


Visual Overview

Abstract
Many spider-venom peptides are known to modulate the activity of the voltage-gated sodium (NaV) subtype 1.7 (NaV1.7) channel, which has emerged as a promising analgesic target. In particular, a class of spider-venom peptides (NaSpTx1) has been found to potently inhibit NaV1.7 (nanomolar IC50), and has been shown to produce analgesic effects in animals. However, one member of this family [µ-TRTX-Hhn2b (Hhn2b)] does not inhibit mammalian NaV channels expressed in dorsal root ganglia at concentrations up to 100 µM. This peptide is classified as a NaSpTx1 member by virtue of its cysteine spacing and sequence conservation over functionally important residues. Here, we have performed detailed structural and functional analyses of Hhn2b, leading us to identify two nonpharmacophore residues that contribute to human NaV1.7 (hNaV1.7) inhibition by nonoverlapping mechanisms. These findings allowed us to produce a double mutant of Hhn2b that shows nanomolar inhibition of hNaV1.7. Traditional structure/function analysis did not provide sufficient resolution to identify the mechanism underlying the observed gain of function. However, by solving the high-resolution structure of both the wild-type and mutant peptides using advanced multidimensional NMR experiments, we were able to uncover a previously unknown network of interactions that stabilize the pharmacophore region of this class of venom peptides. We further monitored the lipid binding properties of the peptides and identified that one of the key amino acid substitutions also selectively modulates the binding of the peptide to anionic lipids. These results will further aid the development of peptide-based analgesics for the treatment of chronic pain.
Introduction
Voltage-gated sodium (NaV) channels are transmembrane proteins that are essential for the initiation and propagation of action potentials in excitable cells. NaV channels are classified into nine different subtypes denoted as NaV1.1–NaV1.9 (Catterall et al., 2005). In recent years, NaV subtype 1.7 (NaV1.7) has emerged as a validated pain target based on several human genetic studies. Gain-of-function mutations in the SCN9A gene encoding the pore-forming α-subunit of NaV1.7 have been shown to cause painful inherited neuropathies (Yang et al., 2004; Estacion et al., 2008; Cheng et al., 2011; Dabby et al., 2011; Theile et al., 2011), whereas loss-of-function mutations in SCN9A result in a congenital indifference to all forms of pain (Cox et al., 2006). This suggests that subtype-selective inhibitors of NaV1.7 are likely to be useful analgesics for treating a broad range of pain conditions (England and Rawson, 2010).
Many venomous animals have evolved venom peptides that modulate the activity of NaV channels. In particular, spider venoms are rich in NaV channel modulators, with one-third of all known ion channel venom peptides from spider venoms acting on NaV channels (Klint et al., 2012). Based on their primary structure and cysteine scaffold, these venom peptides can be classified into 12 distinct families of spider-venom peptides that modulate the activity of NaV channels (NaSpTxs) (Klint et al., 2012).
Members of the NaSpTx1 family of spider-venom peptides have been shown to potently inhibit NaV1.7 (nanomolar IC50), with the best-studied peptide from this family being Huwentoxin IV (µ-TRTX-Hh2a, hereinafter referred to as Hh2a) (Peng et al., 2002). However, one member of this family, Hainantoxin I (µ-TRTX-Hhn2b, hereinafter referred to as Hhn2b), isolated from Haemadipsa hainana shows no inhibition of tetrodotoxin-sensitive or tetrodotoxin-resistant NaV channels expressed in dorsal root ganglia at concentrations up to 100 µM (Li et al., 2003). Hhn2b was subsequently further tested against individual NaV channel subtypes expressed in Xenopus laevis oocytes. Hhn2b displayed weak inhibition of NaV1.2 (IC50 68 µM) and more potent (but still weak) inhibition of the insect neuronal NaV channel, para/tipE (IC50 4.3 µM), with no effect on NaV1.4 and NaV1.5 up to 100 µM (Li et al., 2003). Thus, this venom peptide consistently lacks potency against mammalian NaV channels.
Understanding the molecular basis of the peptide-channel interaction is essential for the rational development of venom-based analgesics. Thus, to explore the unexpected paucity in channel inhibition by the NaSpTx1 peptide Hhn2b, we have here recombinantly produced isotope labeled (13C/15N) Hhn2b in a heterologous expression system, allowing us to determine its high-resolution structure by multidimensional NMR spectroscopy. Consistent with the classification of Hhn2b as a NaSpTx1 member and its sequence identity over amino acid residues that are known to be key determinants for NaV inhibition (Fig. 1) (Klint et al., 2012; Liu et al., 2012; Minassian et al., 2013; Revell et al., 2013), we also found the overall fold of Hhn2b to be in close agreement with other members of the NaSpTx1 family. Based on the three-dimensional (3D) structure and sequence alignment of NaSpTx1 venom peptides, we identified several possible amino acids in Hhn2b that may explain the conspicuous lack of activity of the peptide. Using our recombinant expression system we were able to introduce mutations at the amino acids putatively identified as functionally important. Two of the mutations (G7W and N24S) resulted in a gain of NaV1.7 activity, and remarkably, simultaneous mutation of both these residues resulted in a double mutant that showed nanomolar inhibition of NaV1.7. The high-resolution solution structure of this double mutant was also determined using high-resolution NMR spectroscopy and detailed analyses showed that the mutations cause subtle displacements of the side chains of key pharmacophore residues, thus indirectly perturbing the pharmacophore of the peptide. Further NMR studies of the binding of the peptides to liposomes showed that Hhn2b does not bind strongly to lipids while the G7W/N24S-Hhn2b binds selectively to anionic lipid bilayers. We further show that the G7W mutation alone accounts for this gain of lipid binding. Together, these results provide novel insight into the structural determinants of NaV channel inhibition by NaSpTx1 venom peptides and provide new avenues for fine-tuning of activity and lipid binding through subtle sequence variation.

‘NaV1.7 inhibitor’ is defined as a venom-peptide with IC50 < 500 nM at NaV1.7 [where superscripts a, b and c refer to Li et al. (2003), Revell et al. (2013) and Bosmans et al. (2006) respectively. The effect of the latter on the NaV1.7 subtype is based on unpublished in-house data. Superscripts d and e refer to Liu et al. (2012) and Klint et al. (2015) respectively]. Sequence numbering is with respect to Hhn2b. Residues known to be important for inhibition are shaded in dark purple and the fully conserved serine residue (asparagine in Hhn2b) is shaded in pink. The dashes denote gaps in the sequence alignment.
Materials and Methods
Expression Vector and Mutagenesis
A synthetic gene facilitating periplasmic expression (GeneArt, Invitrogen, Regensburg, Germany) encoding Hhn2b was used as previously described (Klint et al., 2013). Site-directed mutagenesis was performed by the quick-change polymerase chain reaction method using standard protocols with Platinum Pfx DNA Polymerase (Invitrogen, Carlsbad, CA) (Qi and Scholthof, 2008). Mutant cDNA constructs were sequenced to verify the desired mutation and the absence of unwanted spontaneous mutations (AGRF, Brisbane, Australia) before transformation into Escherichia coli [BL21(λDE3)].
Production of Recombinant Wild-Type (WT) Hhn2b and Mutants
Peptides were produced following an optimized protocol described previously (Klint et al., 2013). Peptide purity was assessed by analytical high-performance liquid chromatography and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry, and all peptides used showed >95% purity.
Two-Electrode Voltage-Clamp Recording from X. Laevis Oocytes.
Human NaV subtypes (coinjected with human β1 subunit) constructs were expressed in X. laevis oocytes. Two-electrode voltage-clamp recording techniques (OC-725C, Warner Instruments, Hamden, CT) in a 150-μl recording chamber were used to measure Na+ currents 1–4 days after cRNA injection and incubation at 17°C in ND96 that contained 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, and 1.8 CaCl2, 50 μg/ml gentamycin, pH 7.6 (in mM). Data were filtered at 4 kHz and digitized at 20 kHz using pClamp software (Axon Instruments/Molecular Devices, Sunnyvale, CA). Microelectrode resistances were 0.1–1 MΩ when filled with 3 M KCl. The external recording solution contained 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, and 1.8 CaCl2, pH 7.6 with NaOH (in mM). All experiments were performed at room temperature (∼22°C) and peptide samples were diluted in recording solution with 0.1% bovine serum albumin. Leak and background conductances, identified by blocking the channel with tetrodotoxin, were subtracted for all NaV currents. All chemicals were obtained from Sigma-Aldrich (St. Louis, MO). Off-line data analysis was performed using Clampfit (Axon Instruments/Molecular Devices, Sunnyvale, CA), Origin 7.5 (Originlab, Northampton, MA), and Microsoft Solver (Microsoft Excel).
Automated Patch Clamp.
Human NaV1.7 EZcells division-arrested cells were obtained from ChanTest (Cleveland, OH, Lot #1543). Na+ currents were measured using the automated electrophysiology platform QPatch 16× (Sophion, Ballerup, Denmark) in single-hole configuration. Extracellular Ringers solution of 1 CaCl2, 1 MgCl2, 5 HEPES, 3 KCl, 140 NaCl, 0.1 CdCl2, and 20 TEA-Cl, pH 7.3 (NaOH), and 320 mOsm (in mM), and intracellular Ringers solution of 140 CsF, 1/5 EGTA/CsOH, 10 HEPES, 10 NaCl. pH 7.3 (NaOH), and 320 mOsm (in mM) were used. Cells were positioned on the chip using a positioning pressure of −100 mbar. Gigaseals were obtained by applying negative pressure to the cells between −20 and −130 mbar for 4 minutes at a membrane holding potential of −90 mV. Whole cell configuration was obtained by 1-second suction pulses of increasing intensity from −250 to −500 mbar in 50-mbar increments with 10 seconds between pulses. Whole cell configuration was confirmed by development of a capacitative transient >4 pF.
For measuring NaV1.7 currents, the membrane potential was held at −80 mV then stepped to −120 mV for 100 ms to remove any potential fast inactivated channels, followed by a step to 0 mV for 10 ms to activate NaV1.7. The membrane potential was held at −80 mV between sweeps. Hhn2b and mutants were dissolved in extracellular solution with 0.1% bovine serum albumin (fatty acid free), and after obtaining baseline recordings 5 µl was applied and the effect was assessed after 1-minute peptide incubation with the channel by applying the voltage protocol 25 times at 5-second intervals in order to ensure steady-state inhibition was reached. Data were sampled at 10 kHz and filtered using a 4-pole Bessel filter at 3 kHz. Series resistance was compensated by 75%. Off-line data analysis was performed using Qpatch assay software version 5.0 (Sophion) and Microsoft Excel (for Mac 2011, version 14.3.4). Peak inward current at 0 mV was normalized to cell capacitance (pA/pF) and steady-state current density in response to each sample concentration was normalized to vehicle control (EC + 0.1% bovine serum albumin) and plotted as fractional current. Fraction of inhibition was obtained using Prism 6 (Graphpad Software, San Diego, CA) by column statistics and is given as the mean fraction of inhibition and the S.D. The IC50 values were obtained by nonlinear fit (also by Prism 6) using the following equation: Y=Bottom + (Top − Bottom)/1+10Log(IC50 – x)* HillSlope. The significance of the difference in inhibition of NaV1.7 currents by different peptides was calculated by an unpaired t test using Prism 6 (Graphpad Software). This comparison was only done between peptides that showed inhibition at 1 μM. Peptides showing no inhibition at 1 μM were not included in the t test because much higher (>1 μM) peptide concentrations would be required to produce measurable inhibition.
NMR Structure Determination of WT and G7W/N24S Hhn2b
The structure of WT and G7W/N24S mutants of Hhn2b were determined using heteronuclear NMR. Both samples contained 300 μl of 13C/15N-labeled peptides at 300 μM in 20 mM sodium acetate solution with 5% D2O at pH 5. All spectra were acquired at 298 K on a Bruker Avance II+ 900 MHz spectrometer equipped with a cryogenically cooled triple resonance probe (Bruker, Billerica, MA). Resonance assignments were obtained using two-dimensional (2D) 1H-15N- heteronuclear single quantum coherence (HSQC), 2D 1H-13C-HSQC, 3D HNCACB, 3D CBCA(CO)NH, 3D HNCO, 3D HBHA(CO)NH, and four-dimensional HCC(CO)NH-TOCSY, in which the 3D and four-dimensional spectra were acquired using nonuniform sampling and transformed using maximum entropy reconstruction with the Rowland NMR Toolkit (http://rnmrtk.uchc.edu/rnmrtk/RNMRTK.html) as described previously (Mobli et al., 2007). Interproton distance restraints were obtained from 3D 13C-aliphatic, 13C-aromatic, and 15N nuclear Overhauser effect spectroscopy (NOESY)-HSQC spectra acquired using a mixing time of 200 ms. Spectra were analyzed using CcpNmr Analysis 2.4.1 (Vranken et al., 2005). Dihedral angle restraints were derived from the Talos+ protein backbone dihedral angle prediction program (Shen et al., 2009), with the restraints range for structure calculations set to twice the estimated S.D. The NOESY spectra were manually peak picked and the torsion angle dynamics package CYANA3 (Güntert, 2004) was then used to automatically assign the peak lists, extract distance restraints, and calculate an ensemble of structures. Two hundred structures were calculated for each peptide and the top 30 were selected based on the final CYANA target function value and ranked based on their stereochemical quality as judged by MolProbity (Chen et al., 2010).
Preparation of 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phospho-(1′-rac-Glycerol) (POPG) and 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphocholine (POPC) Liposomes
Stock solution of POPG (25 mg/ml) or POPC (25 mg/ml) (Avanti, Alabaster, AL) in chloroform was dried down under nitrogen gas and left under vacuum overnight. The dried film of lipid was rehydrated in 50 mM sodium acetate, pH 5, 50 mM NaCl, and 5% D2O by shaking the mixture at room temperature for 1 hour. The hydration yielded large multilamellar vesicles. To downsize the large multilamellar vesicles to homogenous, large unilamellar lipid vesicles, the suspension was frozen and thawed repeatedly 20 times, followed by extruding it 21 times through a filter with a pore size of 100 nM (Avanti).
NMR Titrations of Liposomes to Hhn2b Peptides
Prepared liposome solution or buffer solution was added directly to the lyophilized peptide. Each sample contained 50 μM of peptide in 50 mM sodium acetate, 50 mM NaCl, and 5% D2O, pH 5, either with no liposomes, 1.5 mM POPG, or 1.5 mM of POPC. The sample volume was 160 μl, which was contained and run in a 3-mm NMR tube (Norelle, Marion, NC). Spectra were recorded at 298 K on a Bruker Avance II+ 900 MHz spectrometer equipped with a cryogenically cooled triple resonance probe. Spectra were processed and analyzed using Topspin (Bruker).
Results
Effect of Recombinant Hhn2b on Human NaV (hNaV) Channel Subtypes Expressed in Oocytes.
Peptide expression, purity, and identity were assessed by SDS-PAGE, high-performance liquid chromatography, and mass spectrometry (Supplemental Fig. 1). The function of the recombinantly expressed Hhn2b was initially assessed at a concentration of 1 µM against hNaV1.2, hNaV1.5, and hNaV1.7 expressed in X. laevis oocytes. As can be seen from Supplemental Fig. 2, 1 µM Hhn2b inhibited hNaV1.2 by 25% and had no effect on hNaV1.7, hNaV1.3, and hNaV1.5. This is in agreement with the previously reported potency and selectivity of the native peptide (Li et al., 2003) and indicates that the recombinant peptide has the same structural and functional characteristics as the native toxin.
Effect of WT and Mutant Hhn2b on hNaV1.7.
The remaining activity assays were measured by automated patch-clamp electrophysiology against hNaV1.7 stably expressed in Chinese hamster ovary cells. The results are summarized in Fig. 2 and Table 1. In agreement with the initial assessment, WT Hhn2b displays no inhibition of hNaV1.7 (10 µM highest concentration tested).

Effect of WT Hhn2b and mutants on hNaV1.7 in Chinese hamster ovary cells. (A) Representative sodium currents before (black) and after addition of 1 µM (red) or 10 µM (green) of Hhn2b or mutants. Currents were evoked by depolarization from −120 to 0 mV. (B) Fraction of inhibition of sodium currents caused by 1 µM of Hhn2b and mutants. Fraction of inhibition was as follows: G7W 0.74 ± 0.01 N24S 0.79 ± 0.08, G7W/N24S 0.28 ± 0.05, G7W/N24A 0.86 ± 0.08, and G7W/N24S/W29F 0.56 ± 0.05. The double mutant (G7W/N24S) showed significantly increased inhibition at 1 µM compared with either of the G7W or N24S mutation alone (P < 0.0001). (C) Inhibition of sodium current as a function of Hhn2b concentration and the two most active mutants. The IC50 values of the most potent mutants were determined to be 440 ± 4 nM for G7W/N24S and 1.0 ± 0.2 µM (n = 3–6 for each data point; error bars represent S.E.M.). All data were recorded using automated patch clamp (QPatch, Sophion).
Tested mutants of Hhn2b with indication of the mutated residue, the IC50 value obtained by automated patch clamp, the hill slope for the fit of the curve, and the number of repeats for each determined curve
Comparison of the sequence and structure of Hhn2b with other members of this toxin family identified four residues (K4/G7/N24/V32) in proximity to the proposed pharmacophore region that were not conserved in Hhn2b. K4 was mutated to a leucine to remove the positive charge, which is not present in other NaV inhibiting members of this family. G7 was mutated to a tryptophan residue, since this position is often occupied by a residue with an aromatic side chain. N24 was mutated to an otherwise absolutely conserved serine residue. Finally, V32, which is a conserved tyrosine residue, was mutated to a tryptophan residue since this substitution had previously been seen to improve the activity of Hh2a (Revell et al., 2013).
The K4L and V32W point mutations did not result in any gain of activity against hNaV1.7 (Fig. 2B). On the other hand, point mutations of G7W and N24S each resulted in a gain of activity against hNaV1.7 with IC50 values of 2.7 ± 0.4 and 4.0 ± 0.5 µM, respectively. Combining these two mutations in a double mutant, G7W/N24S Hhn2b resulted in potent inhibition of hNaV1.7, with an IC50 value of 440 ± 4 nM (Fig. 2C). To test whether the gain in activity of the N24S mutation resulted from the removal of an unfavorable interaction by the asparagine residue with the channel, a G7W/N24A mutant was also generated. This mutant showed an affinity similar to the G7W mutation alone (IC50 value of 8.6 ± 1.7 µM), suggesting that the gain in activity is due to a specific effect of the serine residue.
Finally, we found that it is possible to remove the inhibitory effect of the G7W/N24S Hhn2b double mutant by mutating the essential Trp29 residue to an alanine (no inhibition at 10 µM). This position has previously also been shown to be critical in the activity of Hh2a (Revell et al., 2013). We further found that a substitution of Trp29 to a less bulky aromatic residue was tolerated but reduced potency, with the triple mutant G7W/N24S/W29F Hhn2b resulting in an IC50 value of 1.0 ± 0.2 µM.
Structural Characterization of WT and G7W/N24S Hhn2b.
Uniformly 13C/15N-labeled WT and G7W/N24S mutant of Hhn2b were produced for structural studies by NMR. 1HN, 15N, 13C′, 13Cα, and 13Cβ resonance assignments for both peptides were obtained by standard triple resonance experiments and a four-dimensional HCC(CO)NH-TOCSY experiment that provides side-chain 1H-13C connectivities (Mobli et al., 2010). Complete chemical shift assignments were deposited to the BioMagResBank (http://www.bmrb.wisc.edu/). The accession numbers for the WT and G7W/N24S mutant Hhn2b are 25031 and 25421, respectively.
The automated nuclear Overhauser effect assignment procedure of CYANA 3.0 was used, and the program assigned 96.2% and 95.1% of all NOESY cross peaks of WT and G7W/N24S mutant of Hhn2b, respectively. The final structures were calculated using 446 unique distance restraints for the WT structure and 437 for the G7W/N24S mutant. Atomic coordinates for the final ensembles were deposited in the Protein Data Bank. The Protein Data Bank identification numbers for the WT and G7W/N24S Hhn2b are 2MQF and 2MXO, respectively. Statistics highlighting the high precision and stereochemical quality of the ensembles are listed in Table 2. Both structural ensembles are highly precise with the backbone and heavy-atom root-mean-square deviation of the WT Hhn2b over the structurally ordered region (residue 3-32) being 0.17 ± 0.05 and 0.63 ± 0.13 Å, respectively, and the backbone and heavy-atom root-mean-square deviation of the structural ensemble of G7W/N24S mutant over the same region being 0.14 ± 0.04 and 1.18 ± 0.27 Å, respectively.
NMR and refinement statistics for 20 structures of WT and G7W/N24S mutant of Hhn2b peptide
Sterochemical quality according to MolProbity (http://helix.research.duhs.duke.edu). Clashscore is the number of steric overlaps >0.4 Å per 103 atoms. All statistics are given as mean ± S.D.
As a member of the NaSpTx1 family, Hhn2b is expected to adopt the inhibitor cystine knot motif, with disulfide connectivities between cysteine residues 1–4, 2–5, and 3–6 (by order in the amino acid sequence). The disulfide connectivity of the WT and G7W/N24S Hhn2b was determined from the preliminary structures calculated without disulfide bond restraints. In both peptides, disulfide linkages were formed between Cys3 and Cys18, Cys10 and Cys23, and Cys17 and Cys30, hence confirming that the peptide adopts the inhibitor cystine knot motif. The structure contains an antiparallel beta hairpin loop and a hydrophobic face, comprised of several hydrophobic residues that are highly conserved among venom peptides in this family (Phe6, Tyr21, Trp29, and Val32 in Hhn2b).
The high-resolution structures of the inactive WT Hhn2b and the most potent mutant (G7W/N24S) allowed direct structural comparison of the two peptides. As illustrated in the overlay of the two structures in Fig. 3, the overall structure of Hhn2b remains similar upon the substitution of Gly7 and Asn24. In the absence of 3D structural data the fold of the peptides can also be evaluated based on backbone chemical shifts, which are sensitive to changes in the secondary structure. Typically, Hα shifts or overlay of 15N HSQC spectra are used, both of which also showed that the G7W/N24S mutant retained a similar fold as the WT Hhn2b (see Fig. 4; Supplemental Fig. 3).

Structure of WT and G7W/N24S mutant of Hhn2b. (A) Cartoon representation of the ensemble of 20 WT (left) and G7W/N24S mutant (right) Hhn2b structures. Hydrophobic residues that make up the hydrophobic face are shown in sticks and the sites of mutations are highlighted in orange. (B) Surface of peptides with the hydrophobic residues highlighted in green and the residues that bear a positively charged side chain highlighted in blue. (C) Overlay of the WT (green) and the G7W/N24S mutant (purple) structure. The Protein Data Bank identification numbers for the WT and G7W/N24S Hhn2b are 2MQF and 2MXO, respectively.

Chemical shift difference between WT and G7W/N24S mutant of Hhn2b. The bar charts show the absolute difference of all 1H (top), 13C (middle), and 15N (bottom) chemical shifts between the WT and the G7W/N24S mutant for each residue.
However, the structural differences between the two peptides lie in subtle changes of the side-chain orientation of certain residues. In contrast to the well-defined aromatic cluster at the hydrophobic surface (Phe6, Tyr21, Trp29), the introduced tryptophan residue at position 7 (G7W) in the mutant was found to have no inter-residue nuclear Overhauser effects, hence this side chain does not adopt an ordered orientation and is likely disordered in solution (Fig. 3A). The high flexibility of the Trp7 side chain is partly responsible for the higher heavy atom root-mean-square deviation of the mutant structure compared with the WT.
Overlaying the structures of the WT and the G7W/N24S Hhn2b also revealed a subtle displacement of the Trp29 residue in the mutant (Fig. 3C). While the aromatic ring of the Trp29 in the WT structure is pointing away from Asp27, Trp29 in the G7W/N24S mutant is tilted and is in closer proximity to Asp27. The repositioning of Trp29 is also consistent with the unusual chemical shifts observed for the atoms of Asp27 in the mutant compared with the WT Hhn2b. Aromatic rings are known to exert ring currents that would account for the observed change in the chemical shift. To quantify the significance of the observed chemical shift changes, we measured the chemical shift differences (Δδ) of all nonmutated atoms of the WT and G7W/N24S Hhn2b (Fig. 4). These results show that Asp27 indeed displays the largest chemical shift perturbations. By comparing the 15N-HSQC and the 2D NOESY spectra of the N24S mutant with those from the WT and G7W/N24S Hhn2b we were able to see that the change in the chemical shifts in Asp27 occurs with the introduction of Ser24 and is further enhanced by introduction of Trp7 (Supplemental Figs. 3 and 4), indicating that both residues contribute to this reorientation.
The second cluster of Δδ outliers include the HE1 and CD1 atoms from the side-chain indole ring of Trp29, and smaller yet consistent changes in the chemical shift of the side-chain atoms of Lys31 (Fig. 4). In particular, we note that the largest changes in the chemical shifts of Lys31 are near the charged amino group of the side chain. The charged group would exert a large electric field component that would significantly affect the chemical shift of nearby atoms, consistent with the changes of chemical shift seen for HE1 and CD1 of Trp29. In the structural overlay we indeed see a change in the orientation of the side chain of Lys31, which appears to move toward Ser24 and the indole ring of Trp29. Thus, the subtle change in the structure appears to be consistent with the chemical shift differences observed. We note that the large shift in HE1 of Trp29 is present in the G7W and the G7W/N24S mutants but not in the N24S mutant, suggesting that the rearrangement of Lys31 is not strongly dependent on Ser24, and largely dictated by the introduction of Trp7.
Interactions of Hhn2b and Mutants with Liposomes.
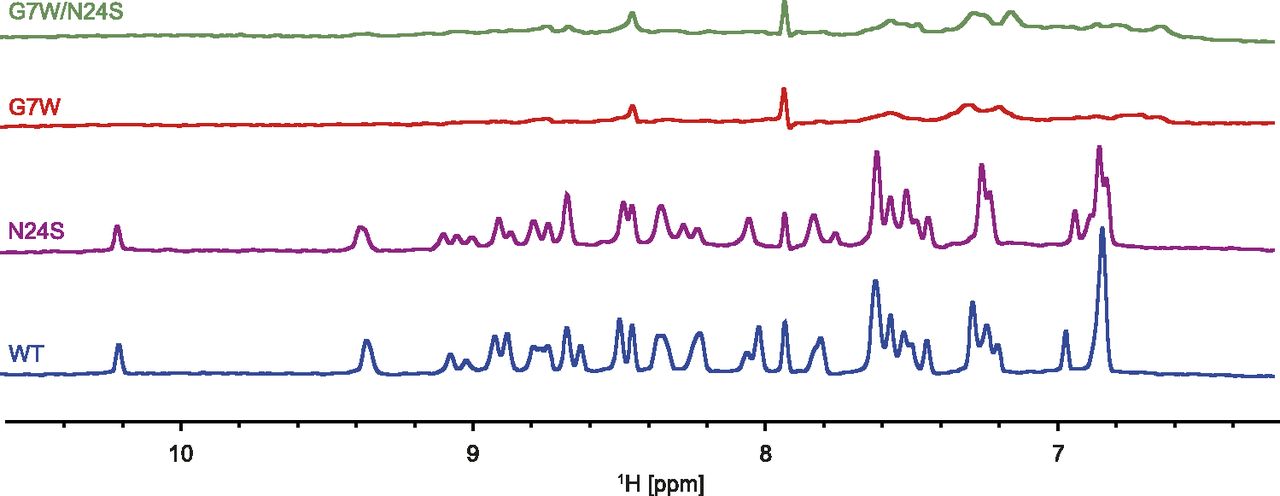
One-dimensional 1H-NMR spectra were acquired for the WT, G7W, N24S, and G7W/N24S mutants of Hhn2b in the presence and absence of anionic POPG or neutral POPC liposomes. In the absence of liposomes, all peptides displayed a well-dispersed 1H spectrum, consistent with the globular fold of the peptides. The NH region (∼6–11 ppm) was monitored, where signals from the buffer and lipids do not interfere with those of the peptide. In the presence of POPC and POPG, the 1H spectra of the WT and N24S mutants show well-resolved and sharp line shapes (Fig. 5; Supplemental Fig. 5). This shows that the molecules retain a fast molecular correlation time, indicating that they do not associate strongly with the much larger liposomes. Similarly, the G7W and G7W/N24S mutants showed no significant change in their 1H spectra in the presence of POPC liposomes, while in contrast the presence of POPG resulted in extensive line broadening (Fig. 5), indicating that the Trp7 containing peptides were bound to the liposomes. These results clearly show that the G7W and G7W/N24S mutants interact with the POPG liposomes with much higher affinity than the WT and N24S mutant.

NH region of one-dimensional 1H-NMR spectra of Hhn2b (WT) and mutants in the presence of an excess amount of POPG liposomes. In the presence of POPG the spectra of WT and N24S mutants remain well resolved, containing well-dispersed sharp lines, comparable to the spectra measured in the absence of liposomes. In contrast to this, almost all signals from the G7W mutant and G7W/N24S double mutant were severely broadened, indicative of binding of the peptide to the liposomes. In all experiments the peptide:POPG ratio was 1:30.
Discussion
Switching on Nav1.7 Activity.
In this study, we have investigated the structural basis of the conspicuous lack of activity exhibited by the spider-venom peptide Hhn2b. This peptide, based on sequence identity and intercysteine spacing, has been classified as part of a family of spider-venom peptides that includes several members known to potently inhibit NaV1.7, including the well-characterized Hh2a venom peptide. Critically, it has been shown that the residues essential for Hh2a activity are Trp30 (Trp29 in Hhn2b) and Lys32 (Lys31 in Hhn2b). Mutation of either of these residues to alanine completely abolishes the activity of Hh2a at NaV1.7. In Hhn2b both of these residues are present, and yet the peptide remains inactive. To understand the structural basis for the apparent loss of NaV activity of Hhn2b we determined its high-resolution structure, which confirms that the overall fold of the peptides is similar to the structure of other members of this family. However, there were a number of amino acid differences, which were found to be in close contact with the important Trp29 and Lys31 residues. Based on this, a series of single, double, and triple mutants were generated to discern the functional importance of these residues. Our results indicated that the most important difference between Hhn2b and other toxins with respect to NaV1.7 activity is the G7W and N24S substitutions in loops 1 and 4, respectively. Individual mutation of either of these residues led to a weakly active peptide, and a double mutant resulted in a nanomolar inhibitor of NaV1.7.
A Structural Basis.
The functional studies of the generated mutants identified the amino acids that account for the lack of Hhn2b activity against NaV1.7. However, this does not explain how the introduced substitutions switch on the observed inhibitory effect. To investigate structural changes that the mutations may have induced that can account for the gain in activity, we also determined the high-resolution NMR structure of G7W/N24S Hhn2b. Comparison of the high-resolution structure of the mutant and the WT peptide showed that the difference in activity cannot be attributed to a change in the overall fold of the peptide. In the absence of further functional or high-resolution structural data, it is tempting to conclude that the residues themselves are interacting with the channel. However, the lack of activity in the triple mutant N24S/G7W/W29A suggests this is unlikely to be the case, and indeed the high-resolution structure of the N24S/G7W mutant shows that the mutations cause several interesting changes in the side-chain orientation of key pharmacophore residues (see Trp29 and Lys31 in Fig. 3).
Comparison of the two structures shows that introduction of Ser24 and Trp7 both contribute to the observed structural changes. The reorientation of the side chain of Trp29 against Ser24 places the indole ring in close proximity to the hydroxyl group of the serine, where it can form favorable electrostatic interactions. Similarly, we find that the disordered Trp7 side chain provides a steric hindrance leading to further packing of Trp29 against the tip of loop 4 (against Asp27). This reorientation further causes a change in the side-chain orientation of Lys31 toward Ser24 and Trp29, where it can form favorable cation-π interactions. The resulting domino effect, which effectively reorients the pharmacophore of the peptide, can be followed both through the nuclear Overhauser effect–based 3D structure (Fig. 3) and the changes in the chemical shifts (Fig. 4).
These data are consistent with the mutations causing a change in the structure of the peptide, albeit in the side chain and not the backbone of the peptide. Our findings highlight that great caution should be exercised in dissecting relative contributions of structure and interaction based on low-resolution structural NMR data such as Hα chemical shift deviations and overlay of 2D 15N-HSQC spectra. We find instead that quantitative chemical shift analysis and high-resolution structural characterization are suitable for structure/function studies in this class of molecules.
Although little is known about how these peptides bind to NaV channels, we note that our data are consistent with the docking model proposed for the binding of Hh2a to the domain II voltage sensor of NaV1.7 (Minassian et al., 2013). In this model the peptide binds to a crevice formed between the four helices of the voltage sensor domain and reveals key interactions between Lys32 (Lys31 in Hhn2b) of the peptide and Glu811 on the channel, as well as a hydrophobic pocket formed in the channel crevice by Met750 and His754, which in the complex accommodates Trp30 (Trp29 in Hhn2b) and Phe6 (Trp7 in Hhn2b mutant) of the peptide. Conversely, the model proposed does not identify any significant interactions between Ser25 (Asn24 in Hhn2b) and the channel. This is consistent with our proposal that this serine residue is part of a structurally important network of intramolecular interactions.
Modulating Lipid Binding.
The amphipathic nature of many spider toxins that bind to voltage-gated ion channels, has led to studies investigating the role of lipid binding in their function (Milescu et al., 2007). It has been shown that spider toxins that act as gating modifiers can bind to lipid membranes (Milescu et al., 2009), and it has been proposed that, together, the protein and lipid interactions prevent the voltage sensor from making the outward movement needed for channel activation (Lee and MacKinnon, 2004; Milescu et al., 2007, 2009; Jung et al., 2010) Furthermore, it has been demonstrated that Hh2a is able to bind to the closed state of NaV1.7 (Xiao et al., 2008), which agrees well with models proposing that the peptide reaches the voltage sensor while it is still embedded in the membrane, thereby blocking the outward movement of the sensor during channel activation (Henrion et al., 2012). These studies suggest that lipid interactions can be directly involved in the mechanism of channel inhibition by venom peptides. However, it is also known that lipid interactions can serve to increase the local concentration of ligands near membrane-embedded receptors (Sykes et al., 2014), and that this has a significant effect on the measured affinity of such ligands. Therefore, lipid binding appears to be a critical determinant of function that must be considered in structure/function studies of ligands that act on membrane-embedded receptors.
Our results show that introducing a hydrophobic residue in loop 1 increases the affinity of Hhn2b toward POPG but not toward POPC liposomes. We note that although POPG is not present in mammalian cell membranes, the presence of other charged groups in the membrane leaflet, as well as charges on the channel are likely to provide the necessary interactions in the relevant hNaV1.7 environment. Since the mutation involved the introduction of a neutral hydrophobic residue, this suggests that the basic nature of these peptides together with their hydrophobicity contribute to their overall lipid binding. An intriguing consequence of this is that these peptides appear to be marginally lipid binding, such that their association with mammalian lipid membranes (dominated by POPC/1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) is transient in the absence of anionic components in the membrane leaflet. In the presence of anionic channel/lipid components their association with the lipid becomes significantly enhanced. In the context of venom evolution, this would appear to be a highly desirable property for an injected venom component because it would allow the peptides to diffuse effectively near the lipid environment—where their target receptors are—without binding so strongly that it would limit their circulation from the site of injection. From a drug design perspective, our findings suggest that lipid binding and selectivity may be modulated through subtle modification of residues in loop 1.
Conclusions
In this study, we have provided novel insight into the molecular determinants of NaV channel inhibition by venom peptides in the NaSpTx1 family of toxins. We have introduced a series of mutations to convert an inactive venom peptide to a nanomolar NaV1.7 inhibitor. Structural studies of both the native and the most potent engineered peptide allowed us to provide a structural basis for the recapitulated activity. The two mutations found to improve activity, N24S and G7W, perturb the side-chain orientation of the critical Trp29 and Lys31 residues. The importance of G7W and N24S in defining the orientation of the critical Trp29 residues was further supported by a triple mutant, G7W/N24S/W29A, which showed complete loss of activity. By performing lipid binding studies we found that the G7W mutation also increased the membrane binding propensity of the peptide. Thus, although both mutations are important for defining the pharmacophore region, the gain in activity by the introduced Trp7 residue may, in addition, be due to an increase in lipophilicity.
Venom peptides in this family have attracted considerable attention as analgesic leads for the treatment of chronic pain, and although our results show that Hhn2b is a potent blocker of NaV1.7 it is not expected to have improved subtype selectivity over other peptides in this family. Future studies will be focused on determining residues that are important for governing subtype selectivity. It is expected that the improved definition of functionally important residues provided here will guide these drug development efforts.
Acknowledgments
The authors are indebted to valuable discussions with Glenn King, Frank Bosmans and Lachlan Rash as well as technical assistance by Carus Lau.
Authorship Contributions
Participated in research design: Klint, Chin, Mobli.
Conducted experiments: Klint, Chin.
Performed data analysis: Klint, Chin, Mobli.
Wrote or contributed to the writing of the manuscript: Klint, Chin, Mobli.
Footnotes
- Received July 13, 2015.
- Accepted September 28, 2015.
↵1 J.K.K. and Y.K.-Y.C. contributed equally to this work.
This research was supported by a National Health and Medical Research Council Project grant [1034958] and an Australia Research Council Future Fellowship [FTl10100925].
↵
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.

Abbreviations
- 3D
- three-dimensional
- 2D
- two-dimensional
- hNaV
- human voltage-gated sodium
- HSQC
- heteronuclear single quantum coherence
- NaV
- voltage-gated sodium
- NaV 1.7
- voltage-gated sodium subtype 1.7
- NOESY
- nuclear Overhauser effect spectroscopy
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- WT
- wild type
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}