difference was not found for the inverse agonists in their competition against 125I-CXCL1/GROα (<2.5-fold change; Table 1, Fig. 2B). The neutral ligands (CXCL5/ENA78, CXCL7/NAP2, and CXCL8/IL8) showed no difference in affinities between wt and [R208H;R212H]-ORF74 (data not shown). In summary, both the agonists and the inverse agonists bound with high affinity to [R208H;R212H]-ORF74, although with a huge decrease in the binding capacities for the agonists. The heterologous binding experiments indicated a constraining in an agonist-impaired conformation for [R208H;R212H]-ORF74 as observed for the signaling (Fig. 1, C and D).
Homologous and heterologous competition binding in wt ORF74-HHV8 and [R208H;R212H]-ORF74
A whole panel of pharmacological different chemokines: agonists (CXCL1-3/GROα, -β, and -γ), and inverse agonists (CXCL10/IP10, CXCL12/SDF-1, and vCCL2/vMIP-II) were tested against 125I-CXCL1/GROα and 125I-CXCL10/IP10 in transiently transfected COS-7 cells as described under Materials and Methods. IC50 values ± S.E.M. are given for all tested chemokines. Fmut indicates the -fold change in affinity between mutant and wt receptor, calculated as IC50 ([R208H;R212H])/IC50 (ORF74-HHV8) for a given ligand measured against 125I-CXCL1/GROα (Ago. Fmut) and against 125I-CXCL10/IP10 (Inv. Fmut). Fligand indicates the -fold change in affinity for a given ligand depending upon applied radioligand, calculated as IC50 (125I-CXCL10/IP10)/IC50 (125I-CXCL1/GROα). The bold values are depicted as curves in Fig. 2.

Homologous and heterologous competition binding in wt ORF74-HHV8 and [R208H;R212H]-ORF74. Transiently transfected COS-7 cells were used for the displacement of 125I-CXCL1/GROα (A and B) and 125I-CXCL10/IP10 (C and D) to wt ORF74-HHV8 (filled symbols) and [R208H;R212H]-ORF74 (open symbols). The homologous competition binding is shown in A and C and the heterologous competition binding in B and D. (n = 3-16).
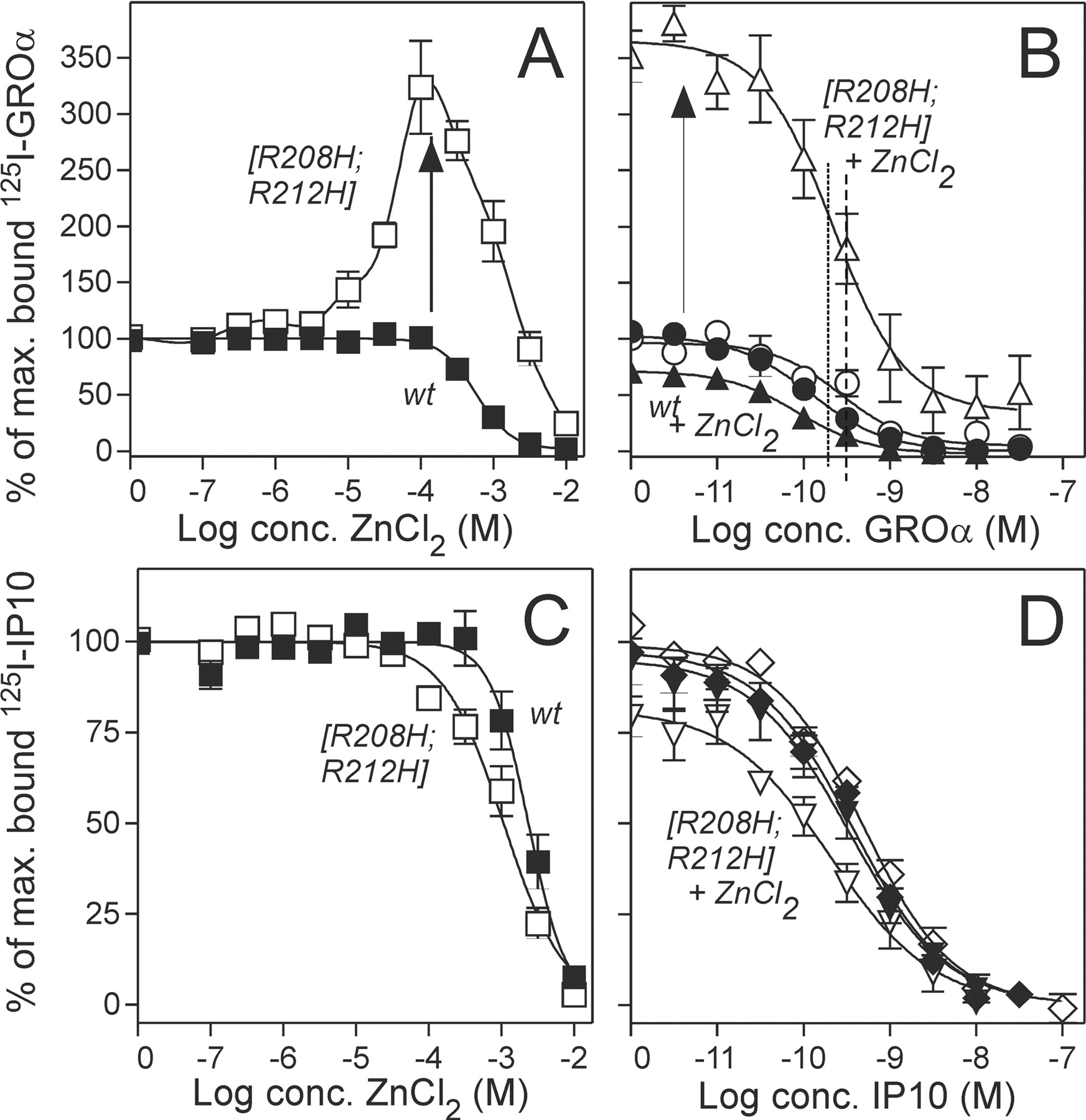
Zn(II) Acts As an Agonist and an Allosteric Enhancer in Addition to Its Role as an Inverse Agonist at Low Concentrations. A thorough examination of the dose-response curve for Zn(II) uncovered a biphasic mode for [R208H;R212H]-ORF74—but not for wt ORF74-HHV8. Thus, agonist property was observed for Zn(II) concentrations >10-fold above the EC50 (1.7 μM) for the inverse agonist activity, with a maximal efficacy of ∼35% of the basal activity reached at 100 μM Zn(II) (Fig. 1B). We decided to characterize the binding profile of Zn(II) in competition against radiolabeled agonist (125I-CXCL1/GROa) and inverse agonist (125I-CXCL10/IP10). It is interesting that the high potency of Zn(II) as an inverse agonist (EC50 of 1.7 μM) was not matched with a similar high affinity—in either competition against agonist or inverse agonist (Fig. 3, A and C). In fact, in competition against 125I-CXCL10/IP10, we observed only a 2-fold increase in the affinity compared with wt ORF74-HHV8 (Fig. 3C). In contrast, a huge increase in the binding capacity of the agonist was observed for Zn(II) concentrations >10 μM, with a peak value at 100 μM Zn(II) of 325% above the maximum specific binding in the absence of Zn(II). (Fig. 3A). Homologous competition binding in the presence of 100 μM Zn(II) revealed a slight increase (1.7-fold) in the agonist affinity for [R208H;R212H]-ORF74 and a major increase (7.1-fold) in Bmax, whereas the effects on wt ORF74-HHV8 were minimal (Table 2, Fig. 3B). The affinity of the inverse agonist increased 2.3-fold upon addition of 100 μM Zn(II) in the case of [R208H;R212H]-ORF74, whereas the Bmax decreased 1.8-fold. The corresponding effects on wt ORF74-HHV8 were again minimal (Table 2, Fig. 3D). Kinetic experiments with determination of on and off rates for CXCL1/GROα in the absence and presence of 100 μM Zn(II) uncovered that the increase in affinity for [R208H;R212H]-ORF74 and the decrease for wt ORF74-HHV8 were determined by changes in the off rate induced by the presence of 100 μM Zn(II) (a slower off rate for [R208H;R212H]-ORF74 and a quicker off rate for wt ORF74-HHV8; data not shown).
Homologous competition binding of agonist and inverse agonist in the presence and absence of 100 μM Zn(II) to wt ORF74-HHV8 and [R208H;R212H]-ORF74
The competition binding was performed in transiently transfected COS-7 cells as described under Materials and Methods with the agonist 125I-CXCL1/GROα and the inverse agonist 125I-CXCL10/IP10 as radioligands. The influence of 100 μM Zn(II) on IC50 and Bmax (± S.E.M.) are shown for both radioligands. FIC50 and FBmax for a given ligand indicate the fold change in affinity and Bmax, respectively, induced by the presence of 100 μM Zn(II).
The highly impaired potency of the agonist on [R208H; R212H]-ORF74 (Fig. 1D) was challenged by the presence of Zn(II). Thus, we measured the potency and efficacy of CXCL1/GROα in the PI-turnover experiments in transiently transfected COS-7 cells in the presence and absence of 100 μM Zn(II) and observed a ∼10-fold increase in the agonist potency for [R208H;R212H]-ORF74 in the presence of Zn(II) (Fig. 4B), whereas the potency for wt ORF74-HHV8 was slightly decreased (2.7-fold, Fig. 4A). Thus, the presence of 100 μM reduced the large difference (>80 fold) in CXCL1/ GROα potency between [R208H;R212H]-ORF74 and wt ORF74-HHV8 (EC50, ∼105 and 1.25 nM, respectively; Figs. 2C and 4, A and B) to a minor difference (3.2-fold; EC50, 10.6 and 3.3 nM, respectively). This apparent stabilization of [R208H;R212H]-ORF74 in an active conformation by 100 μM Zn(II) was confirmed by heterologous competition binding experiments of the agonist (CXCL1/GROα) in competition against radiolabeled inverse agonist (125I-CXCL10/IP10), where we observed an increase in agonist affinity (IC50 from 111 to 16.8 nM; data not shown). Thus, the presence of Zn(II) in activating concentrations (100 μM) increased the binding and signaling properties of the agonist for [R208H;R212H]-ORF74. The increase Bmax and Emax for the agonist was not due to an increase in the actual surface expression of receptors, because we observed no changes in the receptor expression measured by an ELISA-based assay of FLAG-tagged receptors. Thus, [R208H;R212H]-ORF74 and wt ORF74-HHV8 were tagged with the FLAG-tag in the N termini and receptor expression was measured for increasing concentrations of agonist CXCL1/GROα in the absence or presence of Zn(II) in activating concentrations (100 μM) and in inverse agonist concentrations (1 μM) (Fig. 5). An unrelated Flag-tagged receptor (EBI2) with no binding of chemokine or Zn(II) was included in addition to mock-transfected cells, and no changes were observed in the expression of ORF74 receptors or in the controls for the different concentrations of CXCL1/GROα and Zn(II). The FLAG-tagged ORF74 receptor acted like the untagged receptors in terms of ligand binding and activation profile (data not shown).

Competition binding with Zn(II) and chemokines. Transfected COS-7 cells were used for the displacement of 125I-CXCL1/GROα (A and B) and 125I-CXCL10/IP10 (C and D) from wt ORF74-HHV8 (filled symbols) and [R208H;R212H]-ORF74 (open symbols). The competition of radiolabeled chemokine with increasing concentrations of Zn(II) is shown in A and C, whereas the competition of 125I-CXCL1/GROα with CXCL1/GROa in the presence (triangle) and absence (circle) of 100 μM Zn(II) is shown in B. The corresponding competition of 125I-CXCL10/IP10 with CXCL10/IP10 in the presence (triangle) and absence (diamond) of 100 μM Zn(II) is shown in D (n = 3-5).
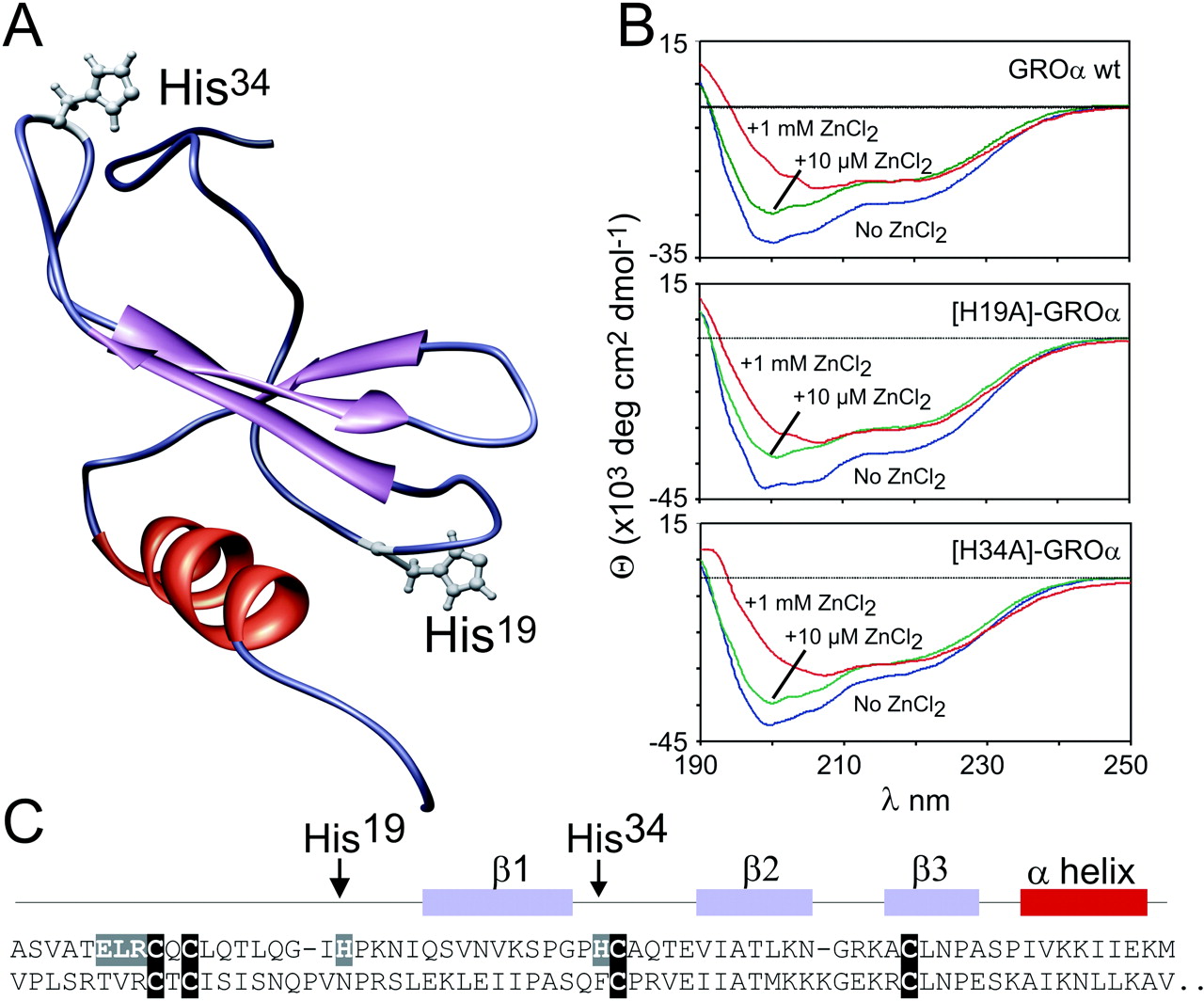
The Enhanced Binding and Action of the Agonist Is Not Due to a Metal-Ion Site between the Agonist and [R208H;R212H]-ORF74. The enhanced binding and action of CXCL1/GROa could, in theory, be due to a metal ion site between ligand and receptor upon introduction of the two His residues in ORF74-HHV8. We therefore designed two CXCL1/GROα analogs in which the two present (and putative metal ion binding) His residues in CXCL1/GROα—His19 and His34—were substituted by alanines (Fig. 5). The H19A and the H34A CXCL1/GROα-analogs were expressed in E. coli, purified by HPLC, and refolded. The chemokines were verified by mass spectroscopy and N-terminal sequencing. Because all the other experiments with agonist and inverse agonists (Fig. 1, 2, 3 and 4) were performed with commercially available CXCL1/GROα, we decided to synthesize wt CXCL1/GROα in parallel with the two CXCL1/GROα-analogs. To study the structure of CXCL1/GROα and analogs, CD spectroscopy was performed (Fig. 6B). The spectra of all three variants were dominated by a negative Cotton effect at 200 nm, indicating a random coil structure. Furthermore a negative Cotton effect at 222 nm, as well as a positive one at 190 nm, indicates the C-terminal α-helix. The addition of zinc ions led to a decrease of the random coil structure monitored by the decrease of the negative Cotton effect at 200 nm. This structural change was further indicated by the increase of the ratio of the Cotton effects at 222 and 207 nm. We were surprised that the observed effect was the same for all three variants with and without the histidine residues situated in loop regions (Fig. 5A). The in-house made H19A, H34A, and wt CXCL1/GROα were iodinated, and similar binding affinities—determined by homologous competition binding—were found, ranging from 0.26 to 0.37 nM for wt ORF74-HHV8 and from 1.7 to 3.6 nM for [R208H-R212H]-ORF74 (data not shown). The specific binding of both analogs was increased by the presence of Zn(II) at concentrations >10 μM, with peak values of agonist binding at 100 μM Zn(II) (data not shown) as observed for wt CXCL1/GROα (Fig. 3). Furthermore, the low potencies of H19A- and H34A-CXCL1 was improved 6- and 8-fold, respectively, upon addition of 100 μM Zn(II), whereas the corresponding potencies for wt ORF74-HHV8 were 1.6- and 1.7-fold decreased (data not shown). The customized wt CXCL1/GROα acted in all cases like the commercially available version. Thus, both CXCL1 analogs acted like wt CXCL1/GROα, indicating that the enhancing effects of Zn(II) on the binding and action of agonist were not due to a metal ion binding site between agonist and receptor.

Zn(II) potentiates the action of CXCL1/GROα. PI turnover experiments in transiently transfected COS-7 cells with CXCL1/GROα mediated activation of wt ORF74-HHV8 (A) and [R208H;R212H]-ORF74 (B) in the absence (filled symbols) and presence (open symbols) of Zn(II) in activating concentrations (100 μM) (n = 3-5).
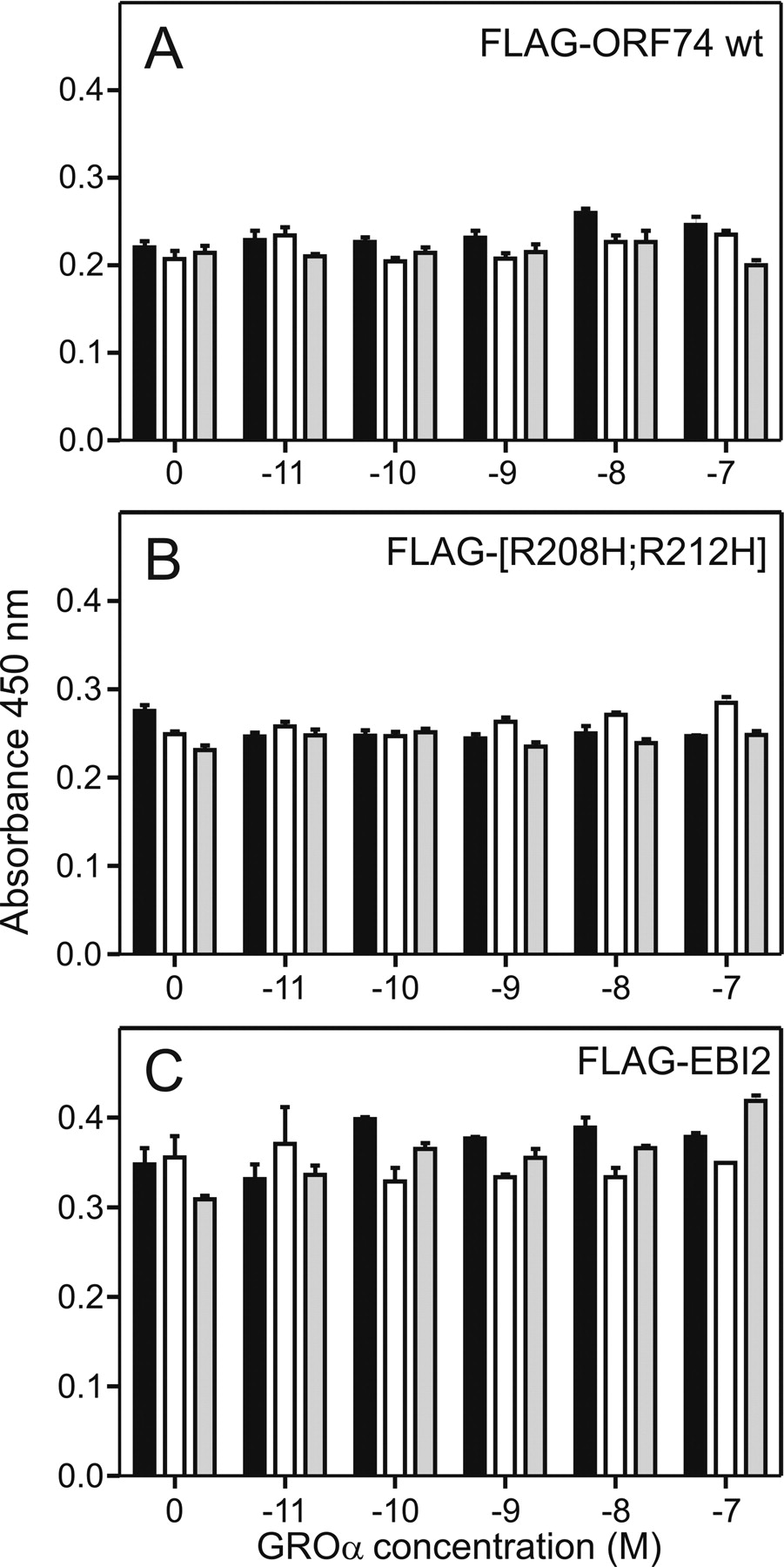
Receptor expression during agonist stimulation in the absence and presence of Zn(II). The ELISA-based surface expression was performed with FLAG-tagged receptors expressed transiently in COS-7 cells in the absence (filled columns), or presence of ZnCl2 (1 μM, open columns; 100 μM, gray columns) for increasing concentrations of CXCL1/GROα (from 10 pM to 100 nM). The results are shown for ORF74-HHV8 wt (A), [R208;R212H]-ORF74 and EBI2 (control receptor with no interaction with CXCL1/GROα or ZnCl2 in the given concentrations). The level of expression for the mock transfected cells was 0.125 (data not shown) (n = 3).
Discussion
In the present study, we characterize a conformational constrained 7TM chemokine receptor created by mutagenesis of two residues located in the top of TM-V (in position V:01 and V:05—facing into the major binding pocket). In the mutated receptor—[R20H;R212H]-ORF74—Zn(II) acted in a bi-phasic manner with a low micromolar inverse agonist potency and activation properties at higher concentrations. Furthermore, the mutated receptor—which was constrained in an agonist-impaired mode with decreased affinity, Bmax, and potency of the agonist compared with wt ORF74-HHV8—could be “unlocked” from this mode by addition of Zn(II) in activating concentrations.
Chemokine Binding to [R208H;R212H]-ORF74. The ORF74-HHV8 receptor is remarkable due to its high constitutive activity and the diverse spectrum of agonists (CXCL1-3/GROα, -β, and -γ), neutral ligands (CXCL5/ENA78, CXCL7/NAP2, and CXCL8/IL8) and inverse agonists (CXCL10/IP10, CXCL12/SDF, and vCCL2/viral macrophage inflammatory protein 2). “Functional” homologous competition binding (agonist versus agonist and inverse agonist versus inverse agonist) revealed an impaired binding of agonists to [R208H;R212H]-ORF74 (Figs. 1, 2, 3 and 4, Table 1). Furthermore, heterologous competition binding (inverse agonist versus agonist and vice versa) showed that the agonists were highly impaired in their competition against radiolabeled inverse agonist for [R208H;R212H]-ORF74 (41-213-fold lower affinity; Fig. 4D, Table 1). The agonist affinities for wt ORF74-HHV8 were not influenced to the same degree (<10-fold difference, Table 1), yet still the “functional” homologous binding revealed the highest affinities. The affinities of the inverse agonists were all found to be highest in competition against inverse agonist (compared with the competition against agonist), yet the differences in affinities were much smaller than for the agonists (1.2- to 7.1-fold). This phenomenon—that affinities may depend upon the employed radio-ligand—has been studied previously in other 7TM receptors (e.g., the tachykinin and opioid receptors) (Rosenkilde et al., 1994; Hastrup and Schwartz, 1996; Hjorth et al., 1996; Sagan et al., 1997). Among the chemokine receptors, a difference of more than 1000-fold in measured affinity was observed for certain CXC-chemokines toward CXCR2 (Ahuja et al., 1996), and a similar difference was observed for the competition of neutral ligands with radiolabeled agonist and inverse agonists in wt ORF74-HHV8 (Rosenkilde and Schwartz, 2000).

Structural properties of CXCL1/GROα wt and analogs. The tertiary structure of CXCL1/GROα with indications of the positions of His19 and His34 (A) was drawn after Protein Data Base entry 1MSG using the program UCSF Chimera. B, the CD spectra were recorded of wt (top), H19A (middle) and H34A CXCL1/GROα (bottom) in the absence (blue line) and in the presence of 10 μM Zn(II) (green line) and 1 mM Zn(II) (red line). Zn(II) induced a structural change indicated by a decrease of the cotton effect at 200 nm and an increase of the ratio of the cotton effect at 222 and 207 nm. The observed effect is the same for all three variants. Furthermore, the primary sequence of CXC1/GROα (upper sequence) and CXCL10/IP10 (lower sequence) are shown in C. The last nine amino acids after the α-helix of CXCL10/IP10 have been deleted because they are not present in CXCL1/GROα.
Correlation between Affinities and Potencies for Chemokine Agonist and Inverse Agonist. The highly impaired agonist potency (EC50 of 105 nM) for [R208H; R212H]-ORF74 was not correlated to the agonist affinity determined against radiolabeled agonist (IC50 of 0.52 nM); instead, it correlated very well to the affinity determined against radiolabeled inverse agonist (EC50 of 111 nM). This was also the case for wt ORF74-HHV8, because the agonist potency (EC50 of 1.25 nM) correlated much better to the affinity determined against radiolabeled inverse agonist (IC50, 0.81 nM) than to the affinity determined against radiolabeled agonist (IC50, 0.09 nM). The potencies of the inverse agonist for wt ORF74-HHV8 and [R208H;R212H]-ORF74 (EC50, 3.6 and 3.9 nM for CXCL10/IP10, respectively) corresponded in both cases best to the affinities determined against radiolabeled agonist (IC50, 2.6 and 5.2 nM, respectively) and not against radiolabeled inverse agonist (IC50, 0.66 and 0.73 nM, respectively). Thus, the affinities in the heterologous (but not homologous) binding correlated very well to the potencies for agonist and inverse agonist. To our knowledge, this correlation has not been described before in any rhodopsin-like 7TM receptor. It is well known, however, that 7TM receptors exist in many conformations, not only the three (active, inactive, and resting) originally described by the ternary complex model (Lefkowitz et al., 1993). For instance, in the NK1 receptor, a very high-affinity state (IC50, 50 pM), as well as a high-affinity state (∼1 nM), has been described for substance P by employing different radiola-beled agonists (Hastrup and Schwartz, 1996; Martini et al., 2002). In the present case, it could be anticipated that the homologous binding “maps” only one conformation (i.e., the conformation with highest affinity), in part determined by the very low radioligand concentration (10-15 pM). In contrast, the apparent affinity determined in heterologous binding may be influenced by kinetic and/or energetic factors related to the conformational interchange and may represent a “mapping” of a broader spectrum of states. Another explanation for the low potency/high affinity for the agonists in their interaction with [R208H;R212H]-ORF74 could simply be that the function of agonists (but not inverse agonists) depends upon an interaction with the Arg residues in the top of TM-V. The is consistent with a proposed model for the interaction between chemokine and receptor, stating that the chemokine-core interacts with the receptor N terminus and extracellular loops while the chemokine N terminus interacts with the helix bundle and promote receptor activation (Schwarz and Wells, 2002), because the agonists all contain an acidic residue in their N termini (Glu in the ELR-motif), whereas this is not the case for the inverse agonists (non-ELR chemokines) (Fig. 6).

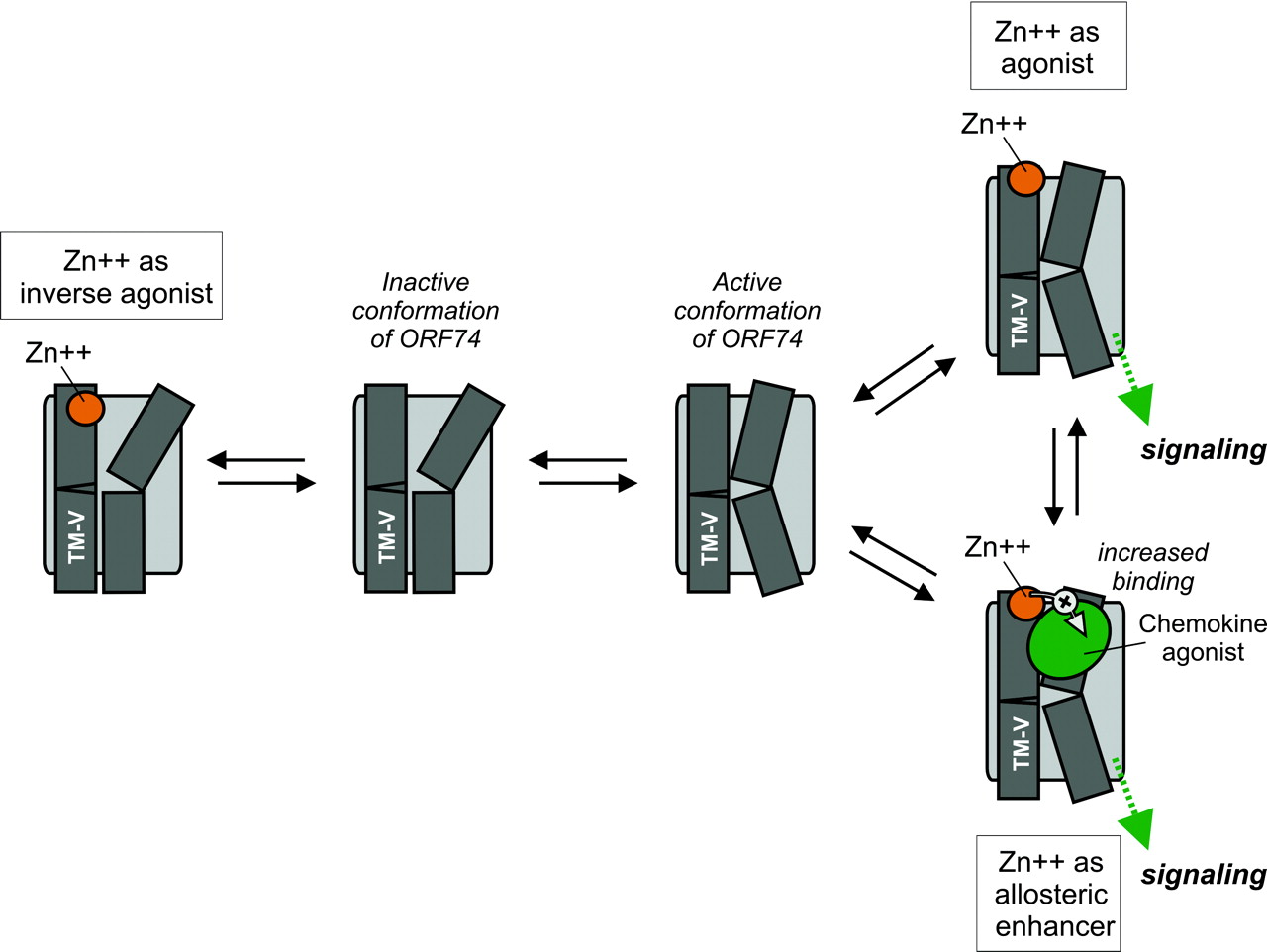
Schematic model of the proposed binding mode of metal ion and chemokine agonist for the active and inactive conformation of ORF74-HHV8. The model is a simplified version of the 7TM receptor activation model [“toggle switch model” presented recently (Schwartz et al., 2006)]. We have chosen to simplify this model and focus on TM-V (which contains the metal-ion binding site in the extracellular end, [R208H; R212H]) and TM-VI (which moves inward toward TM-III at the extracellular end and outward during receptor activation). To the left, the binding of Zn(II) to the inactive conformation of ORF74-HHV8 constrains the receptor in an inactive conformation. Thus, Zn(II) in this concentration, 1 μM, act as an inverse agonist through interaction with the extracellular end of TM-V. To the right, the active conformations of ORF74-HHV8 is given, with the Zn(II), 100 μM, acting as agonist alone (top right) and the action of Zn(II), 100 μM, as allosteric enhancer of chemokine agonist binding and action (bottom right).
Metal Ion Binding and Signaling in ORF74. The bi-phasic dose-response curve of Zn(II) uncovered inverse agonist and agonist properties (Fig. 1B) for [R208H;R212H]-ORF74. In addition, coincubation of chemokine with Zn(II) in activating concentrations (100 μM) uncovered allosteric enhancement of agonist but not inverse agonist binding and action (7.1-fold increase in Bmax; 6.1- and 1.7-fold increases in affinity determined in heterologous and homologous binding, respectively; 9.9-fold increase in potency; and an increase in efficacy from an Emax <2-fold to 4.5-fold above the basal activity). Thus, Zn(II) interacts in a complex manner with [R208H;R212H]-ORF74 in that it functions as an inverse agonist at low concentrations, and as an allosteric enhancer (of agonist binding/function) and an allosteric agonist at higher concentrations (Fig. 7). An allosteric enhancer is defined as an allosteric modulator that increases agonist affinity and/or efficacy, but has no action on its own, whereas an allosteric agonist (“ago-allosteric ligand”) activates the receptor by binding to an allosteric site that is distinct from the orthosteric site (Neubig et al., 2003; May et al., 2004). The introduced metal ion binding site is indeed allosteric, because the orthosteric site for CXC-chemokines (large peptide ligands of 70-90 kDa) is composed of several regions located in the N terminus and in the extracellular loops (Ahuja et al., 1996; Katancik et al., 2000; Rosenkilde et al., 2000). The huge increase in Bmax for agonist deserves further notice, because allosteric modulators that increase Bmax are relatively uncommon in contrast to modulators that decrease Bmax for the orthosteric ligand (May et al., 2004). However, another allosteric enhancer, PD81723, has previously been shown to increase the Bmax of the agonist [3H]cyclohexyladenosine, but not the antagonist [3H]cyclopentyl-1,3-dipropylxanthine 8 dipropyl-2,3 for the adenosine A1 receptor (Kollias-Baker et al., 1997).
What Is the Structural Basis for the Effect of Zn(II)? In theory, the observed allosteric enhancement could be due to a putative metal ion site between agonist and receptor as observed for neurokinin A in the NK2 receptor (M. Lucibello, B. Holst, M. M. Rosenkilde, T. W. Schwartz, unpublished observations). CXCL1/GROα has, in fact, two putative metalion binding residues (that are not present in CXCL10/IP10) located in the flexible N-loop region (His19) that connects the N terminus to the β-sheet region through the single turn of a 310 helix and in the region between the first and second β-sheet (His34) (Fig. 6) (Fairbrother et al., 1994). The N-loop is directly involved in receptor binding, whereas the second region is located distant from the presumed receptor binding site as well as distant from the region being important for the glycosaminoglycan interaction (i.e., between the second and third β-sheets) (Lowman et al., 1996; Fernandez and Lolis, 2002). The fact that both analogs ([H19A] and [H34A]) acted very similarly to the wt CXCL1/GROα excludes a putative, activating metal ion site within the ligand or between the ligand and the receptor. Thus, the structural basis for the complex action of the metal ion should be found within the receptor as such.
A naturally occurring metal-ion site is present at the extracellular end of TM-V in the tachykinin NK3 receptor at the exact place where the two His residues were introduced in ORF74-HHV8 in the present study (V:01 and V:05). At this bis-His site, Zn(II) in physiological concentrations has a positive modulatory effect on the binding and action of the endogenous agonist neurokinin B (Rosenkilde et al., 1998). An allosteric Zn(II) site with negative modulation of antagonist binding has been described at the extracellular end of TM-VI in the dopamine D2 receptor (Liu et al., 2006), and an allosteric Zn(II) site with positive modulation of agonist binding has been described in the intracellular loop 2 of the β2-adrenergic receptor (Swaminath et al., 2003). In the present study, we created a metal ion site in the extracellular end of TM-V (V:01 and V:05) and the observed phenotype (complex action of metal ion, conformational constraining of inverse agonist-preferring conformation that in part could be “unlocked” by metal ion in activating concentrations; Fig. 7) clearly indicates that the exterior end of TM-V is important for 7TM receptor activation, presumably by interfering with the movements of TMs III, VI, and VII during receptor activation (Schwartz et al., 2006). It will therefore be interesting—in future mutagenesis studies of ORF74-HHV8—to elucidate the structural basis for the complex action of the metal-ion.
Acknowledgments
We thank Lisbet Elbak and Inger S. Simonsen for excellent technical assistance, Ann Richmond (Vanderbilt University, Nashville, TN) for the cDNA encoding the wt CXCL1/GROα, and Laura Storjohann for critical reading of and fruitful discussions about the manuscript.
Footnotes
-
↵1 Chemokine names are given according to the novel nomenclature (Murphy et al., 2000) followed by the old names, e.g., CXCL10/IP10. In the figures, we use the old names, but the legends include the new names as well.
-
This study was supported by grants from the Danish Medical Council, the NovoNordisk Foundation, the Vera and Einar Danielsen Foundation and the European Union FP6 (INNOCHEM, grant number LSHB-CT-2005-518167).
-
ABBREVIATIONS: 7TM, seven transmembrane; TM, transmembrane helix; ORF, open reading frame; HHV8, human herpesvirus 8; PI, phosphatidyl inositol; GROα/β/γ, growth-related oncogene; IP10, interferon γ-inducible protein; IL8, interleukin-8; ENA78, epithelial cell-derived activating peptide-78; SDF-1, stromal cell-derived factor-1; HPLC, high-performance liquid chromatography; TBS, Tris-buffered saline; ELISA, enzyme-linked immunosorbent assay; wt, wild type; PD81723, (2-amino-4,5-dimethyl-3-thienyl)(3-(trifluoromethyl)phenyl)-methanone.
- Received June 1, 2006.
- Accepted September 12, 2006.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}