


Abstract
The α7 nicotinic acetylcholine receptor (nAChR), a homopentameric, rapidly activating and desensitizing ligand-gated ion channel with relatively high degree of calcium permeability, is expressed in the mammalian central nervous system, including regions associated with cognitive processing. Selective agonists targeting the α7 nAChR have shown efficacy in animal models of cognitive dysfunction. Use of positive allosteric modulators selective for the α7 receptor is another strategy that is envisaged in the design of active compounds aiming at improving attention and cognitive dysfunction. The recent discovery of novel positive allosteric modulators such as 1-(5-chloro-2-hydroxyphenyl)-3-(2-chloro-5-trifluoromethylphenyl)urea (NS-1738) and 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)urea (PNU-120596) that are selective for the α7 nAChRs but display significant phenotypic differences in their profile of allosteric modulation, suggests that these molecules may act at different sites on the receptor. Taking advantage of the possibility to obtain functional receptors by the fusion of proteins domains from the α7 and the 5-HT3 receptor, we examined the structural determinants required for positive allosteric modulation. This strategy revealed that the extracellular N-terminal domain of α7 plays a critical role in allosteric modulation by NS-1738. In addition, α7-5HT3 chimeras harboring the M2-M3 segment showed that spontaneous activity in response to NS-1738, which confirmed the critical contribution of this small extracellular segment in the receptor gating. In contrast to NS-1738, positive allosteric modulation by PNU-120596 could not be restored in the α7-5HT3 chimeras but was selectively observed in the reverse 5HT3-α7 chimera. All together, these data illustrate the existence of distinct allosteric binding sites with specificity of different profiles of allosteric modulators and open new possibilities to investigate the α7 receptor function.
The α7 nicotinic acetylcholine receptor (nAChR) belongs to the family of ionotropic receptors that share four transmembrane domains as a common structural feature (for review, see Hogg et al., 2005). By virtue of its high expression levels in brain regions involved in learning and memory, such as hippocampus and cerebral cortex, and its unique physiological properties, partly attributed to a high permeability to Ca2+, the α7 nAChR has received considerable attention as drug target for development of drugs intended to treat cognitive/attention disorders underlying neuropsychiatric and neurodegenerative diseases (for review, see Dani and Bertrand, 2007). Furthermore, despite its low sensitivity to acetylcholine, the α7 nAChR has been shown to modulate the release of other neurotransmitters and, in some cases, to contribute directly to signal transmission (for review, see Wonnacott et al., 2006).
Gene knock-out and antisense studies together with pharmacological studies using small-molecule selective agonists and positive allosteric modulators have demonstrated that α7 nAChRs play important roles in aspects of attention, cognition, and neuroprotection relevant to diseases such as schizophrenia and Alzheimer's disease. For example, preclinical studies with α7 nAChR selective agonists including AR-R 17779 (Van Kampen et al., 2004), PNU-282987 (Bodnar et al., 2005), PHA-543613 (Wishka et al., 2006), SSR-180711 (Biton et al., 2007; Pichat et al., 2007), ABBF (Boess et al., 2007), and A-582941 (Bitner et al., 2007) have suggested that augmenting α7 nAChR function is capable of ameliorating cognitive deficits. In addition, genetic analysis has revealed that CHRNA7 is linked to sensory gating deficits that may contribute to the attentional and cognitive deficits observed in schizophrenia (Freedman, 2007). Although selectively activating α7 nAChRs has the potential to prevent or treat a variety of diseases involving cognitive and neurodegenerative deficits, uncertainty exists about whether treatment with agonists in humans might provide suboptimal benefit as a result of sustained receptor activation and desensitization of the nAChR. An alternative approach to reinforce neurotransmission and/or neuromodulation mediated by the α7 nAChR would be to increase their responsiveness to the endogenous transmitter, ACh, via positive allosteric modulation. Positive allosteric modulators can reinforce endogenous cholinergic transmission of ACh increasing the receptor sensitivity or reducing desensitization without directly activating the α7 nAChR (for review, see Bertrand and Gopalakrishnan, 2007)
Initial characterization of the α7 nAChRs and structure-function studies have highlighted the unique properties of this receptor subtype and its modulation by the extracellular Ca2+ ions, which act as a positive allosteric effector (Galzi et al., 1996). Moreover, it was shown that molecules such as ivermectin increase the receptor sensitivity and the response amplitude with little effect on desensitization (Krause et al., 1998). Increase in ACh sensitivity without modification of the response time course was also reported with 5-hydroxy indole (Zwart et al., 2002). More potent positive allosteric modulators (PAMs) have recently emerged (such as, for example, NS-1738) that, like 5-HI or ivermectin, increase ACh sensitivity with only marginal effects on the desensitization kinetics. Although these allosteric modulators caused little or no change in the response time course, other studies have revealed that compounds such as PNU-120596 could evokeprofound effects on receptor desensitizatio, in addition to the increased ACh sensitivity and current amplitudes (Hurst et al., 2005).
Despite identification of structurally different PAMs with specific profiles, little is known about the site of interaction of such compounds at the α7 nAChR. The amino acid position 172 in the extracellular domain was initially identified by Galzi et al. (1996) as the site of action for the calcium modulation, an observation further supported by subsequent site-directed mutagenesis studies (Eddins et al., 2002; McLaughlin et al., 2006). In contrast, little information is available about the site of modulation by molecules such as IVM, 5-HI, NS-1738, PNU-120596, or other PAMs. To elucidate interactions of PAM at α7 nAChR, we decided to take advantage of functional chimeras between α7 and 5HT3 receptors by fusion of the α7 extracellular domain with the transmembrane and intracellular segment of the 5HT3 receptors (Eisele et al., 1993). The strategy was to examine the sensitivity of chimeras hosting progressively increasing amino acid segments of α7 nAChR to different allosteric modulators and assess the domains responsible for the sensitivity of the nAChRs to α7 PAMs, particularly NS-1738 and PNU-120596. Our studies demonstrate, for the first time, distinctions in sites of action of these two phenotypically distinct PAMs, and furthermore, reveal an important role for the M2-M3 segment of the α7 nAChR in channel activation.
Materials and Methods
Molecular Biology. Chimera 1 contains the ligand-binding domain of α7 nAChR and the transmembrane/pore forming region of 5-HT3 receptor. With the use of reverse transcription-polymerase chain reaction, the coding sequence for the N-terminal 224 amino acids of human α7 nicotinic receptor (α7 nAChR; amino acids 1-224 of protein AAA83561) and that for the C-terminal 242 amino acids of human 5-hydroxytryptamine type 3 (5-HT3) receptor (amino acids 243-484 of protein AAP35868) were amplified with overlapping ends. Further amplification using these two overlapping fragments yielded the open reading frame of chimera 1. Primers used to generate the α7 nAChR portion of this chimera were (5′→3′) GCCGCCATGCGCTGCTCGCCGGGAGGCGTCT (A7F, forward) and AGGCTGACCACATAGAAGAGTGGCCTACGTCGGATGACCACTGTGAAGGTACATCG (Chi1R, reverse). Primers used to generate the 5-HT3 portion of this chimera were (5′→3′) GTCAAGCGTACTGCCAGATGGACCAGA (5HT3R, reverse) and CGATGTCACCTTCACAGTGGTCATCCGACGTAGGCCACTCTTCTATGTGGTCAGCCT (Chi1F, forward). Reactions were performed in a Robocycler (Stratagene, La Jolla, CA) using 10 ng of each template and 0.4 μM concentrations of each primer with Platinum Taq DNA Polymerase High Fidelity according to the manufacturer's protocol (Invitrogen, Carlsbad, CA). Recombinant polymerase chain reaction used 1 μl of amplicon directly from each of the two reactions along with 0.4 μM concentrations of primers A7F and 5HT3Rina50-μl reaction. The recombinant polymerase chain reaction product was cloned into the expression vector pcDNA3.1 using Invitrogen's pcDNA3.1 TOPO TA cloning kit and transformed using DH5-α Max Efficiency chemically competent bacteria. Clones were selected on plates containing Luria Bertani agar medium and 100 μg/ml ampicillin. The sequence of the inserted DNA was verified.
Chimeras II-VII were constructed using methods similar to those used for chimera I with select substitutions of the transmembrane-linking loops and C-terminal domains as indicated (Fig. 3). In particular, chimera II had the same amino acid composition as chimera I except that the 10-amino acid loop between transmembrane domain (TM) 2 and TM3 consists of amino acids 280 to 289 of α7 nAChR (AEIMPATSDS) instead of the original amino acids 298 to 307 of 5-HT3 (SDTLPATAIG). Chimera III has the same amino acid composition as chimera II except that the last three amino acids on the C terminus (originally 5-HT3 amino acids 482-484, QYA) have been replaced by the nine C-terminal amino acids of α7 nAChR (VEAVSKDFA). Chimera IV has the same amino acid composition as chimera I except that the loop between TM3 and TM4, originally composed of amino acids 329 to 457 of 5HT3 (TIFIVRLVHKQDLQQPVPAWLRHLVLERIAWLLCLREQSTSQRPPATSQATKTDDCSAMGNHCSHMGGPQDFEKSPRDRCSPPPPPREASLAVCGLLQELSSIRQFLEKRDEIREVARDWLRVGSVLD), was replaced with α7 nAChR amino acids 311 to 468 (TVIVLQYHHHDPDGGKMPKWTRVILLNWCAWFLRMKRPGEDKVRPACQHKQRRCSLASVEMSAVAPPPASNGNLLYIGFRGLDGVHCVPTPDSGVVCGRMACSPTHDEHLLHGGQPPEGDPDLAKILEEVRYIANRFRCQDESEAVCSEWKFAACVVD). This conversion of the TM3-to-TM4 loop from 5HT-3 to α7 nAChR amino acids was repeated on chimera II to generate chimera V. Likewise, this conversion was repeated on chimera III to generate chimera VI. Chimera VII has the same amino acid composition as chimera IV except that the 5HT3 C terminus has been replaced with the α7 nAChR C terminus as in described for chimera III. Chimera VIII, designed as the reverse of chimera I and constructed similarly, contains the ligand-binding domain of 5HT3 (amino acids 1-242 of protein AA-P35868) and the transmembrane/pore forming region of α7 nAChR (amino acids 225-502 of protein AAA83561).
Electrophysiology. All constructs were expressed in Xenopus laevis oocytes using standard conditions, as described previously (Krause et al., 1998). In brief, ovaries were harvested from female X. laevis frogs that were deeply anesthetized and sacrificed according to the animal rights rules established in Geneva, Switzerland. Oocytes were isolated by mechanical and enzymatic dissociation using type I collagenase with gentle mixing for a couple of hours. To minimize heat shock, all preparation and recording procedures were carried out at 18°C. After dissociation, oocytes were then kept in Barth's solution containing 88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 10 mM HEPES, 0.82 mM MgSO4·7H2O, 0.33 mM Ca(NO3)2·4H2O, and 0.41 mM CaCl2·6H2O at pH 7.4, supplemented with 100 unit/ml penicillin and 100 μg/ml streptomycin. On the second day after dissociation, stage 5 to 6 oocytes were manually selected under a binocular microscope and injected into the nucleus with 20 ng of cDNA per oocyte containing either the gene encoding for the α7 nAChR or the desired chimera. Recordings were performed between 2 and 7 days after injection. During recordings, cells were superfused with OR2 medium containing 82.5 mM NaCl, 2.5 mM KCl, 5 mM HEPES, 1.8 mM CaCl2·2H2O, and 1 mM MgCl2·6H2O, pH 7.4. Atropine (0.5 μM) was added to all solutions to block the activity of endogenous muscarinic receptors. Recordings were conducted using a conventional electrophysiological setup. Unless otherwise indicated, cells were held at -100 mV and currents were recorded with a GeneClamp amplifier (Molecular Devices, Sunnyvale, CA) equipped with a virtual ground headstage. Data sampled at 100 Hz were captured and analyzed using proprietary data acquisition and analysis software running under Matlab (Mathworks Inc., Natick, MA).
Concentration-activation curves were fit using the empirical Hill equation Y = 1/1 + (EC50/x)nH, where Y = the fraction of evoked current, EC50 = concentration for 50% activation, nH = the apparent cooperativity, and x = agonist concentration. Average, S.D., S.E.M., and t test results were computed either in Matlab (Mathworks Inc.) for the raw data or in spreadsheets for subsequent computation in Excel (Microsoft Corp., Redmond, WA).
Compounds and Solutions. All chemicals, including ivermectin, were obtained from Sigma-Aldrich (Buchs, Switzerland). PNU-120596 and NS-1738 were synthesized at Abbott Laboratories (Abbott Park, IL).
Results
Studies of allosteric modulators with α7 nAChRs indicate these molecules can affect energy transitions by 1) predominantly affecting the peak current responses (type I profile) or 2) both peak current responses and time course of agonist-evoked response (type II profile; reviewed in Bertrand and Gopalakrishnan, 2007). Thus, the distinction between the profiles of allosteric modulation caused by type I PAMs (e.g., IVM, 5-HI, and NS-1738) and type II PAMs (PNU-120596, 4-naphthalene-1-yl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]-quinoline-8-sulfonic acid amide) suggests that these molecules are probably acting by distinct mechanisms and probably bind to different location on the α7 protein. In this study, we investigated the effects of NS-1738 and PNU-120596, two structurally related urea analogs (Fig. 1) with different α7 nAChR PAM profiles (Hurst et al., 2005; Timmermann et al., 2007). NS-1738 efficiently potentiates the α7 nAChR responses without modifying their time course and does not affect currents at the 5-HT3 receptors (Timmermann et al., 2007), and PNU-120596 that, in addition, exhibits profound effect on the desensitization kinetics (Hurst et al., 2005).
Effects on the current amplitude and time course of the response were obtained after a preincubation with structurally diverse modulators (Fig. 1). Typical α7 currents evoked by a brief ACh test pulse (100 μM, 3 s) were recorded in control, after a preincubation with NS-1738 (10 μM, 20 s) or PNU-120596 (10 μM, 20 s). Recordings of the ACh-evoked currents obtained in a series of ACh concentrations using the same experimental protocol allowed determination of the concentration dependence in the absence of and after exposure to the modulator (Fig. 1, bottom). Plot of the peak inward current as a function of the logarithm of the ACh concentration yielded typical concentration-response curves that are readily fitted by the empirical Hill equation (Table 1). Preapplication of IVM, 5HI, NS-1738, and PNU-120596 yielded a series of concentration-activation curves with typical profiles of positive allosteric modulators—increase in the amplitudes of the peak ACh-evoked currents, leftward shift in concentration-activation curves, and increased steepness of the slope of the concentration response curve. Exposure of oocytes expressing the 5HT3 receptor to NS-1738 or PNU-120596 caused no significant modification of the subsequent response evoked by the agonist (data not shown). The ratio of potentiation obtained for the α7 and 5-HT3 receptor to a fixed exposure to NS-1738 and PNU-120596 are illustrated in Fig. 4.
Coefficients for the best fits of α7 wild type, Chim I, and Chim II
When similar experiments were performed at the α7-5HT3 chimera (Chim I; Table 1 and Fig. 2, top), in which the extracellular domain of α7 is fused at position 201 to the 5HT3 receptor, strikingly different results were observed. Whereas NS-1738 retained a marked potentiation of ACh-evoked current at chimera 1, PNU-120596 failed to elicit any increase in ACh-evoked current. This suggests that energy barriers for the transition from resting to the active state of chimera 1 are different from those of α7 nAChR or that the PNU-120596 failed to bind and modulate this receptor.
Previous studies carried at using the GABAA receptors revealed a critical role of the short amino acid segment between the second (M2) and third (M3) transmembrane segments and that faces the extracellular domain (Kash et al., 2003). The similarity of receptor structure and further work carried out at the nAChRs further underlined the importance of short connecting segment interacting between the ligand-binding domain and the channel domain (Bouzat et al., 2004; Lee and Sine, 2005; Lummis et al., 2005). To examine further the structural determinants of allosteric modulation, effects were tested using chimera II (Chim II), which corresponds to Chim I, but with the M2-M3 segment from the 5-HT3 replaced with a corresponding α7 amino acid segment. Data obtained for the four allosteric modulators (IVM, NS-1738, 5HI and PNU-120596) are illustrated in the lower panel of Fig. 2. Unexpectedly, exposure of Chim II to the NS-1738 alone evoked a significant inward current showing no desensitization over the 20 s of preapplication (Figs. 2 and 3). Furthermore, the subsequent ACh-evoked response was significantly inhibited. A possible hypothesis that could explain both the sustained inward current and inhibition of the ACh-evoked response is that substitution of the M2-M3 segment caused a reduction of the energy barrier between the resting and active state and that further reduction caused by exposure to the positive allosteric modulator NS-1738 is seen as spontaneous opening in absence of ligand. In agreement with this hypothesis, application of the competitive antagonist dihydro-β-erythroidine hydrobromide inhibited the inward current observed during the NS-1738 exposure (data not shown). Activation of a current and subsequent inhibition of the ACh-evoked response observed after exposure to NS-1738 is reminiscent of the properties that would be exhibited by a partial agonist. To evaluate this possibility, further experiments were designed in which cells were exposed to a series of NS-1738 concentrations. Although important variations in the amplitude of the NS-1738-evoked currents were noticed between different cell batches, detectable currents were observed only for concentrations above 3 μM, but further increasing the NS-1738 concentration led to a reduction of this current (data not shown). This indicates either that this compound does not act as a partial agonist or that it causes both activation and blockade of the receptor. While Chim II yielded robust ACh-evoked currents and showed sensitivity to NS-1738, preapplication of the PNU-120596 failed to increase the subsequent ACh-evoked responses (Fig. 4).
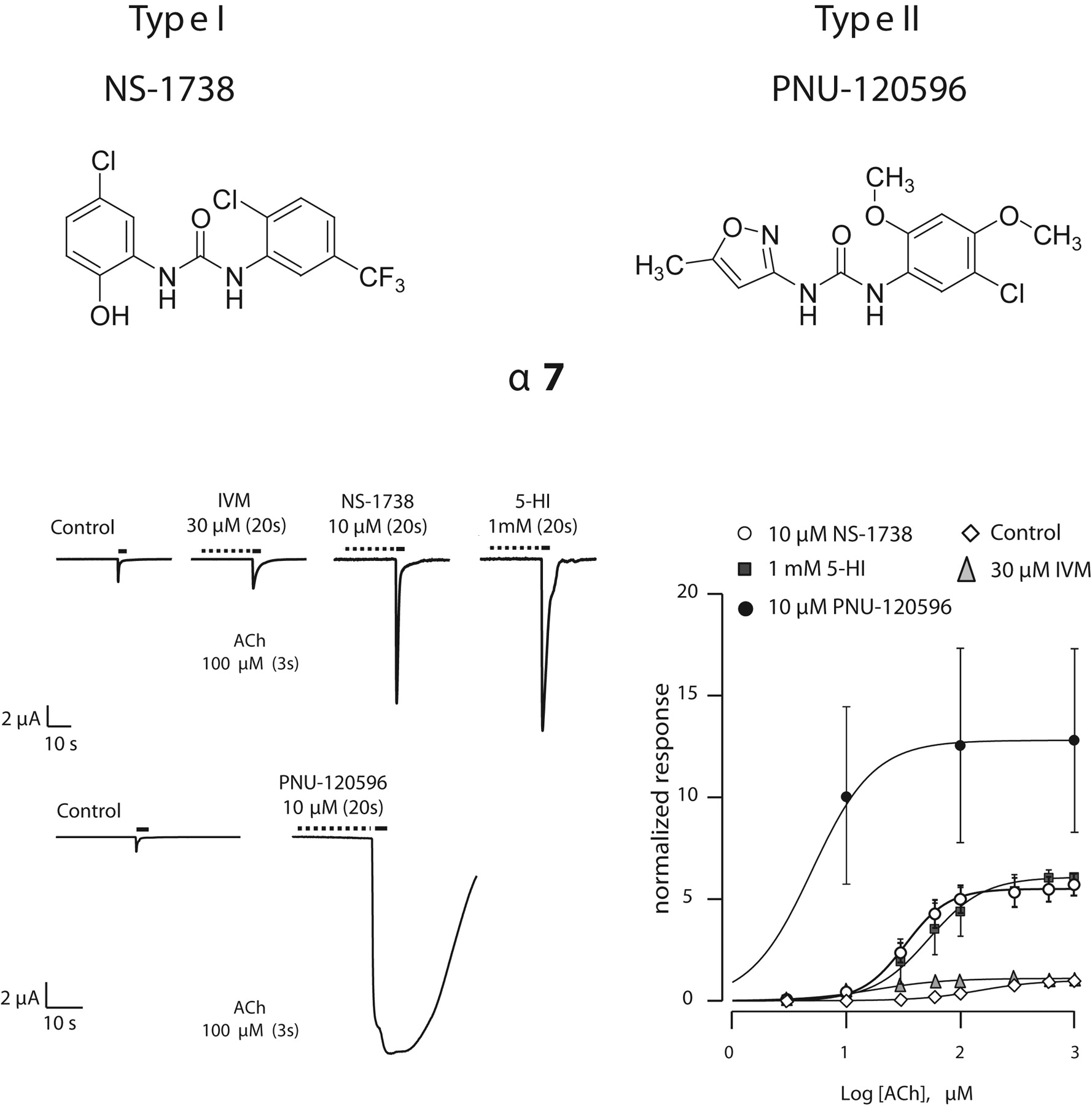
Structures and effects of type I and II positive allosteric modulators of α7 nAChRs. Top, structure of two chemically related positive allosteric modulators of the α7 receptors. Bottom left, effects of NS-1738 and PNU-120596 in an oocyte expressing the α7 nAChR. Currents evoked by brief ACh test pulse (100 μM, 3 s) were recorded at regular intervals. Exposure to IVM, NS-1738, and 5HI caused a substantial increase in the amplitude of the subsequent ACh-evoked current without major modification of the response time course. Full recovery was obtained after wash (not shown). By comparison, exposure to PNU-120596 in the same cell caused both an increase in the amplitude of the ACh-evoked current and a marked slowing of the receptor desensitization. The cell was maintained in voltage clamp at -100 mV. Right, effects of the positive allosteric modulators on the concentration-activation curves. Error bars indicate the S.E.M. for α7(n = 5), α7 IVM (n = 5), α7 5-HI (n = 3), α7 NS-1738 (n = 5), and α7 PNU-120596 (n = 3). Curves through data points are the best fit obtained with the empirical Hill equation (see Materials and Methods). Corresponding coefficients are presented in Table 1.

Effects of four positive allosteric modulators at chimeras I and II. Schematic representation of the α7-5HT3 fusion protein (Chim I; dark line indicates the α7 segment) is illustrated at the top together with the effects of the four modulators on the ACh-evoked currents and on the concentration-activation curves. Note the absence of effects of PNU-120596 and IVM, whereas a positive modulation is still observed with the 5-HI or the NS-1738. Cells were constantly held at -100 mV. Bottom, schematic representation and functional properties of Chim II, in which the N-terminal extracellular domain and the extracellular segment delimited by M2 and M3 have been replaced with that of α7 (dark line). Typical ACh-evoked currents measured first in control and after exposure to allosteric modulators are represented at bottom left and concentration-activation curves measured as for Chim I are shown at bottom right. Curves through data points are the best fits obtained with the empirical Hill equation (see Materials and Methods). Corresponding coefficients are presented in Table 1.

Summary of the modulation caused by the NS-1738 at Chim I-VII constructs. Typical ACh-evoked currents recorded first in control conditions (continuous line) and after 20-s exposure to 10 μM NS-1738 for a series of chimeras are illustrated (dashed lines). Comparable recordings were obtained in at least three cells. Schematic representations of the corresponding chimera constructs (I-VII) are shown above each recording. Dark segments symbolize α7 amino acid sequences; gray lines represent the 5-HT3 segments.
To fully reconstitute the α7 extracellular domain in the α7-5HT3 chimera, another chimera (Chim III) in which the N-terminal domain, the M2-M3 segment, and the M4-C-terminal end from the α7 subunit was constructed and evaluated. Incorporation of the α7 C-terminal fragment, however, caused no substantial differences in the biophysical and pharmacological properties compared with Chim II. PNU-120596 failed to potentiate this chimera, whereas opening of the channels upon application of NS-1738 alone was observed (Fig. 3). It is noteworthy that of the seven chimeras tested, constructs that contained the α7 M2-M3 segments displayed sustained activation when exposed to NS-1738 alone (Fig. 4). Because the ensemble of the protein structure defines properties of the receptor, modifications introduced in the different constructs might result in variation in their respective ACh sensitivities. Determination of the half-activation (EC50) for each of the seven chimeras revealed a range of up to 10-fold differences in ACh sensitivities. To best characterize the effects of the NS-1738 or the PNU-120596, potentiation of currents evoked by ACh concentrations near the respective EC50 values were also determined (Fig. 4, bottom). The similitude observed between data obtained at 100 μM ACh and near the EC50 indicates that results obtained at a single agonist concentration give a good prediction of the overall sensitivity of the receptor to the allosteric modulator. Data presented in Table 2 summarize the shifts in ACh sensitivities caused by exposure to either NS-1738 or PNU-120596. These data confirm the lack of effect of PNU-120596 at the seven chimeras and that chimera II and VI show the largest displacement in the ACh sensitivity caused by the NS-1738.
Effects of the modulators on the ACh sensitivity of the α7 wild-type receptor and the chimeras
In light of the inability to restore potentiation by PNU-120596 in chimeras incorporating the α7 extracellular segments, we hypothesized that PNU-120596 might interact with the intracellular domain of the receptor. To test this hypothesis, Chim IV-VII, comprising the large intracellular loop of the α7 subunit, was designed and constructed (Fig. 4). Although all chimeras yielded functional receptors and displayed robust currents in response to ACh, none of these constructs showed an increase of the peak-evoked response after a 10 μM preapplication with the PNU-120596. Likewise, no significant increase in the response duration was observed in these constructs indicating that structural features of these chimeras prevent the allosteric modulatory action of the PNU-120596.
Many molecules have been reported as allosteric modulators of four transmembrane domain ligand-gated ion channels. For example, it was shown that neurosteroids are potent allosteric modulators at GABAA receptors. In a recent work, Hosie et al. (2006) identified two discrete binding sites that are localized in the transmembrane domain of the receptor and responsible for allosteric modulation. This illustrates that the binding site of the allosteric ligand can be at any place on the receptor and how binding of a molecule at the interface between two adjacent transmembrane domains can affect the receptor function. To examine the putative role of the transmembrane domains, a reverse chimera comprising the 5-HT3 ligand binding domain and the transmembrane domain of α7 was designed and constructed. Expression of the inverse chimera (Chim VIII) proved rather difficult with a percentage of expression of ≤4.8% (n = 41, three batches) versus 30% (n = 76, four batches) for the Chim I. Positive cells responded by significant inward currents in response to 5-HT application (Fig. 5). However, preapplication of either PNU-120596 or NS-1738 using the same protocol as for the α7 receptor caused no detectable modification of the subsequent 5HT-evoked current (Fig. 5, A and B). To best evaluate a putative allosteric modulation, currents evoked by low agonist concentrations were also investigated, but exposure to either NS-1738 or PNU-120596 caused no detectable modification of the response amplitude or time course (Fig. 5B). Because cross talk between α7 and the 5-HT3 receptors has often been reported, we examined the possible effects of ACh and the positive allosteric modulators. We were surprised to find that ACh evoked small, but reproducible, currents that were recorded in Chim VIII and these effects were potentiated by the PNU-120596 but not by NS-1738 (Fig. 5C). Addition of ACh on a low 5-HT concentration ± PNU 120596 was not distinguishable from ACh alone (data not shown).

Allosteric potentiation of Chim I-VII by the NS-1738 and PNU-120596. Plot of the potentiation ratio for the control receptors and seven chimeras. Peak of the ACh-evoked currents recorded after a brief exposure to NS-1738 (10 μM, 20 s, empty boxes) were measured and the ratio versus control responses determined (top). Plot of the average potentiation ratio and S.E.M. are represented. Number of cells tested for each mutant and each condition are indicated in parenthesis. Note the absence of potentiation for the 5-HT3 receptor but significant potentiation in the chimeras. Filled boxes represent the potentiation ratios determined using the same procedure for 1 μM PNU-150596. To best evaluate the potentiation caused by the NS-1738 and PNU-120596, measurements were repeated near the respective ACh sensitivity of each chimera subtype (bottom). Note the lack of potentiation for the PNU-120596 for the 5-HT3 receptors and the seven chimeras.
Discussion
Selective positive allosteric modulation of α7 nAChRs is considered as a viable therapeutic approach for a range of neurological and psychiatric disorders involving cognitive and attention deficits. It is thought that a key advantage of the PAM approach is that modulation is only revealed in the presence of the endogenous agonist acetylcholine, thereby preserving the temporal and spatial integrity of neurotransmission (Hogg et al., 2005). In the case of α7 nAChRs, at least two different profiles of PAMs have been described thus far: type I modulators, which predominantly affect the apparent peak current, agonist sensitivity, and Hill coefficient, and type II modulators, which cause, in addition, a modification of the desensitization profile of agonist-evoked responses (for review, see Bertrand and Gopalakrishnan, 2007; Faghih et al., 2007). For example, compounds such as 5HI, N-4-chlorophenyl-[[(4-chloro-phenyl)amino]methylene]-3-methyl-5-isoxazole acetamide (compound 6 in Ng et al., 2007), and NS-1738 (Timmermann et al., 2007) primarily increase the current amplitude of ACh or choline-evoked α7 currents and belong to the type I class of α7 PAMs. In contrast, molecules such as PNU-120596 and others (Hurst et al., 2005; Grønlien et al., 2007) exhibit type II profile by triggering increases in both α7 current amplitude and evoking a distinct secondary component resulting in prolonged current response to agonists.
To address where and how such molecules interact with the α7 nAChR, we used the capacity of producing functional chimera between this subunit and the 5-HT3 receptor that is potentiated by neither NS-1738 nor by the PNU-120596. Using a strategy of progressive substitution of either the extracellular or intracellular domains of the 5-HT3 receptor, we examined the possibility of circumscribing regions that are indispensable for the effects of positive allosteric modulators. The observation that Chim I is modulated by the NS-1738 indicates that introduction of the α7 N-terminal domain is sufficient to allow potentiation by this compound. On the contrary, the lack of modulation of Chim I by the PNU-120596 indicates that this compound interacts with a distinct protein domain than the NS-1738. Progressive introduction of the extracellular and intracellular domain from α7 into the chimera (I-VII) revealed the critical role played by the short M2-M3 extracellular domain. Introduction of this amino acid segment was sufficient to cause a modification of the sensitivity to the NS-1738 with the apparition of an inward and dihydro-β-erythroidine-sensitive current upon exposure to NS-1738. By comparison, site-directed mutagenesis of the M2-M3 loop previously pointed out the relevance of this short segment in controlling the current amplitude versus amount of α-bungarotoxin binding, and it was hypothesized that this segment could play a role in the receptor gating (Castillo et al., 2006). A simple hypothesis accounting for such observation is that Chim II displays a lower energy barrier between the resting and active state and that further reduction of this barrier causes spontaneous opening that resembled that observed in α7 mutants (Bertrand et al., 1997). Alternatively, it could be postulated that binding of NS-1738 partially activates the Chim II receptor, which could explain the reduction of the subsequent ACh-evoked current by desensitization. Because NS-1738 evoked a small fraction of the ACh response that was variable from batch to batch, it is difficult to make conclusions about the mechanism by which NS-1738 causes opening of the Chim II receptor. The determinant role of the M2-M3 further support previous observations made either on the GABAA and nAChRs (Kash et al., 2003; Lee and Sine, 2005; Lummis et al., 2005). No major modification of the sensitivity either to NS-1738 or to PNU-120596 was observed upon introduction of the α7 C-terminal domain (Chim III). A plausible postulate was that PNU-120596 could interact with the intracellular domain of α7 and that introduction of this amino acid segment might restore its potentiation. Construction of Chim IV-VII, however, failed to identify a segment that would restore PNU-120596. In the view of the high degree of sequence homology observed between the short M1-M2 segments of the α7 and 5-HT3 receptors, it is unlikely that the PNU-120596 effects are mediated through the interaction with this portion of the protein. All together, these data suggest that PNU-120596 probably interacts with one or more transmembrane domain of the receptor, whereas the NS-1738 interacts with the extracellular N-terminal domain. In agreement with this hypothesis, Chim VIII, which comprises the transmembrane domain of α7 but the N-terminal domain of the 5-HT3, shows a potentiation of ACh-evoked current. The relevance of the transmembrane segment in regulating the potentiation of the PNU-120596 is best understood in light of previous work carried out by photoaffinity labeling at the GABAA receptors, which identified the contribution of a single amino acid residue in M1 of the α subunit and another in the M3 of the β3 subunit in controlling the potentiation by the general anesthetic etomidate (Li et al., 2006). This suggests that, as proposed for the etomidate potentiation, the PNU-120596 might interact with the transmembrane segments and alter the energy barriers between the different states. Furthermore, it is important to note that although both NS-1738 and PNU-120596 belong to the biarylurea class of PAMs, there are differences in the substitution patterns on the aryl rings (for example, isoxazole in case of PNU-120596 versus substituted phenyl in case of NS-1738; Fig. 1); accordingly, there are differences in the log P values (∼2.8 versus ∼4.8, respectively). These differences can influence allosteric modulatory interactions of these two compounds at α7 nAChRs. The emergence of structurally distinct PAMs of different in vitro profiles offers valuable tools with which to further define the physiology and pharmacology of α7 nAChR transmission. For example, both types of PAMs have been reported to show in vivo efficacy in animal models of cognition and sensory gating deficit, although the underlying molecular mechanisms remain to be defined (Hurst et al., 2005; Ng et al., 2007). Our studies with α7 nAChRs show that both type I and II modulators cause, as expected for a positive allosteric modulator, a shift of the dose response toward lower ACh concentration, increase in the steepness of the curve (Hill coefficient), and increase in the maximal evoked current. Our investigation of the molecular mechanisms underlying PAM effects demonstrates key distinctions in the sites of action of these types of PAMs. In particular, we show, using recombinant chimeras between extracellular domain of α7 nAChR and 5-HT3 receptors, that NS-1738 and PNU-120596 interact at distinct sites on the receptors and that amino acid residues of the M2-M3 loop control current activation evoked by PAMs such as NS-1738.

Importance of the transmembrane domains for the potentiation by PNU-120596. Construction of the reverse 5HT3-α7 chimera (Chim VIII) yielded 5-HT evoked currents in only a few oocytes injected with this fusion protein. Cartoon represented above the traces illustrates the schematic structure of a single subunit inserted into the membrane. The gray line corresponds to the extracellular 5-TH3 receptor sequence, whereas the dark line indicates the α7 segments. Despite the low yield of expression, significant and reproducible currents were evoked by brief exposure to 5-HT (continuous lines A and B). Exposure to NS-1738 (dashed lines, left) or PNU-120596 (dashed lines, right) caused no modification of the amplitude or time course of the 5-HT evoked currents (dashed lines). A, responses evoked by low 5-HT concentrations that are most susceptible to reveal a putative allosteric modulation. Small, but significant, currents were recorded when these cells were exposed to ACh. C, ACh-evoked currents measured in the same cell as A and B. Exposure to NS-1738 (dashed line) caused no modification of the subsequent ACh response, whereas a clear increase of the current was observed after exposure to PNU-120596 (dashed line).
Footnotes
-
This work was supported by the Swiss National Science Foundation to DB.
-
ABBREVIATIONS: nAChR, nicotinic acetylcholine receptor; PNU-282987, N-((3R)-1-azabicyclo(2.2.2)oct-3-yl)-4-chlorobenzamide hydrochloride; AR-R 17779, spiro(1-azabicyclo(2.2.2)octane-3,5′-oxazolidin-2′-one); PHA-543613, N-(1-azabicyclo(2.2.2)oct-3-yl)furo(2,3-c)pyridine-5-carboxamide; SSR-180711, 4-bromophenyl-1,4-diazabicyclo(3.2.2) nonane-4-carboxylate, monohydrochloride; A-582941, 2-methyl-5-(6-phenyl-pyridazin-3-yl)octahydro-pyrrolo(3,4-c)pyrrole; ACh, acetylcholine; PAM, positive allosteric modulator; NS-1738, 1-(5-chloro-2-hydroxyphenyl)-3-(2-chloro-5-trifluoromethylphenyl)urea; PNU-120596, 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methylisoxazol-3-yl)urea; IVM, ivermectin; 5-HI, 5-hydroxy-indole; TM, transmembrane domain; Chim, chimera receptor.
- Received October 19, 2007.
- Accepted August 1, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}