


Abstract
The evolution of iron chelators from a range of primordial siderophores and aromatic heterocyclic ligands has lead to the formation of a new generation of potent and efficient iron chelators. For example, various siderophore analogs and synthetic ligands, including ICL670A [4-[3,5-bis-(hydroxyphenyl)-1,2,4-triazol-1-yl]-benzoic acid], 4′-hydroxydesazadesferrithiocin, and Triapine, have been developed from predecessors and illustrate potent iron-mobilizing or antineoplastic activities. This review focuses on the evolution of iron chelators from initial lead compounds through to the development of novel chelating agents, many of which show great potential to be clinically applied in the treatment of iron overload disease and cancer.
I. Introduction
Iron chelation therapy involves the use of ligating drugs that avidly bind iron for the treatment of potentially fatal conditions, namely iron overload disease and cancer (for reviews, see Buss et al., 2003c, 2004a; Tam et al., 2003). By coordinating with intracellular and extracellular iron, these ligands promote the excretion and subsequent depletion of this transition metal in biological systems. These iron-chelating agents consist of a range of bidentate, tridentate, and hexadentate ligands in which two, three, or six atoms, respectively, are able to coordinate with iron, forming octahedral complexes (Liu and Hider, 2002a; Tam et al., 2003). Oxygen and nitrogen donor atoms within ligands are able to bind tightly with iron, and this is illustrated within the porphyrin-ring of heme-containing proteins, where Fe-N bonds anchor the iron center in place.
As iron exists in the environment in an insoluble form, microbes have overcome this accessibility problem by excreting low molecular weight ligands, known as siderophores, to specifically sequester iron in a useable form (Reid et al., 1993; Bergeron et al., 2003d; for review, see Neilands, 1995). Many of these siderophores have become lead compounds in the search for more efficient and orally active iron chelators, as these molecules have evolved through natural selection to specifically chelate iron. Currently, the siderophore, desferrioxamine (DFO1), is clinically used for the treatment of iron overload disease, such as β-thalassemia major (for reviews, see Olivieri and Brittenham, 1997; Wong and Richardson, 2003). In addition, DFO has also shown antiproliferative activity against aggressive tumors in clinical trials, including neuroblastoma (NB) and leukemia (for reviews, see Richardson, 1997, 2002; Lovejoy and Richardson, 2003; Buss et al., 2003c, 2004a). However, this hexadentate chelator has many disadvantages owing to it being expensive and orally inactive and having a short serum half-life (Aouad et al., 2002; Chaston et al., 2004). Indeed, the high hydrophilicity of the ligand results in it being poorly absorbed through the gastrointestinal tract (Franchini et al., 2000). As a consequence, DFO treatment requires long periods of subcutaneous infusion, resulting in swelling and pain at the site of injection in a third of patients (Wong and Richardson, 2003). Hence, more orally effective, economical, specific, and efficient iron chelators have been developed as alternatives to DFO for the treatment of iron overload disease and cancer. The evolution of iron chelators from a range of primordial siderophores and aromatic heterocyclic ligands has lead to the formation of a new generation of potent and efficient iron chelators. This review will discuss the evolution of novel iron chelators from their initial lead compounds with regards to their pharmacological actions and structure-activity relationships for the treatment of iron overload disease and cancer.
II. Redox Activity of Iron
Iron is essential for life as it plays an important role in many cellular processes, including energy generation, oxygen transport, and DNA synthesis (for reviews, see Richardson and Ponka, 1997; Andrews, 1999; Lieu et al., 2001; Richardson, 2002; Hentze et al., 2004). The dependence of life upon this transition metal is due to the fact that iron acts as a cofactor within the active site of key enzymes involved in these critical biochemical pathways. This catalytic ability lies in the unique redox activity of iron, which is able to cycle between two stable configurations, the ferric [Fe(III)] and ferrous [Fe(II)] states, allowing it to act as an electron donor and acceptor.
The biological importance of iron and particularly its pathological consequences are due to its participation in the oxidation-reduction process known as the Haber-Weiss reaction (Fig. 1, eq. 3) (Halliwell and Gutteridge, 1989; Lieu et al., 2001). During the iron-catalyzed Haber-Weiss reaction, hydrogen peroxide (H2O2) reacts to form the hydroxyl radical (OH·) and the hydroxide anion (OH-) (Halliwell and Gutteridge, 1989).
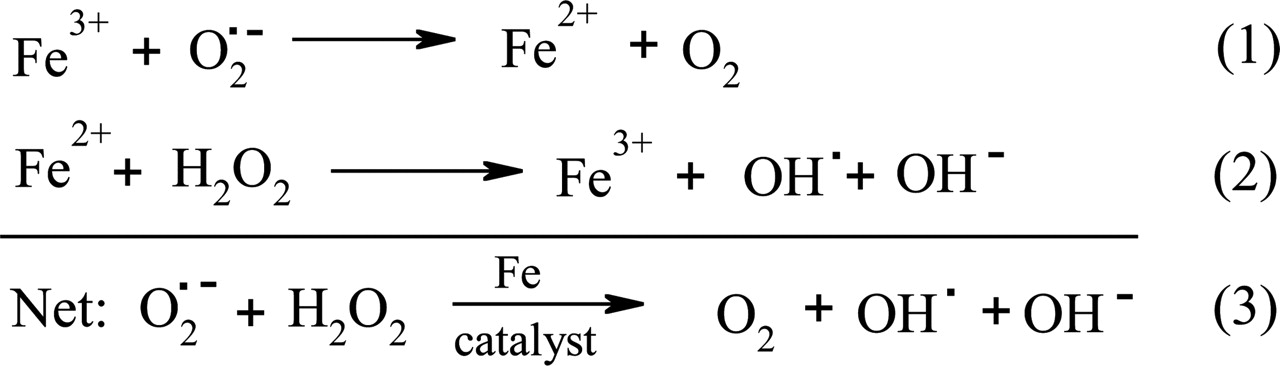
Iron-catalyzed redox reactions of biological importance. Equation 1 shows the reduction of Fe(III) by the superoxide radical ( ). Equation 2 is the Fenton reaction that results in the production of the hydroxyl radical (OH·) and the hydroxide anion (OH-) from hydrogen peroxide (H2O2). Equation 3 shows the iron-catalyzed Haber-Weiss reaction.
). Equation 2 is the Fenton reaction that results in the production of the hydroxyl radical (OH·) and the hydroxide anion (OH-) from hydrogen peroxide (H2O2). Equation 3 shows the iron-catalyzed Haber-Weiss reaction.
Although iron is crucial for life, an excess of iron is toxic. This is due to the deleterious effects of reactive oxygen species (ROS), such as the hydroxyl radical (OH·) which is produced via the Fenton reaction (Fig. 1, eq. 2). The highly reactive OH· radical is able to induce cell death through initiating a series of chemical reactions with many significant biomolecules, resulting in DNA oxidation, mitochondrial damage, and the peroxidation of membrane lipids (Bergeron et al., 2003c; Barnham et al., 2004). In addition, excess free iron can also react with unsaturated lipids to produce alkoxyl and peroxyl radicals (Lieu et al., 2001). These oxidative reactions result in the impairment of cellular functions and lead to damage of cells, tissues, and organs, which is evident in the iron-loading diseases, β-thalassemia and Friedreich's ataxia (FA) (Shinar and Rachmilewitz, 1990; Wong et al., 1999; for reviews, see Alper and Narayanan, 2003; Schrier et al., 2003; Wong and Richardson, 2003). Consequently, iron levels are tightly regulated by specialized proteins, which transport and store iron in a soluble and nontoxic form, thus preventing the deleterious side effects of the Haber-Weiss reaction (Richardson and Ponka, 1997).
III. Iron Metabolism in Normal Cells
A. Iron Absorption from the Gut
The absorption of iron from dietary sources occurs within the small intestine, where enterocytes take up iron in two forms, being inorganic iron and heme (for review, see Conrad and Umbreit, 2002). Inorganic iron is transported across the enterocyte cell membrane via the divalent metal transporter 1 (DMT1), which is also known as the divalent cation transporter, the natural resistance-associated macrophage protein 2 (Nramp2) or solute carrier family 11a member 2 (Slc11a2) (Fleming et al., 1997; Gunshin et al., 1997; Napier et al., 2005). However, as its name suggests, DMT1 can only transport metals in a divalent state, and thus, iron must be in its ferrous form to be absorbed (Hentze et al., 2004). As most dietary nonheme iron is found as ferric complexes, iron must be reduced to the ferrous form prior to absorption. This role is thought to be carried out by the membrane-bound duodenal cytochrome b enzyme, which has been found to possess ferric reductase activity (McKie et al., 2001). However, to date, there has been no direct demonstration that duodenal cytochrome b enzyme activity facilitates iron uptake, and further research is required to confirm the role of this protein in iron transport within enterocytes.
Despite heme being a major source of iron in the human diet, the pathway involved in heme uptake by enterocytes remains unclear. Initial studies suggested that the uptake of heme was believed to occur through passive diffusion, although recent investigations have suggested that a receptor system may be involved (Uc et al., 2004). Considering this, a heme transporter, known as the human feline leukemia virus subgroup C receptor, was recently identified, but its role in heme transportation in the gut remains unknown (Quigley et al., 2004). Heme, once within the enterocyte, is then metabolized by heme oxygenase to liberate iron (Watts et al., 2003).
The precise mechanism of iron transport within enterocytes and its release into the bloodstream is yet to be fully determined. Several proteins have been implicated in the trafficking and release of iron, including hephaestin (Vulpe et al., 1999) and ferroportin 1 (also known as metal transporter 1 or iron-regulated transporter 1; Abboud and Haile, 2000; Donovan et al., 2000; McKie et al., 2000). Hephaestin is a transmembrane ceruloplasmin homolog that is highly expressed in the intestine and was first identified in the sla mouse (Vulpe et al., 1999). A mutation in this protein was found to reduce iron release into circulation, resulting in iron accumulation within enterocytes of the sla mouse (Vulpe et al., 1999). Therefore, it has been suggested that hephaestin may play a role in facilitating iron release in cooperation with the iron transporter, ferroportin 1 (Chen et al., 2004). Mammalian ferroportin 1 has been found to be expressed on the basolateral surface of duodenal enterocytes and is believed to be responsible for iron release from enterocytes into the bloodstream (Donovan et al., 2000).
More recently, a peptide hormone secreted by the liver known as hepcidin has been identified to be critical in iron homeostasis by acting as an iron-regulatory hormone (Nicolas et al., 2001; for review, see Ganz, 2003), regulating enterocyte ferroportin 1 expression (Nemeth et al., 2004). Under conditions of iron overload, hepcidin is highly expressed in the liver (Pigeon et al., 2001). It is thought that hepcidin negatively regulates intestinal iron absorption, maternal-fetal iron transport across the placenta, and iron release from hepatic stores and macrophages (Ganz, 2003). Once in the circulation, hepcidin may bind to ferroportin 1 on cell membranes, leading to its internalization and degradation (Nemeth et al., 2004). This results in reduced iron efflux from enterocytes and completes a homeostatic loop, whereby iron regulates hepcidin secretion that then affects ferroportin-1 expression on the cell surface (Nemeth et al., 2004). In addition to these molecules, the serum protein ceruloplasmin probably is also involved in iron efflux (Lee et al., 1968; Owen, 1973; Richardson, 1999). Patients suffering from aceruloplasminemia were found to have a genetic defect within the ceruloplasmin gene, resulting in iron loading (Harris et al., 1995). Therefore, it is possible that the ferroxidase activity of ceruloplasmin is necessary for the removal of iron from transport elements on the cell surface (Lee et al., 1968; Oshiro et al., 1993; Richardson, 1999).
B. Iron Uptake
In serum, iron is found in a highly soluble form, bound to the iron transport protein, transferrin (Tf) (for reviews, see Morgan, 1981; Richardson and Ponka, 1997). Tf is able to bind two atoms of Fe(III) with high affinity at sites located in the N and C termini of the protein (Morgan, 1981; Richardson and Ponka, 1997). Two molecules of diferric Tf then avidly bind to the transferrin receptor 1 (TfR1) homodimer located on the cell surface, whereas iron-free Tf (apo-Tf) cannot (Fig. 2). A second Tf receptor known as TfR2 has been discovered, although its role in iron metabolism remains unclear (Kawabata et al., 1999, 2000). The Tf-TfR1 complex formed is internalized into an endosome via receptor-mediated endocytosis (Morgan, 1981; Klausner et al., 1983). Within the endosome, iron is released from Tf following the decrease in intravesicular pH mediated by an endosomal membrane-bound proton pump (Morgan, 1981). Recently, Cheng et al. (2004) observed that a conformational change occurs in Tf after binding to TfR1, which is thought to promote iron release where the lobes open, possibly allowing for the access of other proteins to iron (Cheng et al., 2004; Richardson, 2004). Such a direct transfer of iron from Tf to other proteins including DMT1 would prevent iron precipitation within the endosome. Although Fe(III) binds to Tf, it is released in the Fe(II) state, but it is not known whether Fe(III) is reduced before or after its release from Tf. It is speculated that perhaps a membrane-bound ferrireductase is able to associate with the Tf-TfR1 complex, subsequently reducing iron to its Fe(II) state (Cheng et al., 2004; Richardson, 2004). DMT1 then transports this reduced iron across the endosomal membrane and into the cytosol. Another structural change is believed to occur in Tf after the release of its iron, thus resulting in a decreased affinity for TfR1 (Cheng et al., 2004; Richardson, 2004). This freed apo-Tf is released into the plasma via exocytosis, whereby TfR1 is returned to the cell surface (Fig. 2) (Morgan, 1981; Klausner et al., 1983).
Although TfR1-mediated endocytosis remains the major pathway of iron uptake, cells can also internalize iron from small molecular weight complexes such as ferric citrate through a membrane-based iron transport system (Sturrock et al., 1990; Kaplan et al., 1991; Richardson and Baker, 1991; Richardson and Ponka, 1995). However, under physiological conditions, most iron is bound to Tf, and the presence of low molecular weight iron is only appreciable in patients with iron overload disease (Hershko et al., 1978; Grootveld et al., 1989; Hider, 2002a). Under these conditions, iron uptake from low molecular weight iron complexes may be significant and could contribute to the pathological iron-loading observed (Gutteridge et al., 1985; Halliwell and Gutteridge, 1986).
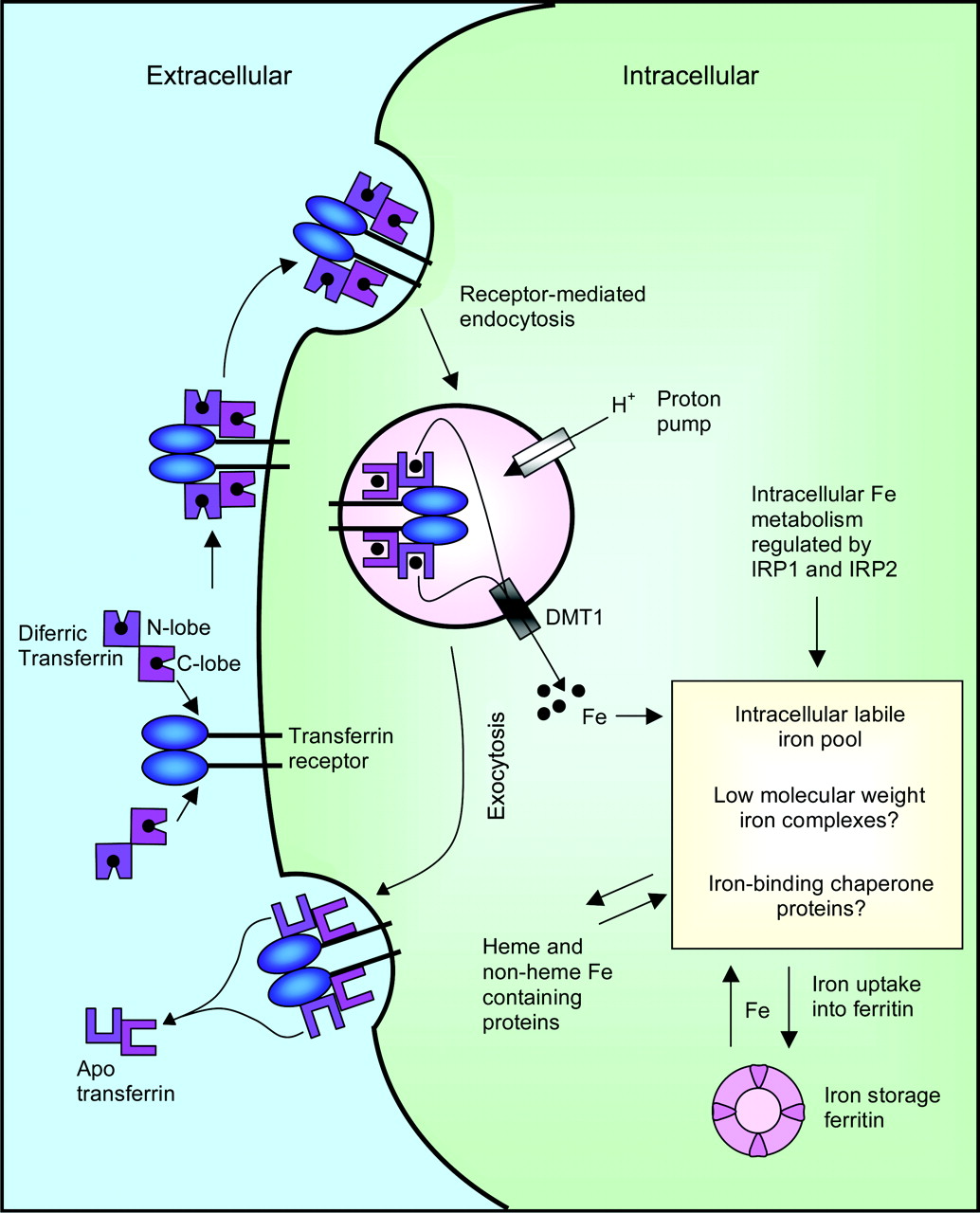
Schematic diagram illustrating the mechanisms involved in iron uptake. Diferric Tf undergoes endocytosis after binding to TfR1. Iron is released from Tf mediated by a decrease in pH and is exported out of the endosome by DMT1, where it enters the LIP. Iron in the LIP can subsequently be incorporated into ferritin for iron storage or into iron-containing proteins.
C. The Labile Iron Pool and Iron Storage
Upon release of iron from the endosome, this transition metal then enters the labile iron pool (LIP), a poorly characterized compartment believed to comprise small molecular weight iron complexes or high molecular weight intermediates. Although the identity of LIP substituents remains elusive, it has been suggested that iron in this pool is bound to amino acids, nucleotides, and sugars (Jacobs, 1977). However, little low molecular weight iron is present within cells, suggesting that such a pool must exist in very low concentrations or may not be present (Vyoral et al., 1992; Richardson et al., 1996). In fact, several models of intracellular iron transport have been proposed that do not involve the presence of low molecular weight species of iron (Zhang et al., 2005).
Iron chelators are believed to bind iron from the LIP, and, hence, it is also known as the chelatable iron pool (Konijn et al., 1999). Iron is distributed from the LIP to specific compartments within the cell, implying that a defined iron-trafficking system exists, used for iron storage or for the inclusion of iron into the active sites of proteins (Richardson, 2002; Buss et al., 2003c). For example, iron is targeted to the mitochondrion in erythroid cells for heme synthesis (Richardson et al., 1996; Ponka, 1997; Zhang et al., 2005).
Iron in the LIP can be targeted to the iron storage protein, ferritin, which is composed of two subunits, heavy and light, forming a protein able to accommodate >4500 iron atoms (Harrison and Arosio, 1996; Buss et al., 2003c). The synthesis of this protein is up-regulated under conditions of iron abundance, whereas the expression of ferritin is reduced when iron availability is low, contributing to overall iron homeostasis (Lieu et al., 2001). Storage of iron within ferritin is critical for the prevention of ROS formation, protecting the cell from oxidative damage.
D. Iron Homeostasis
As iron is required for a variety of cellular processes, a balance between iron uptake, usage, and storage must be maintained. This is achieved by regulating the expression of proteins at the post-transcriptional level via the intracellular iron concentration (for reviews, see Hentze and Kühn, 1996; Hentze et al., 2004). Two mRNA-binding molecules, iron-regulatory proteins 1 and 2 (IRP1 and IRP2), regulate the expression of crucial proteins involved in iron homeostasis. This is attained by the binding of the IRPs to hairpin-loop structures known as iron-responsive elements (IREs) located in the 5′- or 3′-untranslated regions of their mRNAs (Hentze and Kühn, 1996; Hentze et al., 2004). The binding of IRP1 and IRP2 to the IRE is controlled by intracellular iron levels. This iron-mediated regulatory feedback mechanism allows for cells to achieve and maintain a desired intracellular iron level (Hentze and Kühn, 1996; Hentze et al., 2004).
1. Iron-Regulatory Proteins 1 and 2.
Sequence analysis of IRP1 has shown that this protein has high homology to mitochondrial aconitase, being the cytosolic equivalent (Hentze and Kühn, 1996; Hentze et al., 2004). Under high intracellular iron levels, IRP1 contains a [4Fe-4S] cluster, which results in the loss of IRE-binding ability, imparting aconitase activity (Hentze and Kühn, 1996; Hentze et al., 2004). In contrast, IRP1 of iron-depleted cells does not contain this [4Fe-4S] cluster and hence is able to bind to IREs.
IRP2 has binding affinity to IREs similar to that of IRP1, although this protein does not have a [4Fe-4S] cluster. This protein is rapidly degraded in iron-depleted cells via the proteosome (Hentze et al., 2004).
2. Iron-Regulatory Protein Binding: Control of Iron Homeostasis.
As mentioned previously, IRPs are able to bind to IREs located at the 3′ or 5′ end of mRNA, either increasing mRNA stability or inhibiting translation and consequently regulate protein expression (Hentze and Kühn, 1996; Richardson and Ponka, 1997; Hentze et al., 2004). Under conditions of iron deficiency, IRPs are able to bind to IREs located at the 3′ end of mRNA-encoding iron-uptake proteins, protecting the molecule from exonuclease activity and hence improving mRNA stability (Hentze and Kühn, 1996; Richardson and Ponka, 1997; Hentze et al., 2004). This increases the expression of TfR1 and other proteins involved in iron uptake, thus elevating intracellular iron levels. In iron-replete cells, IRPs bind to IRE within the 5′ untranslated region of ferritin mRNA, sterically hindering translation, which allows the cell to use the iron that is present (Hentze and Kühn, 1996; Hentze et al., 2004).
On the other hand, when iron is abundant, IRPs cannot bind to IREs located at the 3′ end of mRNA of iron-uptake proteins, allowing for mRNA degradation and subsequently decreases in intracellular iron levels (Hentze and Kühn, 1996; Hentze et al., 2004). Simultaneously, under high iron levels, IRP can no longer bind to the 5′ end of ferritin mRNA, increasing ferritin expression and levels of iron in storage.
IV. Iron Overload Disease
Although a vast network of proteins are involved in the metabolism of iron, no efficient mechanism of iron excretion exists, and, hence, problems can arise when errors occur within this metabolic pathway. Excess iron, which cannot be bound by ferritin, is able to participate in the Haber-Weiss reaction and in turn induces oxidative stress. This production of free radicals can result in severe damage to tissue and organs and can be fatal if left untreated (Bergeron et al., 2003a). Iron chelation therapy represents an avenue of treatment for these conditions, preventing oxidative effects by removing catalytically active iron (for review, see Chaston and Richardson, 2003a).
A. β-Thalassemia
Two α- and β-globin chains combine to form the essential heme-containing protein, hemoglobin, that is vital for oxygen transport (Nick et al., 2003). Mutations affecting the β gene result in a lack of effective hemoglobin synthesis and leads to β-thalassemia (for reviews, see Cao, 1995; Olivieri and Brittenham, 1997; Wong and Richardson, 2003). Approximately 200 point mutations present within patients are responsible for the reduction in or lack of β-chain synthesis (Wong and Richardson, 2003). The most prevalent point mutation leads to an alteration in the splice junction, creating aberrant mRNA. Others occur within the promoter region, resulting in a decrease in RNA polymerase binding, and within exons, leading to shifts in the mRNA reading frame which creates a premature stop codon (Wong and Richardson, 2003). The subsequent imbalance between α- and β-chain production creates unstable α-chain aggregates that precipitate, affecting erythrocyte membrane plasticity. This causes ineffective erythropoiesis and leads to anemia (Wong and Richardson, 2003).
The first step in treatment of β-thalassemia involves repeated blood transfusions to reverse the anemic state, and this, in combination with iron absorption from the gastrointestinal tract, leads to iron overload (Nick et al., 2003). Potentially fatal tissue damage and fibrosis are seen in the iron-overloaded heart and liver, which is typical of this condition, due to oxidative reactions initiated by the redox activity of iron (Olivieri and Brittenham, 1997; Schrier et al., 2003). Iron chelation therapy offers a secondary route of treatment to relieve the iron burden of these cells.
B. Friedreich's Ataxia
FA is an inherited condition characterized by neurodegeneration and cardiomyopathy (for reviews, see Pandolfo, 2002, 2003; Napier et al., 2005). In this condition, iron overload affects the central nervous system, which results in a gradual loss of motor skills leading to paralysis and death (Puccio and Koenig, 2000; Lieu et al., 2001). This form of iron overload disease is due to the decreased expression of frataxin, a small, mitochondrial protein believed to play a role in mitochondrial iron homeostasis (Puccio et al., 2001; for reviews, see Pandolfo, 2002; Alper and Narayanan, 2003). Frataxin is encoded within a gene known as FRDA, located on chromosome 9q13 (Lodi et al., 2002). In the majority of FA patients, a GAA triplet expansion occurs within the first intron of the FRDA gene, resulting in a reduction of mature frataxin mRNA which in turn decreases frataxin levels (Campuzano et al., 1996; Lodi et al., 2002; Alper and Narayanan, 2003).
The mitochondrion is of great importance to the cell as it plays a pivotal role in energy production through oxidative phosphorylation. This organelle is also responsible for heme and [Fe-S] cluster biosynthesis, and hence, any mitochondrial damage has a large impact on the function of a cell (Cooper and Schapira, 2003). Although the exact function of frataxin has not been determined, it is thought to take part in mitochondrial iron metabolism and [Fe-S] cluster synthesis (Puccio et al., 2001; Becker et al., 2002; Muhlenhoff et al., 2002; for reviews, see Pandolfo, 2002; Alper and Narayanan, 2003; Napier et al., 2005). This correlates with iron accumulation seen in the liver, heart, and spleen of FA patients in a pattern consistent with mitochondria (Sachez-Casis et al., 1976; Lamarche et al., 1993), whereas a decrease in respiratory chain activities of the [Fe-S] cluster-containing complexes I, II, and III is also observed (Rotig et al., 1997; Puccio et al., 2001; for reviews, see Becker and Richardson, 2001; Lodi et al., 2002; Cooper and Schapira, 2003). The lack of productive oxidative phosphorylation is thought to lead to a decrease in intracellular ATP levels, resulting in cellular dysfunction and death (Ristow et al., 2000).
Alternatively, the reduction in activity of these respiratory chain complexes may also be, in part, due to their sensitivity to free radical damage induced by excess iron accumulated within the mitochondrion (Cooper and Schapira, 2003). Excess iron can generate free radicals within the mitochondria, creating oxidative damage that can be at least partially inhibited by antioxidants (Rustin et al., 1999; for reviews, see Alper and Narayanan, 2003; Richardson, 2003). Iron-mediated oxidation leads to the loss of mitochondrial DNA, greatly impairing the function of this organelle (Rotig et al., 1997; Lodi et al., 2002). This free radical damage is believed to be a secondary event after the disruption of iron homeostasis within the mitochondria. Therefore, iron chelators that are able to penetrate the mitochondria and facilitate iron removal provide a potential route of treatment for this iron overload condition (Richardson et al., 2001; Richardson, 2003).
V. Targeting Iron in Cancer Therapy: Iron Metabolism in Neoplastic Cells
The iron chelation field also extends to the treatment of cancer by targeting the enzyme involved in the rate-limiting step of DNA synthesis, ribonucleotide reductase (RR) (Kicic et al., 2001a; for reviews, see Richardson, 1997; Buss et al., 2003c, 2004a; Dayani et al., 2004; Richardson, 2005). This enzyme mediates the conversion of all four ribonucleotides to their deoxyribonucleotide counterparts, providing the precursors necessary for DNA synthesis (Thelander and Reichard, 1979; Kolberg et al., 2004). RR comprises two subunits, R1 and R2, of which R1 is responsible for the binding of ribonucleotides and allosteric factors (Shao et al., 2004). Iron is essential for the catalytic activity of RR and stabilizes the tyrosyl radical located within the R2 subunit (Thelander et al., 1983), making iron an obvious target for chemotherapeutic agents (Thelander and Gräslund, 1983).
Cancer cells have a higher requirement for iron than normal cells as they rapidly proliferate. Hence, iron metabolism is altered within these cells. This is reflected by the fact that tumor cells have higher numbers of Tf receptors on their cell surface, mediating a high rate of iron uptake (Richardson and Baker, 1990, 1992; Trinder et al., 1996; for reviews, see Kwok and Richardson, 2002; Le and Richardson, 2002). Production of the iron storage protein, ferritin, is also increased in tumor tissue (Vaughn et al., 1987). In addition, RR is up-regulated in cancerous cells, facilitating the production of deoxyribonucleotides for DNA replication and subsequent cell division (Elford et al., 1970). Therefore, depleting iron from rapidly dividing cancer cells through the implementation of iron chelators deprives these cells of the DNA precursors necessary for replication.
A. Antiproliferative Activity and Lipophilicity
Chelators with high lipophilicity theoretically possess advantages over less lipid-soluble ligands as these can easily enter cells and subsequently deplete iron from intracellular pools necessary for iron incorporation into the R2 subunit. Alternatively, these ligands can interact directly with the hydrophobic iron center of RR (Hodges et al., 2004). Previous evidence has demonstrated an increase in antiproliferative activity with the elevation in chelator hydrophobicity (Johnson et al., 1982; Baker et al., 1985; Richardson et al., 1995; Hodges et al., 2004). This trend is attributed to the ability of lipophilic ligands to pass through membranes and thus gain access to intracellular iron pools critical for cellular proliferation (Baker et al., 1985; Richardson et al., 1995).
B. Antiproliferative Activity and Redox Activity
In addition, chelators with the highest antiproliferative activity not only chelate iron, decreasing the available iron in the LIP, but may also enhance its redox activity (Fig. 3). Examples of these ligand types include thiosemicarbazones such as Triapine (Chaston et al., 2003; Yuan et al., 2004) and some aroylhydrazones such as those of the 2-pyridylketone isonicotinoyl hydrazone (PKIH) class (Chaston et al., 2004). Ligands that are able to bind both Fe(II) and Fe(III) have the potential to redox cycle (Hider, 2002b; Liu and Hider, 2002a; Bernhardt et al., 2003). For instance, chelators containing “soft” donor atoms, such as nitrogen, possess lower redox potentials and as such the iron core can be enzymatically reduced under biological conditions (Fig. 3) (Liu and Hider, 2002a). The resulting Fe(II) can catalyze the generation of damaging oxygen radicals, leading to oxidative damage and subsequent cell death. In contrast, “hard” coordinating groups such as the hydroxamate oxygens of the hexadentate siderophore, DFO (Fig. 4), have a large affinity for Fe(III) over Fe(II) (Hider, 2002b). With such hexadentate ligands, the coordination sphere is fully occupied, preventing direct access of hydrogen peroxide or oxygen with the complexed iron center. On the other hand, bidentate and tridentate complexes are kinetically labile and are able to form partially dissociated complexes, producing 2:1 and 1:1 complexes, respectively. These partial complexes render the cation surface exposed and as a consequence can facilitate the production of damaging radical species. Such properties enable iron chelators to promote the redox activity of this metal ion, initiating ROS production and consequently leading to cell death. This provides an alternative form of treatment in the fight against cancer in which tumor resistance to standard chemotherapeutics remains an ongoing problem.

Iron complex redox cycling. The Fe(III) complex can be reduced enzymatically to produce the corresponding Fe(II) complex. The resulting Fe(II) can subsequently generate reactive oxygen species such as the superoxide radical.
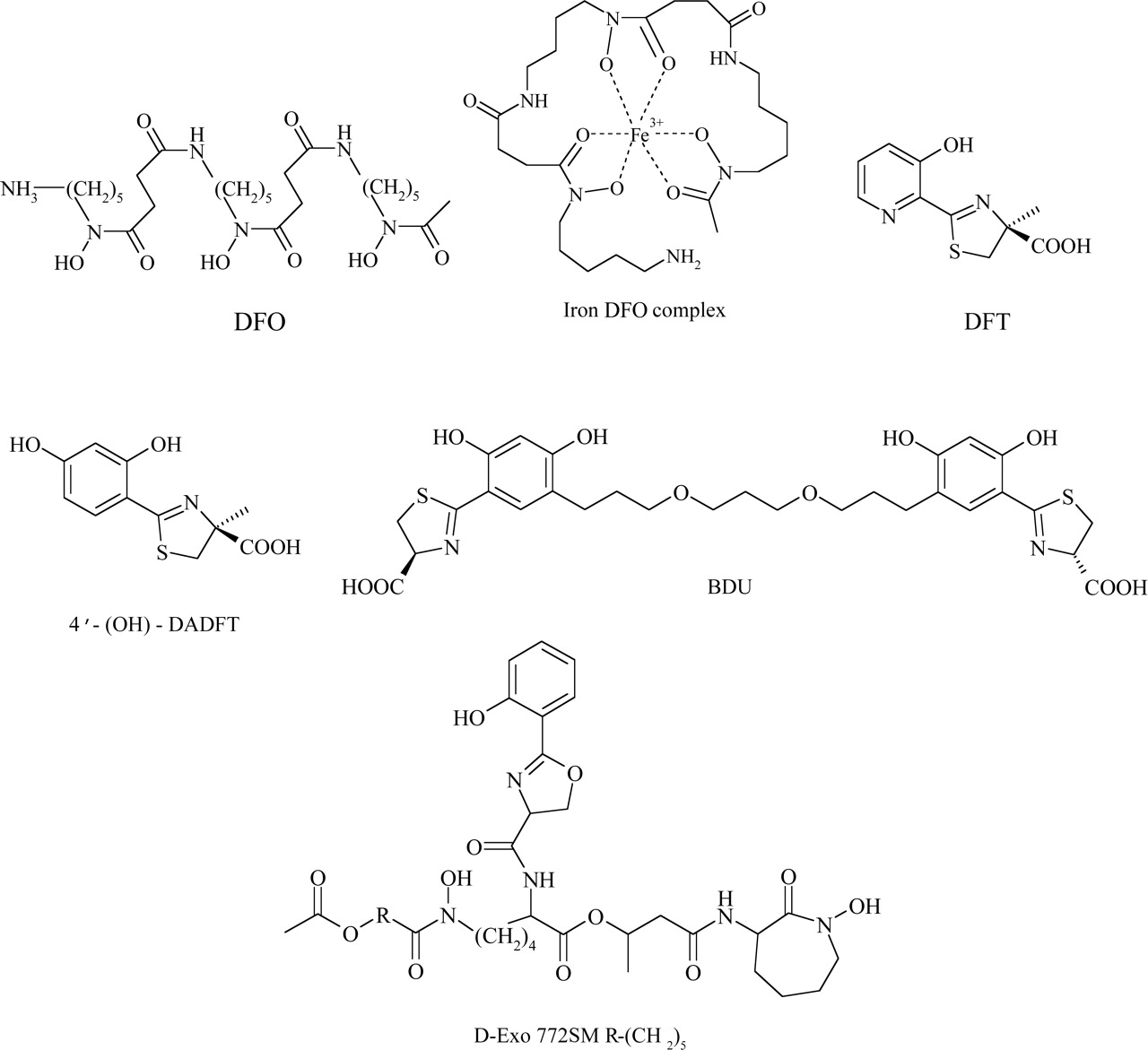
Chemical structures of significant siderophore ligands and their derivatives, including DFO and its iron complex, DFT, 4′-(OH)-DADFT, BDU, and D-Exo 772SM.
VI. The Evolution of Iron Chelators
In the quest to develop effective iron chelators, three main considerations must be taken into account, including the route of administration, the iron chelation efficiency, and the toxicity profile of the ligand. Ideally a drug should be orally active, economical and highly specific, while exhibiting no adverse side effects. Although a large variety of chelators designed to be orally active have been created, further research is being conducted to increase the efficiency and reduce the toxicity associated with these drugs. These parent molecules act as a starting point for the development of a new generation of potent iron chelators. Below we describe the evolution of some of the most effective lead compounds through to the formation of a novel range of efficient iron chelators.
A. Siderophores
Siderophores are natural product iron chelators, secreted by microbes in response to the insoluble nature of iron in the environment (for review, see Neilands, 1995). These low molecular weight ligands were selectively evolved to sequester this transition metal in a soluble form, allowing microorganisms to take up these complexes via specific transport systems. These molecules were created specifically for chelating iron; thus, siderophores represent a range of selective lead compounds. Some of these have been examined and structurally altered in the search for novel, specific, and nontoxic chelators for clinical use.
1. Desferrioxamine.
DFO (Fig. 4), a hexadentate siderophore isolated from Streptomyces pilosus, is the current clinical chelator of choice for the treatment of iron overload diseases such as β-thalassemia (Aouad et al., 2002; Brittenham, 2003). This ligand has validated iron chelation therapy as an effective form of treatment by showing an improvement in the survival and well-being of iron-overloaded patients (Olivieri and Brittenham, 1997; Hershko et al., 2003). The high affinity of this siderophore for Fe(III) renders iron bound in the resulting 1:1 complex metabolically inactive, preventing the production of ROS. Consequently, DFO is able to decrease the oxidative stress in iron-overloaded cells, alleviating the symptoms associated with iron overload disease (Olivieri and Brittenham, 1997). However, the highly hydrophilic nature of DFO limits the efficiency of this ligand, imparting poor absorption from the gastrointestinal tract and a short plasma half-life of 12 min due to rapid drug metabolism (Aouad et al., 2002). As a result, DFO must be administered via subcutaneous infusion for extensive periods to achieve a negative iron balance, ranging from 8 to 12 h, five to seven times per week at a daily dosage of 20 to 60 mg/kg (Hershko et al., 2003). Apart from the cumbersome administration route and high cost, a third of patients treated with DFO experience pain and swelling at the injection site, cumulatively leading to poor patient compliance (Olivieri and Brittenham, 1997; Wong and Richardson, 2003).
Previous studies and clinical trials have illustrated that aggressive tumors, including NB and leukemia, are sensitive to iron chelation therapy with DFO (for review, see Buss et al., 2003c). After a 72-h exposure to 60 μM DFO, a >80% reduction in viability of NB cell lines was evident, demonstrating some cytotoxic effect (Blatt and Stitely, 1987). In addition, DFO has been shown to inhibit DNA synthesis in a NB cell line after a 4-h incubation (Blatt et al., 1988). The cytotoxic effects of DFO were prevented upon the addition of iron or iron-saturated DFO (Blatt and Stitely, 1987), suggesting that the antiproliferative action of this siderophore can be attributed to iron depletion and subsequent inhibition of RR.
Apart from in vitro assessment of antitumor activity, an Italian clinical study of nine NB patients given a single course of DFO, administered by an 8-h continuous infusion at 150 mg/kg for 5 days, resulted in a response in seven of nine patients (Donfrancesco et al., 1990). A decrease in bone marrow infiltration was observed as well as a 48% decrease of tumor size in one patient. At this dosage level, no significant side effects were evident (Donfrancesco et al., 1990).
Although DFO has shown pronounced antiproliferative activity during in vitro studies and clinical trials, the high hydrophilicity of this ligand limits its membrane permeability and efficiency. There have been numerous attempts to improve upon the efficacy of DFO. These include the generation of a high molecular weight form of the chelator coupled to hydroxyethyl starch. In a clinical trial, high plasma concentrations of hydroxyethyl starch-DFO were obtained (3 mM) after a 4-h intravenous infusion (Dragsten et al., 2000), and this treatment was well tolerated and led to substantial urinary iron excretion (Dragsten et al., 2000). However, the major problem with DFO was not overcome, that is, the need for long infusions. More lipophilic DFO analogs have been prepared by reacting the terminal primary amino group with fatty and aromatic acid chlorides or anhydrides (Ihnat et al., 2000). However, to date, none of these have been reported to be more effective than DFO administered via the subcutaneous route.
More recently, combinatorial approaches have been used to generate libraries of structural DFO analogs including nonamide analogs, C-terminal modified analogs, reverse-amide analogs, and hybrid analogs (Poreddy et al., 2004). However, none of these compounds have been evaluated in animal models. To the present day, the evolution of DFO analogs has not resulted in chelators with advantages over the original siderophore. Hence, other orally active alternatives have been sought in the quest to develop effective and specific iron chelators, which are discussed in detail below.
2. Desferrithiocin.
Desferrithiocin (DFT) [2-(3-hydroxypyridin-2-yl)-4-methyl-4,5-dihyrothiazole-4-carboxylic acid; Fig. 4] is a tridentate siderophore from the bacterium Streptomyces antibioticus DSM 1865 (for reviews, see Bergeron et al., 2002; Brittenham, 2003; Nick et al., 2003). This ligand binds iron through its phenolic oxygen, carboxylate oxygen, and thiazole nitrogen donor atoms (Brittenham, 2003). Desferrithiocin has a formation constant of 4 × 10-29 M with Fe(III) and was among the first orally active siderophores to be discovered (for review, see Tam et al., 2003). DFT was found to be orally active in a bile duct-cannulated rodent study (Bergeron et al., 1991). In an iron-overloaded Cebus apella primate model, when orally administered at 150 μmol/kg, DFT was 3 times more active than subcutaneously administered DFO (Bergeron et al., 1993).
Although DFT showed much promise as a chelating agent for the treatment of iron overload disease, a major drawback was that it exhibited severe nephrotoxicity (Bergeron et al., 1993). This is believed to be due to the cytotoxic effects of the desferrithiocin-iron complex (Baker et al., 1992a). However, the high oral activity of DFT made this chemical backbone a promising scaffold warranting further structural investigations, and as such, this siderophore has acted as a lead compound in the pursuit of novel orally active iron chelators. Structure-activity relationship (SAR) studies have been performed to develop less toxic derivatives of this pharmacophore, and these investigations are described below.
a. A structure-activity relationship examination of the desferrithiocin scaffold.
Due to the nephrotoxicity of DFT, studies have been carried out to identify which structural components confer this adverse side effect. These studies lead to the synthesis of (S)-desmethyldesferrithiocin (DMDFT; Fig. 5), an analog discovered to retain iron clearing activity in the bile duct-cannulated rat model (Bergeron et al., 1993). In contrast, a decrease in iron chelation efficacy of DMDFT was observed in the Cebus apella primate model compared with DFT (Bergeron et al., 1993). Although the efficiency of this analog was below that of DFT, this ligand was found to exhibit no toxic side effects when orally administered on a daily basis to rodents in a 30-day trial (Bergeron et al., 1999b). Interestingly, this dramatic shift in toxicity was simply due to the removal of the thiazoline methyl group. However, the fact that this ligand still retained the pyridyl moiety reduced its application as a lead compound, as the pyridine ring does not lend itself to electrophilic aromatic substitution. Hence, other DFT analogs, including (S)-desazadesferrithiocin (DADFT; Fig. 5) and (S)-desazadesmethyldesferrithiocin (DADMDFT; Fig. 5), were also synthesized, containing a carbon in place of the pyridyl nitrogen (Bergeron et al., 1999a,b). Unfortunately, both DADFT and DADMDFT were found to elicit severe gastrointestinal toxicity during rodent trials (Bergeron et al., 1993, 1999b). Despite their adverse side effects, DADFT and DADMDFT have acted as critical intermediate structures during the SAR examination of this series of iron chelators.
Subsequent structural investigations into the distances between the donor groups of DADMDFT revealed that this scaffold could not be altered without the loss of iron-binding ability (Bergeron et al., 1999b). The addition of a methylene group between the two rings of the scaffold or either one or two methylene groups between the carboxyl and thiazoline ring resulted in poor orally active analogs (Fig. 5). Even when administered subcutaneously, the efficiency was not observed to increase (Bergeron et al., 1999b). From these results it can be hypothesized that the poor activity of these analogs was not due to bioavailability but rather that these modifications altered the optimum spacing needed for chelation, reducing their iron-binding capabilities.
Further structural studies examining the effect of thiazoline ring alterations on iron clearing activity were also conducted on DMDFT and DADMDFT (Fig. 5). Various thiazoline modifications, including oxidation to a thiazole, reduction to a thiazolidine, expansion by a methylene group, and substitution of the thiazoline sulfur with oxygen, nitrogen, or a methylene group resulted in a loss of activity (Bergeron et al., 1994, 1999b). In all cases, these alterations lead to a drastic reduction in iron chelation efficiency (Bergeron et al., 1994, 1999b). Upon modification of the thiazoline ring, it would be expected that the iron-binding properties of these analogs would be altered, but the complete lack of activity was difficult to explain. From these results, it can be concluded that the thiazoline ring must remain intact for iron clearing activity.
Finally, changing the C-4 stereochemistry (Fig. 5) showed an interesting effect on iron clearing efficiency in the primate model. By retaining the same stereochemistry as DFT, the (S)-enantiomers of DMDFT and DADMDFT were more effective than their corresponding (R)-enantiomers in promoting iron clearance (Bergeron et al., 1993, 1999a,b).
b. A structure-activity relationship examination of chemical parameters.
In the hope of increasing residence time and thus efficiency, the lipophilicities of the (R)- and (S)-enantiomers of DMDFT and DADMDFT were increased by incorporating an additional aromatic ring (Fig. 6). However, overall, the addition of lipophilic features did not increase iron clearing efficiency, with these analogs showing less activity during the primate model than the parent compounds, DMDFT and DADMDFT (Bergeron et al., 1996, 1999b).

Flow diagram illustrating the SAR examination of DFT. The alteration of the DFT scaffold has lead to the formation of three crucial intermediate structures: DMDFT, DADFT, and DADMDFT. Subsequent structural modifications have established that the addition of methylene groups between chelating centers and alteration of the thiazoline ring reduce iron-clearing efficiency. Investigations into C-4 stereochemistry have revealed that (S)-enantiomers generally have higher iron-clearing efficiency.
Another chemical parameter investigated involved altering the electronic effects within the aromatic ring of the parent compound, DADMDFT, by the addition of electron-withdrawing groups (Fig. 6). For example, a hydroxyl or methoxyl group was added to the 3-position of the aromatic ring to confer an inductive electron-withdrawing effect on the thiazoline nitrogen. Also a carboxylic acid group at the 4-position was used to electronically confer the same effect. Interestingly, these electron-withdrawing analogs were found to have diminished iron clearing efficiency when orally administered in the rodent model (Bergeron et al., 1999b). The methoxy analog was found to have the highest efficiency of these latter analogs, albeit lower then that of the parent compound, DADMDFT (Bergeron et al., 1999b). It seemed that the electron-withdrawing effect induced on the thiazoline nitrogen reduced iron clearing efficiency.

Flow diagram showing the evolution of DFT through to the promising iron chelator, 4′-(OH)-DADFT. A decrease in iron-clearing efficiency was observed after an electron-withdrawing group or a further aromatic ring was added. However, an increase in iron-clearing ability was found to occur with the addition of electron-donating groups, leading to the discovery of the nontoxic iron chelator, 4′-(OH)-DADFT.
Alternatively, electron-donating groups were also used to alter the redox activity of the aromatic ring. In this case, a hydroxyl group placed at the 4-position was able to induce a donating effect, electronically, to the thiazoline nitrogen. This had a profound effect on both the efficiency and toxicity of the compound. The 4′-hydroxylated analog of DADMDFT (4-hydroxydesazadesferrithiocin [4-(OH)-DADMDFT]; Fig. 6) was found to induce iron clearance using both the rodent and primate models (Bergeron et al., 1999b). Although this was not as active as DADMDFT in the primate model, toxicity trials over 10- and 30-day treatment periods illustrated that the gastrointestinal toxicity of the parent compound was eliminated (Bergeron et al., 1999b). All histopathologies seemed to be normal compared with the control (Bergeron et al., 1999b). In addition, the 4′-hydroxylated analog of the 5,5-dimethyl thiazoline DADMDFT derivative (Fig. 6) was found to have good iron chelation efficacy in the primate model (Bergeron et al., 2003c). The addition of a hydroxyl group at the 4-position of DADFT [4′-(OH)-DADFT; Fig. 6] led to an extremely effective iron chelator in the primate model (Bergeron et al., 1999a). Due to the remarkable level of iron clearance, 4′-(OH)-DADFT was further analyzed for its toxic effects. During a 10-day trial, rats treated with this chelator demonstrated no severe side effects, with the compound being well tolerated and almost all histological assessments were normal (Bergeron et al., 1999a). This analog represents one of the most active and nontoxic iron chelators of this series.
In relation to the electron-donating substituents, it has been speculated that the formation of a quinone structure (Fig. 7) may occur on complexation (Bergeron et al., 1999b). In this process, the electrons are redistributed around the aromatic ring, donating a lone pair of electrons onto the thiazoline nitrogen. As a result, a negative charge is located on the nitrogen, enhancing donation to Fe(III) (Bergeron et al., 1999b). Although the effect of the 4′-hydroxy group on the stability of the complex is yet to be determined, the proposed intermediate structure certainly begins to explain the decrease in activity observed with the electron-withdrawing analogs, which remove electron density from the thiazoline nitrogen.
In conclusion, the systematic SAR studies performed by Bergeron and colleagues have led to the discovery of 4′-(OH)-DADFT. This highly orally effective iron chelator is able to promote levels of iron excretion in the primate model at almost 3 times that of subcutaneously administered DFO (Bergeron et al., 1999a). Preclinical animal studies have illustrated no toxic effects at levels to be used clinically, and as such, 4′-(OH)-DADFT is ready for human phase I evaluation.
c. Hexadentate desferrithiocin analog.
Generally, it is known that hexacoordinate ligands form more stable complexes than those of lower denticity (Liu and Hider, 2002a). Due to their additional stability, the majority of natural siderophores are hexadentate. By fully occupying the coordination sphere of iron, hexadentate chelators can prevent the interaction of iron with surrounding molecules, avoiding the formation of ROS through Fenton chemistry (Liu and Hider, 2002a; Bergeron et al., 2003b; Chaston et al., 2004). Owing to these advantages, Bergeron's group synthesized a hexacoordinate chelator based upon the nontoxic tridentate ligand, 4′-(OH)-DADMDFT, named (S,S)-1,11-bis[5-(4-carboxy-4,5-dihydrothiazol-2-yl)-2,4-dihydroxyphenyl]-4,8-dioxaundecane (BDU; Fig. 4). This design was based on linking two tridentate units by a polyether tether and Job's plot of BDU confirmed the hexadentate nature of this ligand (Bergeron et al., 2003b).

Electron distribution in the 4′-hydroxyl analog of DADMDFT. The electron-donating effects of the 4′-hydroxyl group allow for a negative charge to be placed on the thiazoline nitrogen, enhancing donation to Fe(III).
In vivo studies in the noniron-overloaded, bile duct-cannulated rodent model demonstrated that the hexadentate chelator had little iron chelation efficiency when administered orally (Bergeron et al., 2003b). This was found to be within the experimental error of the tridentate unit, 4′-(OH)-DADMDFT, at equivalent iron-binding doses. However, when administered subcutaneously, the iron clearing efficiency of BDU was substantially increased, being 3 times that of the tridentate unit when administered via the same route (Bergeron et al., 2003b). Unfortunately, trials in the iron-overloaded Cebus apella monkey showed a decrease in iron clearing efficiency of BDU when subcutaneously administered. This was found to be only half as effective as the tridentate unit given subcutaneously at the equivalent iron-binding capacity (Bergeron et al., 2003b). Thus, no great advantage was found by coupling two 4′-(OH)-DADMDFT units together. The lack of activity of the hexadentate ligand suggests that this large chelator may be less membrane permeable than the smaller tridentate unit, thus decreasing intracellular chelator levels.
d. Desferrithiocin as a selective antiproliferative agent.
Although the studies on DFT described above mainly concentrated on the efficacy of the chelator for the treatment of iron overload disease, several studies have assessed the antiproliferative activity of this ligand in vitro (Kicic et al., 2001b, 2002). These investigations showed that DFT was more effective than DFO at inhibiting proliferation (IC50 = 40 μM) although its activity was far less than that of other chelators in current development, for example, those of the di-2-pyridylketone thiosemicarbazone (DpT) class for which IC50 values of 0.03 μM were reported (Yuan et al., 2004) (see section VI.B.5.c.). Importantly, the antiproliferative effect of DFT on normal cells (hepatocytes and fibroblasts) was far less than that found in hepatoma cells and their antitumor activity could be diminished by saturating the ligand with iron (Kicic et al., 2002).
3. Desferri-exochelin.
Desferri-exochelins (D-Exo; Fig. 4) are a group of hexadentate siderophores released by Mycobacterium tuberculosis, which use oxygen donor atoms to avidly bind iron (Pahl et al., 2000). Their balance of lipophilicity and hydrophilicity allows them to readily enter cells and complex iron, rendering their bound iron redox-inactive (Hodges et al., 2004). A previous study demonstrated the ability of these siderophores to directly remove iron from both human Tf and horse ferritin (Gobin and Horwitz, 1996). Upon incubation of D-Exo with 95% iron-saturated Tf, D-Exo was observed to become iron saturated after just 1 min (Gobin and Horwitz, 1996). Similarly, D-Exo was demonstrated to remove iron from horse ferritin, although at a much slower rate than with Tf, requiring 3 days to reach saturation (Gobin and Horwitz, 1996). These results demonstrate the iron scavenging ability of this siderophore and illustrate its potential application to iron overload disease.
Further research has illustrated the potential use of D-Exo 772SM (Fig. 4) as an antiproliferative agent. An in vitro study examining the effect of 20 μM D-Exo 772SM on MCF-7 breast cancer cells illustrated potent antineoplastic activity where all cells were killed after 4 days of treatment (Pahl et al., 2001). No cytotoxicity was evident when normal human mammary epithelial cells were treated at 5 or 20 μM, although a growth arrest was seen (Pahl et al., 2001). These effects were prevented when either cell line was incubated with the iron-loaded D-Exo, indicating that the iron-binding ability of this ligand was responsible for its antiproliferative activity (Pahl et al., 2001). Further studies examining D-Exo 772SM on DNA synthesis demonstrated a 95% to 97% reduction of [3H]thymidine incorporation in both the normal and cancerous breast cell lines after a 6-h incubation (Pahl et al., 2001). A 40-fold higher concentration of DFO was necessary to inhibit [3H]thymidine incorporation to a similar extent (Pahl et al., 2001). These results indicated that the increased antiproliferative effects of D-Exo 722SM resulted from its greater lipophilicity and enhanced membrane permeability compared with DFO, allowing access to critical intracellular iron pools required for DNA replication (Pahl et al., 2001). Recently, the ability of D-Exo 772SM in inhibiting RR was examined by measuring the tyrosyl radical signal using electron spin resonance spectroscopy in cell lysates (Hodges et al., 2004). After treatment with 50 μM D-Exo 772SM at 37°C, the electron spin resonance signal of RR was shown to rapidly decline after 60 min (Hodges et al., 2004). Further work on the antiproliferative activity of this chelator is required, particularly in relation to the high activity of aroylhydrazones and thiosemicarbazones of the PKIH and DpT series, respectively (Becker et al., 2003; Yuan et al., 2004) (see sections VI.B.4.e. and VI.B.5.c., respectively).
B. Synthetic Iron Chelators
In contrast to the naturally occurring siderophores described above, a vast array of chelators have been artificially designed and synthesized in the search for an ideal iron chelator. To attain maximum iron clearance or antiproliferative activity, a large range of chelators have been synthesized, forming a generation of lead compounds with great potential. Below we discuss the development of novel synthetic iron chelators from lead compounds to potent ligands.
1. ICL670A.
ICL670A (Fig. 8) is a tridentate chelator that uses a triazolyl nitrogen and two phenolic oxygens as donor groups. This ligand, representing one of >700 compounds designed through computer modeling, belongs to the bis-hydroxyphenyl-triazole class of iron chelators possessing oral activity intended for the treatment of β-thalassemia (Nick et al., 2003). Of central importance is the ability of these chelators to selectively bind Fe(II) and/or Fe(III) and not other metal ions of biological importance such as Cu(II) and Zn(II). By using hard donor atoms such as oxygen, ICL670A can bind Fe(III) specifically, avoiding disturbances to other significant transition metals (Heinz et al., 1999).
The remarkable iron clearing activity of ICL670A has been illustrated through several in vivo trials, including those performed using rats (Hershko et al., 2001) and primates (Sergejew et al., 2000). When administered orally during a rodent study, ICL670A was able to induce iron clearance 4 to 5 times greater than that of DFO, reaching a plateau between a dose of 100 and 200 mg/kg (Hershko et al., 2001). In addition, using DFO and ICL670A simultaneously resulted in a “shuttle effect,” in which iron exchange was evident between the two chelators (Hershko et al., 2001). This effect may be mediated by the more hydrophobic ICL670A entering cells more readily, binding intracellular iron and then transferring it to intra- or extracellular DFO (Fig. 9).
ICL670A has already completed phase I and II clinical trials in which a dose-dependent increase in iron excretion was observed in iron-loaded patients (Nick et al., 2003; Nisbet-Brown et al., 2003). These trials have confirmed the ability of ICL670A to induce iron clearance while no major adverse side effects were observed (Nisbet-Brown et al., 2003). Recently, phase III clinical trials were commenced, and this ligand illustrates the potential of a synthetic orally active iron chelator to replace the cumbersome DFO in the treatment of iron overload disease.
2. Deferiprone and Hydroxypyridinone Analogs.
Deferiprone (1,2-dimethyl-3-hydroxypyridin-4-one, L1, CP20; Fig. 8), is a member of the bidentate hydroxypyridinone family of iron chelators that uses two vicinal oxygen atoms to selectively chelate tribasic metal cations over dibasic species. This bidentate ligand is the only orally active treatment of β-thalassemia available in Europe and some other countries but not the United States (Kontoghiorghes et al., 2004). This is due to conflicting clinical trials related to the safety and iron clearing efficiency of this ligand, and consequently its use remained controversial for may years (for review, see Richardson, 2001). One study monitoring the safety of long-term deferiprone administration found a worsening of liver fibrosis in thalassemia patients (Olivieri et al., 1998). However, it was suggested that this conclusion was skewed due to the inclusion of patients with hepatitis C, a condition known to induce liver fibrosis (Wong and Richardson, 2003). Other long-term clinical trials of deferiprone administration have found no significant increase in liver fibrosis (Maggio et al., 2002; Wanless et al., 2002), implying that deferiprone treatment did not induce this condition. In addition, one study using a small group of patients reported that deferiprone was more efficient at removing myocardial iron than DFO (Anderson et al., 2002). In contrast, a larger trial found no difference in cardiac iron levels between DFO and deferiprone-treated patients (Maggio et al., 2002).

Chemical structures of significant synthetically derived iron chelators, including ICL670A, deferiprone, CP94, CP502, TREN-(Me-3,2-HOPO), Pr-(Me-3,2-HOPO), tachpyridine, PIH, SIH, 311, 5-hydroxypyridine-2-carboxaldehyde thiosemicarbazone (5-HP), and Triapine.
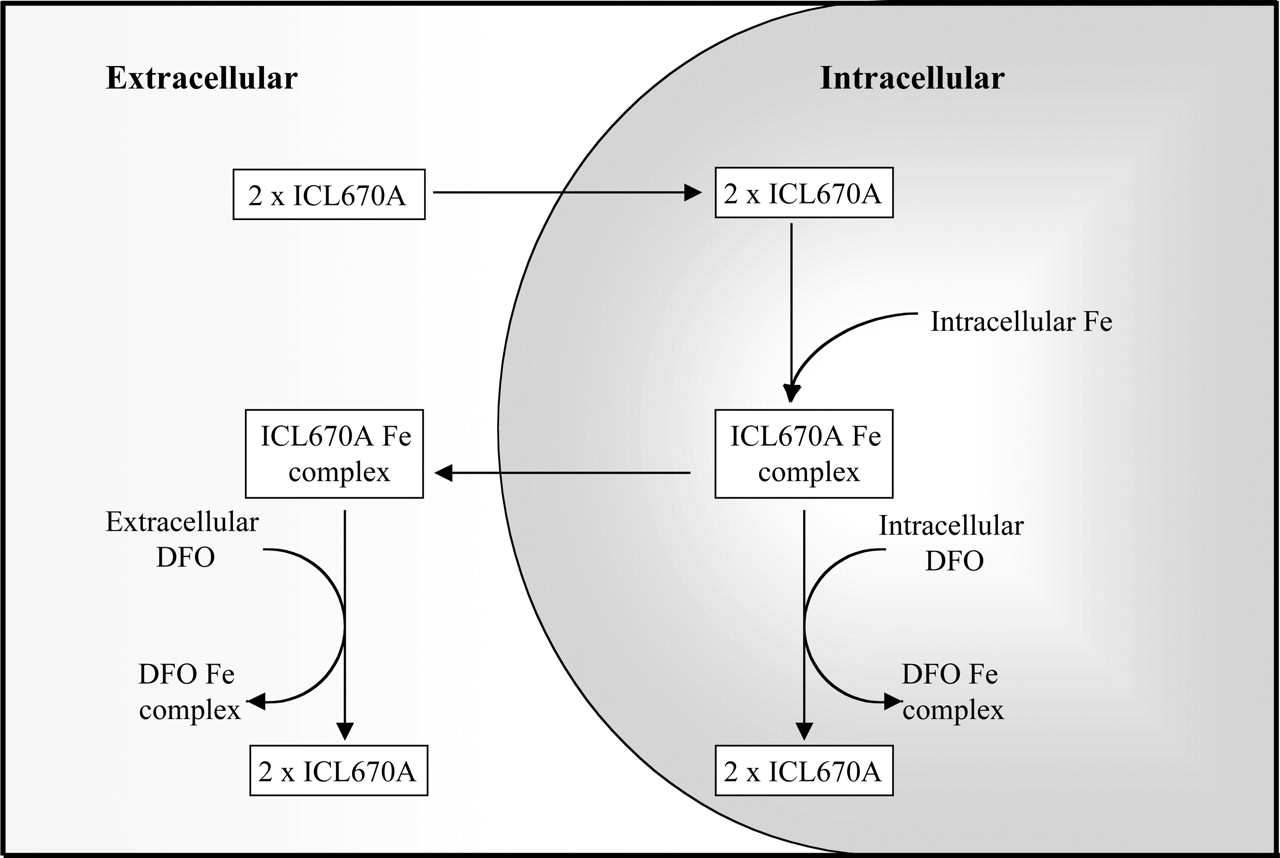
Possible mechanisms involved in the iron-mobilizing shuttle effect between ICL670A and DFO. The more membrane-permeable ICL670A can readily enter cells, thus binding intracellular iron and transferring it to intra- or extracellular DFO.
The mixed reports of deferiprone toxicity have led to investigations of the redox activity and speciation of deferiprone complexes. As deferiprone is bidentate, three molecules are necessary to occupy the coordination sites of iron and such tris complexes, [Fe(deferiprone)3], dominate the speciation profile in the millimolar concentration range at neutral pH (Motekaitis and Martell, 1991). However, a strong dilution effect is evident and the bis complex, [Fe(deferiprone)22H2O]+, accounts for almost 40% of Fe(III) at micromolar concentrations at pH 7 (Motekaitis and Martell, 1991). Clearly, the incomplete coordination of iron by deferiprone in biological systems would enable access of reductants to the iron core and may potentially lead to ROS production. This was illustrated in vitro where an increase in oxygen radical generation was observed at deferiprone/iron ratios <3:1 (Cragg et al., 1998). Again, such findings could question the safety of deferiprone, and to overcome this problem combination therapy with DFO and deferiprone has been assessed (see below).
a. Combination therapy: deferiprone and desferrioxamine.
In an attempt to increase iron excretion and reduce the possible side effects of DFO and deferiprone treatment, combination therapy trials of these chelators have been performed in humans (Balveer et al., 2000; Kattamis et al., 2003). Such treatment has been found to enhance median iron urinary excretion levels in thalassemia patients by almost 2-fold compared with monotherapy with DFO or deferiprone (Kattamis et al., 2003). As seen with ICL670A, deferiprone was thought to participate in the shuttle effect, in which this smaller and more hydrophobic chelator mobilizes intracellular iron stores, subsequently transferring it to extracellular DFO. It has also been suggested that these chelators may target different iron pools (Kattamis et al., 2003). Combination therapy offers a new regimen of treatment to those overload patients who show adverse effects to single-drug therapy. Although these preliminary results show substantial promise, further investigation is needed to determine the dosage levels necessary for optimal iron clearance.
b. Deferiprone metabolism.
To maintain a negative iron balance in overload patients, deferiprone must be administered at a high daily dosage of 75 mg/kg (Balfour and Foster, 1999). The limited efficiency of deferiprone is due mainly to extensive phase II drug metabolism in the liver where the hydroxyl group, essential for chelation and iron clearance, undergoes glucuronidation (Fig. 10A). Analysis of human urinary metabolites demonstrated that more than 85% of deferiprone administered was converted to the nonchelating 3-O-glucuronide (Singh et al., 1992; Kontoghiorghes et al., 2003). The metabolic inactivation of deferiprone has sparked research into the synthesis of other hydroxypyridinones that do not undergo this form of modification in vivo. This has resulted in a wide variety of deferiprone analogs, which are discussed below.
c. CP94.
Of the deferiprone analogs synthesized, CP94 (Fig. 8), has been extensively analyzed. This more hydrophobic analog was found to have higher iron clearing efficiency than that of deferiprone in both rodent and primate models (Porter et al., 1990; Bergeron et al., 1992). During one rodent study, CP94 was found to have an iron clearing efficiency that was almost 6 times greater than that of deferiprone and approximately 2.5 times greater than that of subcutaneous DFO (Bergeron et al., 1992). Similar efficiencies were observed during a primate study, in which CP94 was able to keep these animals in a negative iron balance (Bergeron et al., 1992).

The drug metabolism of deferiprone and its analogs. A, deferiprone metabolism. This ligand participates in extensive phase II metabolism, in which the chelating hydroxyl group undergoes glucuronidation. B, metabolism of CP94. In rodents, CP94 was found to undergo phase II glucuronidation and phase I oxidation. The corresponding phase I oxidative product, CP365, does not undergo further phase II metabolism; thus, this metabolite retains iron-chelating ability. C, hydrolysis of ester prodrugs. The hydrophobic prodrugs CP117 and CP165 are easily absorbed from the gastrointestinal tract and are rapidly hydrolyzed in the hepatocyte by esterase enzymes. This yields the more hydrophilic chelators CP102 and CP41, respectively. These ligands in turn can bind hepatic iron, which is removed via the bile or efflux into the circulatory system to remove extracellular iron via the kidneys. CP41 undergoes an additional phase I metabolic step via alcohol dehydrogenase, forming the even more hydrophilic chelator, CP38.
The underlying reason for the increase in iron clearing efficiency observed during the animal trials described above is attributed to the difference in drug metabolism between deferiprone and CP94 (Fig. 10B). During a rodent study, CP94 was also found to form a 3-O-glucuronide metabolite (Singh et al., 1992). However, such deactivating modifications occurred to 13.8% of the administered dose compared with 44% with deferiprone (Singh et al., 1992). In addition, CP94 was found to undergo oxidative phase I metabolism, a route not evident with deferiprone, resulting in the formation of metabolite CP365 (Fig. 10B) (Singh et al., 1992). The glucuronide conjugate at either hydroxyl position of CP365 was not found; thus, the chelator retained iron chelation efficacy (Singh et al., 1992).
Although the unusual oxidative pathway of CP94 helped to preserve the iron-chelating activity of this analog in rodents, these promising results were not reflected during human studies (Porter et al., 1994). As with deferiprone, the 3-O-glucuronide of CP94 was the major metabolite, accounting for >90% of the administered dose in thalassemia patients (Porter et al., 1994). Unfortunately, these results severely limited the clinical application of CP94 as an orally active iron chelator.
d. Hydroxypyridinone ester prodrugs.
In the search for a new generation of deferiprone analogs with favorable in vivo drug metabolism, a range of hydroxypyridinone ester analogs have been designed especially to target the hepatocellular low molecular weight iron pool (Hider, 1995; Hider et al., 1996; Liu and Hider, 2002a). These highly hydrophobic prodrugs can easily be absorbed through the gastrointestinal tract to then be extracted by the liver, hydrolyzing the ester linkage (Fig. 10C) (Hider, 1995). This results in the formation of a more hydrophilic analog, which is able to scavenge iron in the hepatocyte, and then subsequently be eliminated via the bile, or able to pass into circulation, binding iron from the extracellular iron pool. The resultant increase in hydrophilicity limits the membrane permeability of these analogs, and this was suggested to reduce the potential toxicity associated with these chelators (Liu and Hider, 2002a). Indeed, such pivaloyl ester prodrugs, including CP117 and CP165 (Fig. 10C), and their hydrophilic metabolites, CP102 and CP41 (Fig. 10C), respectively, have been found to increase 55Fe biliary excretion in both normal and iron-loaded rats without any toxic effects (Zanninelli et al., 1997). In fact, the results achieved with these chelators exceeded those obtained with deferiprone, illustrating their superior ability to promote biliary iron excretion from hepatocytes (Zanninelli et al., 1997). Metabolic and pharmacokinetic studies of CP117 in rodents revealed that the hydrophilic metabolite, CP102, was the major product, although a small proportion of phase II glucuronidation was found to occur (Choudhury et al., 1997). In conclusion, the addition of the ester linkage results in the greater efficiency and lack of toxicity of these analogs than the lead compound, deferiprone.
e. Increasing pFe(III) values of hydroxypyridinones.
As previously described, one downfall of bidentate hydroxypyridinones is the kinetic lability associated with their iron complexes. Therefore, to reduce drug toxicity and improve chelation efficiency, a range of hydroxypyridinone analogs that contain high pFe(III) values have been produced. This property allows such chelators to effectively scavenge iron at lower concentrations and also dissociate less easily (Hider et al., 2000; Liu and Hider, 2002b). Consequently, a lower amount of the partially coordinated complexes are formed, reducing the potential for ROS production.
An example of a high pFe(III) chelator is CP502 (Fig. 8), a 2-amido-3-hydroxypyrinidin-4(1H)-one containing a pFe(III) value of 21.7 compared with deferiprone at 19.4 (Hider et al., 2000; Liu et al., 2001). This novel ligand was observed to have enhanced iron chelation activity in a [59Fe]ferritin-labeled rat model, being more effective than deferiprone (Liu et al., 2001). In addition, bile cannulation studies of CP502 metabolism in rats demonstrated only a small amount of phase II glucuronidation, a factor at least partly responsible for this chelator's higher efficacy than that of deferiprone (Liu et al., 2001). The incorporation of the 2-amido moiety has a dramatic effect on the properties of this chelator. The pKa values associated with the chelating groups of CP502 are lower than those of deferiprone and CP94 (Liu et al., 2001). This is due to the stabilizing effects the amido group confers on the ionized species (Fig. 11). The intramolecular hydrogen bonding between the 3-hydroxy group and the 2-amido moiety, combined with the electron-withdrawing effects induced by the 2-amido function are factors that stabilize the negative charge (Liu et al., 2001). These contribute in lowering pKa values of chelating groups and thus substantially increase the pFe(III) value of CP502. Analysis of speciation plots illustrated this effect, indicating that the CP502-iron complex would dissociate less readily than the deferiprone-iron complex, leading to lower concentrations of partial complexes at pH 5.5 to 7.0 (Liu et al., 2001). CP502 shows great potential for the clinical treatment of iron overload disease and a small number of related compounds have been selected for preclinical development by the pharmaceutical company Apotex Inc. (Weston, ON, Canada) (Liu and Hider, 2002a).
f. Hexadentate hydroxypyridinone analogs.
In another attempt to reduce the production of redox active partial complexes and increase complex stability, various hexadentate hydroxypyridinone analogs have been synthesized and analyzed (Streater et al., 1990; Cohen et al., 2000; Xu et al., 2002). One hexadentate ligand known as TREN-(Me-3,2-HOPO) (Fig. 8) was determined to be a more efficient iron chelator than its bidentate parent compound, Pr-(Me-3,2-HOPO) (Fig. 8), during an in vivo rodent study. When orally administered, TREN-(Me-3,2-HOPO) induced biliary iron excretion in iron-loaded rats, this result being markedly higher than that of Pr-(Me-3,2-HOPO (Yokel et al., 2000). These results illustrate the clinical advantage that a hexadentate hydroxypyridinone analog may have over its corresponding bidentate counterpart, as the increased iron chelation efficiency allows for the treatment of iron overload disease at lower and safer ligand concentrations.

The amido function of CP502 stabilizes the ionized species by two mechanisms. Intramolecular hydrogen bonding between the 2-substituent and the 3-hydroxyl function and the electron-withdrawing effect of the amido function stabilize the ionized species, thus lowering the associated pKa value of the chelating groups.
3. Tachpyridine.
Tachpyridine (Fig. 8) is a hexadentate chelator based on a cis,cis-1,3,5-triaminocyclohexane scaffold that uses three pyridyl nitrogens and three secondary amine nitrogens to bind iron (for reviews, see Buss et al., 2003c, 2004a). Under aerobic conditions tachpyridine was found to bind Fe(II) more effectively than Fe(III) (Planalp et al., 2002). In addition, tachpyridine has been shown to bind Fe(III) and subsequently reduce it to the Fe(II) state (Park et al., 1998). Such reduction of the iron complex is believed to be relevant to the biological cytotoxicity of tachpyridine against cancer cells, as iron exists in both the Fe(II) and Fe(III) states (Park et al., 1998). Interestingly, under aerobic conditions, iron-tachpyridine complexes undergo iron-mediated ligand oxidation, forming an inseparable mixture of mono- and diimino Fe(II) complexes (Park et al., 1998) (Fig. 12A). This iron-mediated ligand oxidation was also evident during in vitro cell culture studies (Zhao et al., 2004).
In addition, the reduced metal specificity of this chelator is evident by the fact that various metal tachpyridine complexes have been synthesized or characterized by X-ray crystallography. These include Zn(II), Cd(II), Hg(II) (Park et al., 2000), Cu(II) (Ma et al., 2002) Ga(III), and In(III) (Hilfiker et al., 1997). The ability of tachpyridine to bind other biologically important metal ions has been implicated in its cytotoxic effects (Zhao et al., 2004).
When MBT2 or T24 bladder cancer cells were treated in vitro with tachpyridine, profound cytotoxicity was observed (Torti et al., 1998). This chelator was found to have an IC50 value of 4.6 μM, being more potent than DFO with an IC50 value of 70 μM (Torti et al., 1998). To probe the cytotoxic effect of this ligand, various tachpyridine metal complexes were assessed, including Fe(II), Ca(II), Mn(II), Mg(II), Cu(II), and Zn(II) complexes. The Fe(II) and Zn(II) complexes of tachpyridine were found to result in no cytotoxic effect, which is consistent with the notion that the cytotoxicity of tachpyridine arises from its ability to deplete both Fe(II) and Zn(II) from cells (Torti et al., 1998). This is further confirmed by an in vitro study of human cervical HeLa cells and SUM149 breast cancer cells treated with tachpyridine (Zhao et al., 2004). The concentrations of free ligand and metal complexes in cells and media were analyzed by sensitive reversed-phase high-performance liquid chromatography, establishing the fact that the primary targets of tachpyridine were Zn(II) and Fe(II) (Zhao et al., 2004). In addition, pretreatment of cells with Zn(II) or Fe(II) was sufficient to protect them from the cytotoxic effects of tachpyridine (Zhao et al., 2004). Therefore, these studies suggest that the ability of tachpyridine to bind and remove intracellular Zn(II) and, more importantly, Fe(II) lies at the center of its antiproliferative effects.
Another mechanism that is believed to be responsible for the cytotoxicity of tachpyridine involves the redox cycling of the resultant iron complex (Samuni et al., 2003). After forming the Fe(III) complex, the superoxide radical or tachpyridine itself can induce the reduction of the ferric center, which consequently promotes ROS such as OH· through the Haber-Weiss reaction (Samuni et al., 2003). Thus, the antiproliferative effect of tachpyridine is a culmination of its ability to induce Fe-deprivation and oxidative stress (Samuni et al., 2003).
Tachpyridine has also been found to inhibit ferritin synthesis (Torti et al., 1998) and induce apoptosis via a p53-independent pathway (Abeysinghe et al., 2001). This is important, as >50% of human tumors contain a defective p53 gene, and gives tachpyridine an advantage in antitumor therapy (Abeysinghe et al., 2001).
a. Novel tachpyridine analogs.
A small number of tachpyridine analogs have been synthesized, incorporating methyl or ethyl substituents onto the amine groups, which results in tertiary amine derivatives (Park et al., 1998) (Fig. 12B). These N-alkyl analogs have been shown to be essentially nontoxic in human and mouse bladder cancer cells (Torti et al., 1998). The lack of effect on cell viability is attributed to the inability of these N-alkylated analogs to strongly interact with iron in either the ferric or ferrous states (Torti et al., 1998). This is attributed to steric effects, where the added alkyl groups interfere with the ability of the ligand to chelate iron (Park et al., 1998; Torti et al., 1998).
Another series of tachpyridine analogs have recently been designed to incorporate a linker, which can be conjugated with a monoclonal antibody that could be specific for tumor cell antigens (Chong et al., 2004). Such coupling will probably enhance the selective targeting of iron chelators to tumor cells but also increase the biological half-life and lower potential side effects. The linker design incorporates a maleimide group that is able to undergo Michael addition with nucleophilic thiol groups of the antibody (Chong et al., 2004) (Fig. 12C). Of the different linker analogs created and tested, derivatives containing a side chain added to the pyridyl ring (Fig. 12C) showed the highest in vitro activity against HeLa cells (Chong et al., 2004). These analogs showed a minimal reduction in cytotoxicity compared with the parent compound, tachpyridine. The tert-butoxycarbonyl-protected derivative in particular showed highest antitumor activity (Chong et al., 2004). Analogs containing a linker attached to a secondary amine (Fig. 12C) resulted in a decrease in antiproliferative activity (Chong et al., 2004). Therefore, it can be inferred that the chelating atoms must remain intact and that the linker is best placed on the pyridyl group. The tert-butoxycarbonyl-protected derivative was further converted to contain a maleimide linker, ready for conjugation with thiolated monoclonal antibodies (Chong et al., 2004). Although to date no work has been published on the antiproliferative activity of this derivative bound to antibodies, these analogs show potential as a specific and selective method of antitumor therapy.

A, ferrous tachpyridine complexes undergo iron-mediated ligand oxidation under aerobic conditions, resulting in the formation of mixtures of the mono- and diimino ferrous complexes. B, N-alkyl tertiary amine analogs of tachpyridine. The addition of a methyl or ethyl group decreases the ability to bind iron sterically. C, tachpyridine analogs containing a linker at the secondary amine and pyridyl ring and a bifunctional maleimide-containing derivative ready for antibody conjugation.
4. Aroylhydrazones: Pyridoxal Isonicotinoyl Hydrazone and Analogs.
a. 100 series analogs.
Pyridoxal isonicotinoyl hydrazone (PIH, also known as 111; Fig. 8) is an orally active, tridentate chelator, which binds iron octahedrally through the carbonyl oxygen, imine nitrogen, and phenolic oxygen (Fig. 13A) (for reviews, see Richardson and Ponka, 1998; Buss et al., 2002b). PIH is synthesized from pyridoxal and isonicotinic acid hydrazide via a simple Schiff base condensation (Ponka et al., 1979a,1979b). The tridentate nature of the ligand was demonstrated through the synthesis of its iron complex, which demonstrated a ligand/iron ratio of 2:1 (Ponka et al., 1979a). Since this preliminary investigation, a range of PIH analogs (Fig. 13B) have been synthesized and those that use the pyridoxal as the aldehyde are known as the 100 series (Edward et al., 1988; Ponka et al., 1988; Baker et al., 1992b). A study examining the iron-binding affinity of these ligands revealed that PIH and its analog, pyridoxal benzoyl hydrazone (analog 101; Fig. 13B), have high affinity for Fe(III), comparable to that of DFO (Vitolo et al., 1990). Interestingly, PIH was also found to bind Fe(II) although with lower affinity (Vitolo et al., 1990). The ferrous complex of PIH was found to be sensitive to oxidation, forming ferric complexes in the presence of oxygen (Avramovici-Grisaru et al., 1985; Vitolo et al., 1990). The ability of PIH to bind Fe(II) is believed to be responsible in its role of protecting plasmid pUC-18 DNA against OH·-mediated strand breaks by scavenging Fe(II) and enhancing its autooxidation rate (Hermes-Lima et al., 1998). This results in limiting Fe(II) levels available to catalyze Fenton generated ROS (Hermes-Lima et al., 1998).
Further analysis of the complexation of the divalent metals Ca(II), Mg(II), and Zn(II) with the PIH, 101, 107, and 109 ligands (Fig. 13B) illustrated that the chelation of Zn(II) was greater than that for Ca(II) and Mg(II), which were found to bind weakly (Richardson et al., 1989). However, the affinity for these latter metal ions was much lower than that for Fe(III) (Richardson et al., 1989). This indicates that in biological systems, the chelation of Fe(III) and to a much lesser extent that of Zn(II) are expected by the 100 series over other biologically important metal ions (Richardson et al., 1989).
Initial studies of the ability of PIH to mobilize iron from cells used reticulocytes in which heme synthesis was inhibited in the mitochondrion, leading to marked iron accumulation within this organelle (Ponka et al., 1979a). PIH was identified to be highly effective in permeating the plasma and mitochondrial membranes to chelate the iron (Ponka et al., 1979a). Subsequent trials have also demonstrated the efficiency of the 100 series for mobilizing iron from various cell types (Baker et al., 1985, 1992b; Richardson et al., 1995). In hepatocyte cell culture experiments, 101 and PIH were found to be as effective as DFO in preventing 59Fe uptake from transferrin, decreasing to 41, 45, and 48% of the control, respectively (Baker et al., 1985). In addition, 59Fe mobilization studies from prelabeled primary hepatocyte cultures demonstrated that the more hydrophobic analog, 101, was able to induce cellular iron mobilization to a greater extent than PIH and DFO (Baker et al., 1985). This suggests that the greater lipophilicity of 101 and its corresponding iron complex contributes to increased iron mobilization activity (Baker et al., 1985). The iron chelation activities of the 100 series analogs have also been analyzed using the human SK-N-MC neuroepithelioma cell line (Richardson et al., 1995). The PIH ligand was the most effective of the 100 series in reducing 59Fe uptake, followed by analogs 101, 107, 109, and 115 (Fig. 13B), all of which showed greater activity than DFO (Richardson et al., 1995). Similarly, the majority of the 100 series were more effective than DFO in increasing 59Fe release from prelabeled SK-N-MC cells, with 101, 107, 114, and 115 being more effective than PIH (Richardson et al., 1995).
In vitro experiments have also demonstrated the ability of the 100 series to inhibit the cellular proliferation of human SK-N-MC cells (Richardson and Ponka, 1994; Richardson et al., 1995). An investigation into the effect of these chelators proved that analogs 101, 107, and 109 were more effective than both PIH and DFO in preventing [3H]thymidine uptake (Richardson and Ponka, 1994). In a more comprehensive study, of the 15 members of the 100 series examined, only 3 exhibited higher antiproliferative effects than DFO, and this highlights the suitability of the 100 series for the treatment of iron overload disease (Richardson et al., 1995). Also the ability of the PIH iron complex to support hemoglobin synthesis (Ponka et al., 1982) and cell proliferation (Brock and Stevenson, 1987; Forsbeck et al., 1986) supports the data regarding the low antiproliferative activity of PIH (Buss et al., 2002b). The most lipophilic derivative of the 100 series, namely the ligand 106 (p-tert-butyl-substituted), demonstrated increased antiproliferative effect over other more hydrophilic members, such as the p-hydroxy substituted chelator 102 (Fig. 13B) (Richardson et al., 1995). The antiproliferative effect of these ligands was shown to be caused by their ability to bind iron as the addition of a soluble iron source, namely ferric ammonium citrate, was found to prevent their cytotoxic effects (Richardson et al., 1995). Subsequent analysis of the relationship between the lipophilicity of the PIH analogs and their antitumor effects illustrated a weak linear relationship (Richardson et al., 1995). An earlier study of aroylhydrazones also demonstrated a correlation between lipophilicity and the cytotoxicity of the ligand (Johnson et al., 1982).

A, comparison between the iron-binding modes of PIH and thiosemicarbazones. The tridentate chelator, PIH, uses an imine nitrogen and carbonyl and phenolic oxygens to bind iron. In contrast, the tridentate iron-binding of a general thiosemicarbazone involves imine and pyridyl nitrogens and a sulfur donor atom to coordinate iron. R1 and R2 can be hydrogen atoms or hydrocarbon groups. R3 can be a hydrogen or a pyridyl ring. B, PIH analogs illustrating the 100, 200 and 300 series backbone and their corresponding R groups. C, relationship between the antiproliferative activity of members of the 300 series in SK-N-MC cells plotted against the aromatic substituent hydrophobicity constant of each derivative. A general trend is observed in which the activity increases as the substituents become more hydrophobic.
In vivo studies have also been performed, highlighting the ability of these ligands to induce iron excretion when administered orally in animals (Blaha et al., 1998; Link et al., 2003) and man (Brittenham, 1990). The oral administration of PIH to rodents at doses between 25 and 100 mg/kg led to increased fecal iron excretion of up to 8 times greater than the basal level (Hoy et al., 1979). In addition, when administered intraperitoneally or orally, PIH was shown to increase the biliary excretion of 59Fe from labeled rats as soon as 15 min after administration (Cikrt et al., 1980). In addition, a decrease in 59Fe radioactivity was evident in the liver and kidney of rats following repeated PIH administration (Cikrt et al., 1980). In addition to rat studies, the effects of PIH treatment have also been examined during a phase I clinical trial in both normal and iron-overloaded patients (Brittenham, 1990). No sign of toxicity was evident when patients were treated daily with PIH at 30 mg/kg (Brittenham, 1990). The iron excretion promoted in overload patients treated with this latter dose of PIH, three times a day for 3 weeks, achieved a negative iron balance in nontransfused subjects (Brittenham, 1990). However, this dose would not be sufficient to achieve a negative iron balance in transfused patients and it has been suggested that the oral administration of the chelator needs to be optimized (Brittenham, 1990).
Other members of the 100 series have also been subjected to in vivo animal trials, including the analogs 101, 107, 109, 114, and 115 (Fig. 13B) (Blaha et al., 1998). Of these analogs, 109 was the most effective, increasing biliary iron excretion by approximately 150-fold over the basal level after a single dose administered intraperitoneally at 0.2 mmol/kg in rats (Blaha et al., 1998). This was in contrast to DFO, which was found to increase the biliary iron concentration by 20- to 30-fold under the same treatment regimen (Blaha et al., 1998). Interestingly, both 107 and, in particular, 109 were more effective than PIH, the lead compound, in promoting iron excretion (Blaha et al., 1998).
Subsequent investigations have been launched to analyze the mechanisms of action of PIH and its analogs. An acid dissociation constant study of PIH, 101, 107, and 109 revealed that at physiological pH, the majority (∼80%) of each chelator is present in a neutral state, with a small proportion being found as a singly charged anionic species (Richardson et al., 1990). These results, in combination with their lipophilicity (Baker et al., 1985), indicate that PIH and its analogs are able to readily permeate through cell membranes, allowing absorption in the small intestine where the basic pH facilitates the formation of neutral species (Richardson et al., 1990).
More recent studies have confirmed the importance of the ligand and subsequent complex lipophilicity on the iron mobilization activity of these chelators (Buss et al., 2002a, 2003a). One study hypothesized that the release of complexes from cells was kinetically limited by the high affinity of hydrophobic complexes for cell membranes (Buss et al., 2002a). Interestingly, the addition of bovine serum albumin, known to have high affinity for hydrophobic species, to the extracellular medium resulted in an increase in iron mobilization from reticulocytes, presumably by acting as an extracellular sink for the lipophilic iron complexes (Buss et al., 2002a, 2003a).
Another factor believed to be associated with the limited toxicity of this series of chelators lies in the oxidative stress mediated by the resultant iron complexes (Buss et al., 2002c). Although studies have shown the iron-PIH complex not to be toxic below its solubility limit (Buss et al., 2002c, 2003b), a recent study suggested that oxidative stress imparted by the formation of intracellular iron complexes can be detected (Buss et al., 2002c). When K562 cells loaded with eicosapentaenoic acid, a readily oxidized fatty acid, were treated with the iron complex of 101, lipid peroxidation was evident (Buss et al., 2002c, 2004b). However, the iron-PIH complex did not induce oxidative damage in this model (Buss et al., 2002c). In addition, the treatment of glutathione-deprived Jurkat T lymphocytes with PIH or 101 resulted in increased sensitivity to their oxidative effects (Buss et al., 2002c, 2004b). Although the damage induced by PIH was to a lower extent than that of its analogs, the iron complex is not totally inert (Buss et al., 2002c, 2004b). Thus, the ability of PIH and other analogs of the 100 series to promote ROS plays a role in oxidatively damaging cells (Buss et al., 2003a), although the extent of this effect is many magnitudes less than that found for cytotoxic chelators, such as Triapine or those of the PKIH series (Chaston et al., 2003; Yuan et al., 2004) (see section VI.B.5. below). Further studies suggested that the inability of more lipophilic iron complexes to efflux from cells (Buss et al., 2002a) results in an increase in intracellular iron complexes that may account for their greater cytotoxicity within this series of analogs. Finally, again it must be noted, that the cytotoxicity of the 100 series of ligands is far less than that of the related 200 and 300 series of chelators (Richardson et al., 1995). Indeed, many of the 100 series chelators have high potential for the treatment of iron overload disease, having only limited antiproliferative activity, that is comparable or even less than that of DFO (Richardson et al., 1995).
b. 200 series analogs.
To further investigate the structure-activity relationships of PIH analogs, a new range of derivatives were synthesized (Edward et al., 1988), replacing the pyridoxal moiety with the more hydrophobic salicylaldehyde group, creating the 200 series (Fig. 13B). These tridentate ligands use the same backbone and chelating groups as the 100 series (Baker et al., 1992b; Richardson et al., 1995). The mechanisms of action of the parent compound, salicylaldehyde isonicotinoyl hydrazone (SIH; Fig. 8) and its analogs have been examined both in vitro and in vivo for their iron-binding and cytotoxic effects. As seen with PIH, the ligands SIH and salicylaldehyde benzoyl hydrazone (201; Fig. 13B) have been shown to bind both ferric and ferrous forms of iron (Vitolo et al., 1990; Dubois et al., 1992). It is interesting to note that the structural alteration involving the change of the pyridoxal group to the salicylaldehyde group resulted in increased Fe(III) binding affinity of SIH over the lead compound PIH (Vitolo et al., 1990). Hence, the electron-withdrawing effects of the pyridoxal nitrogen on the phenolate group of PIH greatly affect the binding affinity of this ligand (Richardson et al., 1990; Vitolo et al., 1990). As seen with a number of analogs of the 100 series, 86% of SIH remains in its neutral state at physiological pH, enhancing membrane permeability (Richardson et al., 1990).
In vitro analysis of the iron-chelating properties of SIH and its analogs have illustrated a reduced capacity to mobilize intracellular iron from SK-N-MC cells compared with that of the 100 series (Richardson et al., 1995). During iron efflux experiments, all of the 200 series were found to mobilize iron to a greater extent than DFO, and eight showed activity similar to or higher than that of PIH (Richardson et al., 1995). In addition, all members of the 200 series were shown to be more efficient than DFO in preventing iron uptake from Tf, and seven had efficiency comparable to or greater efficiency than that of PIH (Richardson et al., 1995). This shows the potential ability of these aroylhydrazones to replace the cumbersome use of DFO in the clinical treatment of iron overload disease. In addition, the iron-SIH complex has been shown to donate iron to reticulocytes for heme synthesis to a greater extent than that of saturating concentrations of diferric Tf (Ponka and Schulman, 1985). On this note, the great potential of SIH and its analogs as agents for the treatment of iron overload disease is evident, especially as a more recent in vivo trial has illustrated the lack of toxicity of this ligand (Klimtova et al., 2003). When SIH was administered at 50 mg/kg once a week over a 10 week period to rabbits, no adverse effects were evident, again emphasizing the promising potential of this range of ligands in iron chelation therapy (Klimtova et al., 2003). However, further investigations must be performed to investigate in vivo iron mobilization of this class of compounds and their efficacy in clinical trials.
The capacity of SIH and its analogs to induce antiproliferative activity against various cancer cell types in vitro have been demonstrated (Richardson et al., 1995). In contrast to the decreased iron-mobilizing ability of this range of analogs in comparison to that of the 100 series, an increase in antiproliferative activity was seen (Richardson et al., 1995). An in vitro study involving human melanoma, bladder, and lung epithelial carcinoma cell lines illustrated the capability of both SIH and ligand 201 to inhibit DNA synthesis, more so than their corresponding 100 series analogs, PIH and 101 (Johnson et al., 1982). More importantly, the Cu(II) complexes of these ligands, in particular that of analog 201, were found to contain higher activity than the chelators alone (Johnson et al., 1982). The effect of this complex was not only inhibitory but also cytotoxic in action, emphasizing the importance of metal complexes in the toxic effects of chelating agents (Johnson et al., 1982; for review, see Lovejoy and Richardson, 2000). Further in vitro tests on the antiproliferative effects on SK-N-MC cells again illustrated the increased cytotoxic effects of the 200 series in comparison to the 100 series of ligands (Richardson et al., 1995). Of the 11 200 series analogs examined, 10 possessed higher antiproliferative activity than their 100 series counterparts (Richardson et al., 1995). This study also demonstrated a clear linear relationship between antiproliferative activity and the ability of the chelator to either prevent iron uptake from Tf or induce iron mobilization from prelabeled cells (Richardson et al., 1995). Further examination of the effects of one of the most lipophilic members of the 200 series, namely ligand 206 (Fig. 13B), illustrated the ability of this cytotoxic chelator to inhibit [3H]thymidine, [3H]leucine and [3H]uridine incorporation (Richardson and Milnes, 1997). This ligand also showed antiproliferative activity that was better than or comparable to that of three clinically used cytotoxic agents, namely cisplatin, bleomycin, and doxorubicin (Richardson and Milnes, 1997).
As determined with PIH and ligand 101, the iron complex of SIH was found to induce a cytotoxic effect on K562 cells loaded with eicosapentaenoic acid (Buss et al., 2002c). However, the mechanism behind the cytotoxicity of the SIH complex differs from that of 101, as no lipid peroxidation was evident (Buss et al., 2002c). In addition, the increased lipophilicity of the 200 series in comparison to that of the 100 series demonstrates the importance of this factor on antiproliferative ability. This relationship is further emphasized by studies with the 300 series chelators described below.
c. 300 series analogs.
Another range of PIH analogs developed, namely the 300 series (Fig. 13B), have even higher lipophilicity than that of the 100 and 200 series by incorporating a 2-hydroxy-1-naphthaldehyde group. Again, this range of ligands use two oxygen atoms and a nitrogen to bind iron in a meridional manner, forming a distorted octahedral complex with high spin Fe(III) (Richardson and Bernhardt, 1999). The planar nature of these ligands leads to preorganization of the donor atoms in a geometry that allows iron binding (Richardson and Bernhardt, 1999).
The ability of the 300 series to induce iron mobilization has been studied in vitro using SK-N-MC neuroepithelioma cells (Richardson et al., 1995). Iron efflux studies demonstrated that 8 of the 10 ligands tested were more effective than DFO and 6 showed iron-mobilizing activity similar to or greater than that of PIH (Richardson et al., 1995). Of this series, 311 was found to be the most effective, mobilizing 44% of 59Fe compared with 15% released by DFO (Richardson et al., 1995). Two of the more hydrophilic analogs, 302 (p-hydroxy substituted) and 305 (p-amino-substituted; Fig. 13B) were found to have less activity than DFO in preventing iron uptake from Tf (Richardson et al., 1995). Again, 311 was also the most effective in preventing iron uptake (Richardson et al., 1995). Interestingly, in contrast to the 200 series of ligands, analysis of the relationship between IC50 and the ability of the 300 series chelator to mobilize iron or inhibit iron uptake showed no correlation (Richardson et al., 1995).
The cytotoxic effects of the 300 series were also analyzed in vitro using the SK-N-MC neuroepithelioma cell line (Richardson et al., 1995). This study demonstrated that from all of the 100, 200, and 300 series of analogs, the latter group of chelators had the highest lipophilicity and greatest antiproliferative activity (Richardson et al., 1995). All members of the 300 series were found to have antiproliferative activity markedly higher than that of DFO (ID50 = 22 μM), with ID50 values ranging from 1 to 8 μM (Richardson et al., 1995). Interestingly, complexation of 311 (Fig. 8) with iron prior to treatment resulted in the inhibition of its cytotoxic effects, indicating that its antitumor activity relies on its ability to bind Fe (Richardson and Bernhardt, 1999). The effects of 311 were also studied in CCRF-CEM, breast, bladder, and head and neck cancer cell lines, and the chelator was also shown to inhibit growth (Green et al., 2001).
As previously mentioned, the 300 series were shown to possess the most potent antiproliferative activity of all of the three series of PIH analogs assessed (Richardson et al., 1995). This effect is attributed to the increased lipophilicity of this range of chelators, for which the 2-hydroxy-1-naphthaldehyde portion is responsible. Analysis of the various aromatic substituents of the hydrazide portion in this series (Fig. 13B) also demonstrated the importance of lipophilicity on the activity of the 300 series. By implementing the hydrophobicity constant (π), a measure of a substituent's lipophilicity relative to hydrogen derived from log P values (Patrick, 2001), the examination of these aromatic substituents illustrates a trend in which the antiproliferative activity increases as the substituents become more hydrophobic (Fig. 13C). Considering the relationship between lipophilicity and proliferation, it can be speculated that the more lipophilic analogs may potentially bind iron from different iron pools than their hydrophilic counterparts, specifically pools that are necessary for vital functions such as RR activity.
Since the more preliminary investigations, which demonstrated the potent antiproliferative effects of 311, various experimental techniques have been used to examine the biological aspects that are responsible for its antitumor activity. Cyclic voltammetry conducted on both the free ligand and ferric complex of 311 suggested that redox cycling does not play a major role in its antiproliferative activity (Richardson and Bernhardt, 1999). Indeed, 311 prevented ascorbate oxidation and benzoate hydroxylation and its iron complex seemed to be redox-inactive in both experiments (Chaston et al., 2003). Other studies have illustrated the ability of 311 to affect proteins involved in iron metabolism. For example, 311 is able to increase the RNA-binding activity of the IRPs far more effectively than DFO (Darnell and Richardson, 1999). This in turn resulted in a marked increase of TfR1 mRNA and protein levels (Chaston et al., 2003).
Experiments examining [3H]thymidine, [3H]uridine, and [3H]leucine incorporation in a number of tumor cell lines demonstrated a marked decrease in cells treated with 311 consistent with marked inhibition of DNA, RNA, and protein synthesis (Richardson and Milnes, 1997). Subsequent experiments demonstrated a 58% decrease in the electron paramagnetic resonance signal generated by the tyrosyl radical of RR in CCRF-CEM cells treated with 311 (Green et al., 2001). SK-N-MC cells treated with 311 have also been reported to show a 48% decrease in RR activity by electron paramagnetic resonance (Chaston et al., 2003). These results indicate that the iron-binding activity of 311 results in a decrease in intracellular iron pools that are necessary for RR activity, leading to inhibition of cellular proliferation (Green et al., 2001; Chaston et al., 2003).
In molecular biology studies, the effects of 311 on the expression of molecules necessary for cell cycle progression have also been assessed. For instance, the effect of the chelator on the expression of the WAF1 (wild-type p53 activating fragment 1) and GADD45 (growth arrest and DNA damage) genes were examined, as these are critical for DNA repair and cell cycle regulation (Darnell and Richardson, 1999). In fact, the protein product of the WAF1 gene, p21CIP1/WAF1, is involved in the universal inhibition of cyclin-dependent kinases (cdks) that are proteins responsible for interacting with the cell cycle proteins, the cyclins (El-Deiry et al., 1993; Harper et al., 1993). The formation of cyclin-cdk complexes are critical for progression through the cell cycle, via the phosphorylation of proteins such as the retinoblastoma protein that is necessary for G1/S progression (for review, see Le and Richardson, 2002). The function of p21 is to regulate G1/S progression, and it can induce cell cycle arrest, whereas GADD45 is involved in nucleotide excision repair, and it can also arrest the cell cycle (El-Deiry et al., 1993; Levine, 1997). It is important to note that the expression of these genes can be regulated via both p53-dependent and -independent pathways (Michieli et al., 1994; Zeng and El-Deiry, 1996). More detailed studies have shown that chelators up-regulated WAF1 mRNA expression by 1) a transcriptional mechanism that was p53-independent and 2) via a post-transcriptional mechanism that was sensitive to cycloheximide (Liang and Richardson, 2003).
Treatment of different cell lines with 311 has been shown to increase WAF1 and GADD45 mRNA expression, whereas the iron complex had no effect (Darnell and Richardson, 1999). This seemed to occur via a p53-independent pathway, as the cell lines examined in this study, for example, K562 erythroleukemia, did not contain functional p53 (Bi et al., 1992; Davidoff et al., 1992; Darnell and Richardson, 1999). However, despite the marked increase in WAF1 mRNA after iron chelation (Darnell and Richardson, 1999; Gao and Richardson, 2001), the nuclear levels of its protein product, p21CIP1/WAF1, were found to decrease (Le and Richardson, 2003; Liang and Richardson, 2003), illustrating the inhibition of its translation or its increased degradation after iron chelation. Incubation with ferric ammonium citrate was observed to reverse the effects of this chelator, indicating that p21CIP1/WAF1 protein levels are dependent upon the intracellular iron concentration (Le and Richardson, 2003).
Iron chelation by 311 or DFO was also found to result in a decrease in the expression of cdk2 and also cyclin A, B1, D1, D2, and D3 (Gao and Richardson, 2001). These effects were completely inhibited upon incubation with the 311-iron complex (Gao and Richardson, 2001). More recent investigations have highlighted other cellular targets that are controlled by intracellular iron levels (Le and Richardson, 2004). For example, gene array studies have demonstrated that N-myc downstream regulated gene 1 (Ndrg1) is specifically up-regulated by the effects of iron chelation (Le and Richardson, 2004). Although the exact function of this gene remains unclear, it is thought to act as a potent metastasis suppressor (Bandyopadhyay et al., 2003). Incubation with 311 resulted in an increase in both mRNA and protein levels, a result not induced by incubation with the iron complex (Le and Richardson, 2004). These results suggest that Ndrg1 may act as a novel link between the effect of iron chelation therapy by 311 and the inhibition of cellular proliferation (Le and Richardson, 2004). Collectively, the results above clearly demonstrate the many molecular targets of iron chelators.

A, backbone structure of the PCIH series illustrating their various R groups, including PCBH, PCHH, PCBBH, PCIH, PCAH, and PCTH. B, iron-catalyzed oxidation of PCIH to isonicotinoyl(picolinoyl)hydrazine after formation of the Fe(III) complex in aerobic solution. C, iron-catalyzed oxidation products of the PCIH series.
Knowledge derived from the assessment of the biological activity of the 100, 200, and 300 series of chelators was subsequently used in the generation of a variety of new ligand classes that have potential for the treatment of iron overload disease or cancer. These are described in the sections below.
d. 2-Pyridylcarboxaldehyde isonicotinoyl hydrazone analogs.
By analyzing the structure of the thiosemicarbazone series of ligands (see section VI.B.5. below), a novel range of aroylhydrazone iron chelators were designed to contain the 2-pyridylcarboxaldehyde moiety (Becker and Richardson, 1999). Thus, the 2-pyridylcarboxaldehyde isonicotinoyl hydrazone (PCIH) analogs (Fig. 14A) were generated (Becker and Richardson, 1999). This tridentate range of chelators incorporate the same hydrazone bridge used by the 100, 200, and 300 series, binding iron through imine and pyridyl nitrogens and a carbonyl oxygen (Bernhardt et al., 2001).
Based on the activity of some thiosemicarbazones (Liu et al., 1992) (see section VI.B.5. below), inclusion of the 2-pyridylcarboxaldehyde moiety into the aroylhydrazone structural backbone was expected to result in a ligand with high antitumor activity. Surprisingly, the in vitro assessment of the PCIH analogs demonstrated the low antiproliferative activity of these ligands and highlighted their potential to replace DFO in the treatment of iron overload disease (Becker and Richardson, 1999; Richardson et al., 2001). Iron efflux experiments illustrated the ability of PCIH analogs to mobilize intracellular iron. Of the synthesized analogs, 2-pyridylcarboxaldehyde thiophenecarboxyl hydrazone (PCTH), 2-pyridylcarboxaldehyde benzoyl hydrazone (PCBH), and 2-pyridylcarboxaldehyde m-bromobenzoyl hydrazone (PCBBH) (Fig. 14A) demonstrated activity similar to that of PIH, resulting in the release of 40 to 42% of cellular 59Fe from SK-N-MC cells (Becker and Richardson, 1999). These same analogs were also found to be the most effective in preventing iron uptake from Tf, showing levels similar to that of PIH (Becker and Richardson, 1999). The 2-pyridylcarboxaldehyde p-aminobenzoyl hydrazone (PCAH) and 2-pyridylcarboxaldehyde p-hydroxybenzoyl hydrazone (PCHH) chelators (Fig. 14A) were consistently the poorest of the series in mobilizing cellular iron. In addition, further studies using a model of mitochondrial iron overload, that is, reticulocytes treated with the heme synthesis inhibitor, succinylacetone, demonstrated the potential application of these ligands for the treatment of FA (Richardson et al., 2001). These experiments illustrated the superior capability of PCIH to increase iron release compared with that of other PCIH analogs (Richardson et al., 2001). PCTH was found to have an effect similar to that of PIH, whereas DFO was found to have little effect on mobilizing mitochondrial iron (Richardson et al., 2001).
Due to the promising results of PCTH during the in vitro examinations, an in vivo trial was recently completed (Wong et al., 2004). No signs of toxicity were evident in mice orally treated with PCTH at 50 and 100 mg/kg twice daily for >3 weeks (Wong et al., 2004). Also a dose-dependent increase in fecal iron excretion was observed, and the level of iron excretion induced by 200 mg/kg of PCTH administered twice daily was comparable to that induced by the same dose of PIH and deferiprone (Wong et al., 2004). This illustrates the ability of PCTH to induce substantial levels of iron excretion when orally administered to mice, and consequently PCTH may be a candidate to replace DFO for the treatment of iron overload.
As briefly alluded to above, these ligands do not show potent antiproliferative effects, making them ideal candidates for the treatment of iron overload disease. Two of these analogs, PCBBH (IC50 = 42 μM) and PCBH (IC50 = 50 μM) were found to have an antiproliferative effect on SK-N-MC cells similar to that of DFO (IC50 = 47 μM), whereas all analogs were far less active than 311 (IC50 = 2 μM) (Becker and Richardson, 1999). Furthermore, all analogs showed much lower activity than 311 in preventing [3H]thymidine, [3H]leucine, and [3H]uridine incorporation (Becker and Richardson, 1999). In summary, these findings emphasize the ability of the PCIH series to mobilize intracellular iron without inhibiting cellular proliferation.
The crystal structures of some of the PCIH series of ligands and iron complexes have been reported (Richardson et al., 1999; Bernhardt et al., 2001; Armstrong et al., 2003), and, interestingly, studies undertaken to solve the crystal structures of their iron complexes serendipitously identified an unprecedented oxidization step catalyzed by Fe(III) (Bernhardt et al., 2001). In this process, PCIH was oxidized to isonicotinoyl(picolinoyl)-hydrazine once complexed with Fe(III) (Fig. 14B), and thus, the biological activities of this novel range of oxidation products (Fig. 14C) were analyzed to assess their role in the ability of the PCIH range to mobilize iron (Bernhardt et al., 2001). The diacylhydrazine product of PCIH, namely isonicotinoyl(picolinoyl)hydrazine, showed little iron mobilization activity. This is believed to be a result of its limited membrane permeability, as this analog remains charged at physiological pH, although its resulting iron complex is neutral (Bernhardt et al., 2001). Hence, it can be suggested that the parent compound, PCIH, can readily enter cells where it binds iron, and, after oxidation, the neutral iron diacylhydrazine complex may diffuse from the cell (Bernhardt et al., 2001). In contrast to isonicotinoyl(picolinoyl)hydrazine, other diacylhydrazine analogs that were neutral at physiological pH demonstrated high iron chelation efficacy (Bernhardt et al., 2001). These results emphasize the importance of charge on the ability of ligands to access intracellular iron.
PCIH has also been demonstrated to bind Fe(II), resulting in an octahedral complex, although the Fe(II)-binding affinity was not particularly great (Armstrong et al., 2003; Bernhardt et al., 2004). The ability of the PCIH and PCAH to bind other transition metals, including Zn(II), Mn(II), Co(II), Ni(II), and Cu(II), has also been analyzed (Armstrong et al., 2003). PCIH was found to form a range of structurally diverse complexes in which the isonicotinoyl nitrogen also coordinated with the transition metals (Armstrong et al., 2003). These ligands, like all other iron chelators, were found to form a variety of metal complexes, and such results demonstrate that they might potentially target other metals in biological systems (Armstrong et al., 2003). However, the extent of chelation of other metals will depend upon their relative concentrations, which are very low in comparison to the concentration of iron found in iron overload disease.
In light of the high iron-mobilizing ability of this range of chelators, the ability of their resulting iron complexes to undergo redox reactions must be considered if these ligands are to be used as a form of therapy in iron overload disease. Such oxidative reactions are not desired in the treatment of iron overload disease, in which the oxidative stress needs to be relieved. Therefore, the redox activity of the resultant iron complexes of the PCIH analogs were examined (Chaston and Richardson, 2003b). Importantly, the oxidation of ascorbate in the presence of Fe(III) was not enhanced by the addition of PCIH analogs. In addition, the amount of benzoate hydroxylation in the presence of Fe(III) was not increased by the PCIH series (Chaston and Richardson, 2003b). Furthermore, no significant level of DNA damage was observed during experiments on intact human cell lines treated with these ligands (Chaston and Richardson, 2003b). These results indicate that the corresponding complexes do not induce harmful redox reactions and thus confirm the potential of the PCIH series to be applied in treatment of iron overload disease.
e. Di-2-pyridylketone isonicotinoyl hydrazone analogs.
In an attempt to generate a novel series of chelators with antiproliferative activity, a range of PCIH analogs that featured additional lipophilic groups were synthesized. As previously mentioned, this property seems to increase the cytotoxic activity of the ligand (Richardson et al., 1995). Consequently, the PKIH analogs (Fig. 15A), which contain an additional pyridyl ring, were synthesized (Becker et al., 2003). As seen with the PCIH analogs, this series uses the same system of donor atoms to bind iron, namely a carbonyl oxygen and imine and pyridyl nitrogens, which was confirmed through crystallography studies (Bernhardt et al., 2003). This latter investigation also highlighted the ability of the PKIH series to form octahedral complexes with the Fe(II) ion (Bernhardt et al., 2003). In addition, the PKIH series predominately remain neutral at physiological pH, facilitating their ability to cross cell membranes (Bernhardt et al., 2003).
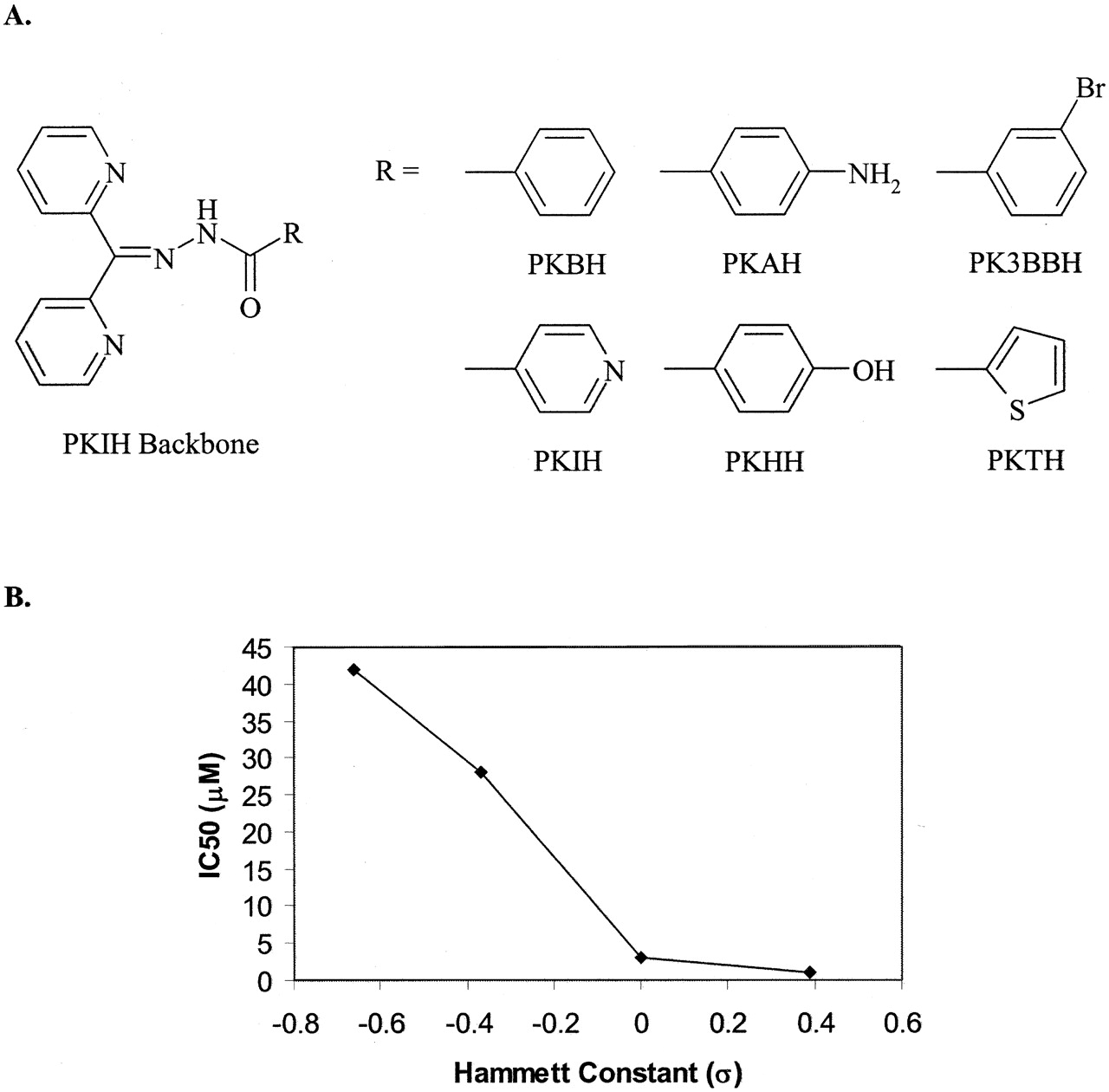
A, backbone of the PKIH series featuring their various R groups, including di-2-pyridylketone benzoyl hydrazone (PKBH), PKAH, PK3BBH, PKIH, PKHH, and di-2-pyridylketone thiophenecarboxyl hydrazone (PKTH). B, relationship between the Hammett constant and the antiproliferative effects of the PKIH series. As the substituents become increasingly electron-withdrawing, the cytotoxicity of the ligand is observed to increase.
During in vitro studies performed on SK-N-MC cells, all PKIH analogs apart from di-2-pyridylketone 3-bromobenzoyl hydrazone (PK3BBH; Fig. 15A) were found to be more efficient than their corresponding PCIH analogs in both increasing iron efflux from prelabled cells and preventing iron uptake from Tf (Becker et al., 2003). More significantly, this series of analogs was found to be highly cytotoxic, more so than their lead compounds, the PCIH series. Of the chelators examined, PKIH, di-2-pyridylketone thiophenecarboxyl hydrazone, di-2-pyridylketone benzoyl hydrazone, and PK3BBH (Fig. 15A), were found to have the highest antiproliferative activity against SK-N-MC cells having activity similar to that of 311 (Becker et al., 2003). The analogs with electron-donating substituents such as di-2-pyridylketone p-aminobenzoyl hydrazone (PKAH) and di-2-pyridylketone p-hydroxybenzoyl hydrazone (PKHH) (Fig. 15A), were far less effective. Interestingly, the corresponding iron complexes showed antiproliferative activity similar to that of the uncomplexed PKIH ligand, suggesting that other factors besides iron depletion were contributing to their mechanism of action (Becker et al., 2003). In addition, this series of ligands showed selectivity toward tumor cells as their activity against MRC-5 fibroblasts was much less pronounced (Becker et al., 2003).
Investigations on the effect of PKIH analogs on cellular processes were also conducted. This series of chelators were found to decrease [3H]thymidine, [3H]leucine, and [3H]uridine incorporation to a greater extent than the PCIH analogs (Becker et al., 2003). Unlike the PCIH analogs, the PKIH series were found to have a substantial effect on the cell cycle, increasing the expression of both GADD45 and WAF1 mRNA levels, vital for G1/S arrest, to a higher extent than 311 (Becker et al., 2003).
To further understand the biological activity of the PKIH series and their corresponding iron complexes, an effort to characterize their solution chemistry and redox activity was initiated (Bernhardt et al., 2003; Chaston et al., 2004). For instance, the PKIH analogs were found to stimulate benzoate hydroxylation in the presence of Fe(II) and H2O2 (Chaston et al., 2004). Furthermore, the ferrous complexes were found to enhance DNA plasmid degradation in the presence of H2O2 (Bernhardt et al., 2003). In addition, experiments using 2′,7′-dichloro-fluorescein-diacetate as a redox-sensitive probe proved that PKIH markedly increased the generation of intracellular ROS (Chaston et al., 2004). These results indicate that the redox cycling of the resultant iron complex plays an important role in the cytotoxicity of the PKIH analogs.
Investigations into the SAR of this series of ligands demonstrated no strong correlation between log P values and the antiproliferative activity (Becker et al., 2003). In addition, PKIH, the most hydrophilic ligand of the series showed high levels of cytotoxicity and effectively prevented [3H]thymidine incorporation into DNA (Becker et al., 2003). However, this effect may be due to the fact that this analog contains an electron-withdrawing pyridyl substituent on the hydrazide portion. Interestingly, those analogs with electron-donating substituents, including PKAH and di-2-pyridylketone p-hydroxybenzoyl hydrazone, were found to be less cytotoxic than their electron-withdrawing counterparts, such as PK3BBH and PKIH (Becker et al., 2003). Electrochemistry investigations of the iron complexes illustrated the inductive effects that the electron-withdrawing and -donating groups conferred on the iron center. For instance, more electron-withdrawing substituents influence the Fe(III)/(II) redox potential, which undergoes an anodic shift (Bernhardt et al., 2003). On the other hand, the electron-donating effects of the 4-amino substituent of PKAH result in its complex containing the lowest metal-centered redox couple (Bernhardt et al., 2003). This electron-donating effect acts to stabilize the trivalent iron state and consequently PKAH is a relatively poor oxidant of ascorbate (Bernhardt et al., 2003). However, two of the most electron-withdrawing analogs, PKIH and PK3BBH demonstrated the highest ability to induce ascorbate oxidation (Bernhardt et al., 2003). When the Hammett constant (σ), a measure of an aromatic substituent's ability to confer an electron-donating or -withdrawing effect in relation to hydrogen, is plotted against the antiproliferative activity, a general trend emerges in which electron-withdrawing groups are able to enhance the antiproliferative activity of the ligand (Fig. 15B). Thus, the electronic and inductive effects induced by the various hydrazide substituents play an important role in the biological activity of this series of ligands.
Collectively, from the assessment of all the aroylhydrazones described above, it can be seen that a wide variety of ligands have been generated, many of which may be useful in the future for various clinical applications from the treatment of iron overload disease to cancer. From the initial development of the PCIH series, which were found to be an effective range of iron-mobilizing agents, the potent antineoplastic PKIH analogs have evolved. This series results in iron complexes that mediate at least part of the cytotoxic effects generated by these ligands, and information obtained from the synthesis of these compounds have led to the development of the novel thiosemicarbazones described below.
5. Thiosemicarbazones.
Thiosemicarbazones were among the first chelators to be comprehensively assessed as anticancer agents (Sartorelli and Booth, 1967; Sartorelli et al., 1971; Antholine et al., 1977; Agrawal and Sartorelli, 1978; Finch et al., 2000). These ligands have a high affinity for iron and for other metal ions such as Cu(II), Ga(III), Co(II), and Zn(II). Indeed, thiosemicarbazone-metal complexes often have greater antitumor activity than the free chelators (Sartorelli et al., 1971; Antholine et al., 1977). The pronounced antitumor activity of these ligands has been widely attributed to inhibition of RR (Sartorelli and Booth, 1967; Sartorelli et al., 1971; Antholine et al., 1977; Agrawal and Sartorelli, 1978). Indeed, the α-(N)-heterocyclic carboxaldehyde thiosemicarbazones (HCTs) have been reported to be among the most effective RR inhibitors yet identified (Agrawal and Sartorelli, 1978; Finch et al., 2000).
Thiosemicarbazone ligands bind iron in a tridentate manner that is similar to that of the PIH analogs (see section VI.B.4.a. and Fig. 13A) (Antholine et al., 1977; Avramovici-Grisaru et al., 1985; Richardson and Bernhardt, 1999; Garcia-Tojal et al., 2002). However, thiosemicarbazones feature a sulfur donor atom in the coordination sphere, whereas in hydrazones the analogous donor atom is a carbonyl oxygen (Fig. 13A). The implications of this structural difference, in terms of the stability and redox properties of iron complexes in cells, are important and are discussed further below.
Early reports suggested that thiosemicarbazones inhibited RR by binding iron at the active site of the enzyme or by forming an iron complex that directly destroyed the tyrosyl radical in the R2 subunit (Sartorelli and Booth, 1967; Sartorelli et al., 1971; Antholine et al., 1977; Agrawal and Sartorelli, 1978). However, recent studies with other iron chelators, including structurally related aroylhydrazones, have demonstrated that inhibition of RR may also occur by depleting iron pools critical for enzyme activity (Nyholm et al., 1993; Cooper et al., 1996; Green et al., 2001). Hence, potentially chelators may act on RR at several sites by direct interaction with the protein or via interception of iron prior to its being incorporated into the molecule. The crystal structure of mouse RR is known (Kauppi et al., 1996), but molecular modeling studies have yet to be used to assess these possibilities.
Initially, one of the most active of the HCTs identified was 5-hydroxypyridine-2-carboxaldehyde thiosemicarbazone (Fig. 8) (Creasey et al., 1972). This ligand did increase iron excretion from patients, but it was glucuronidated and rapidly excreted (DeConti et al., 1972; Krakoff et al., 1974). New HCTs that are resistant to glucuronidation and rapid elimination have been developed; one of the most promising of these chelators is 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (Triapine, Vion Pharmaceuticals, New Haven, CT; Fig. 8) (Liu et al., 1992; Finch et al., 2000). The properties and studies of this compound are described in the section below.
a. Triapine.
i. Biochemical properties.
One of the best characterized properties of Triapine both in vitro and in vivo is that it acts as a potent RR inhibitor (Finch et al., 2000; Chaston et al., 2003). Mammalian RR is a tetramer consisting of two nonidentical homodimers, R1 and either R2 or p53R2, which are considered to be involved in DNA replication and repair, respectively. The p53R2 enzyme is a p53 target gene that has only recently been discovered to be transactivated upon DNA damage (Nakano et al., 2000; Tanaka et al., 2000). Interestingly, examination of structural homology suggested that like R2, p53R2 possesses an iron-binding site that is crucial for enzymatic activity and thus could potentially be inactivated by iron chelators (Le and Richardson, 2002). Initial studies measured total cellular RR activity but did not assess the ability of the chelator to inhibit the two forms of the enzyme involved in DNA replication and repair, respectively.
Recent studies by Shao et al. have shown that Triapine was equally potent on both the R2 and p53R2 subunits in vitro. In contrast, p53R2 was 158-fold more sensitive to DFO than the R2 subunit despite these enzymes having similar iron contents (Shao et al., 2004). This is of interest since Triapine has been shown to possess activity similar to that of DFO but was far less efficient than 311 in inducing iron release from cells and preventing iron uptake from Tf (Chaston et al., 2003). All three chelators increased IRP RNA-binding activity, suggesting chelation of intracellular iron pools. Together, these results suggest that patients undergoing Triapine treatment should be monitored for iron depletion. Indeed, studies using mice have shown that Triapine administered at a dose of 12 mg/kg resulted in splenic expansion and hematopoiesis in nude mice bearing tumor xenografts (M. Whitnall, J. Howard, and D. R. Richardson, unpublished observations).
Further studies comparing Triapine to DFO and 311 showed that in contrast to the iron complexes of DFO and 311, the Triapine-iron complex showed cytotoxicity similar to that of the free ligand (Chaston et al., 2003). These results suggested that the Triapine-iron complex may play a role in the cytotoxicity of the ligand. Four other lines of evidence also indicated that the Triapine-iron complex was redox-active: 1) Triapine increased the oxidation of ascorbate in the presence of Fe(III), whereas DFO and 311 prevented it; 2) Triapine increased benzoate hydroxylation in the presence of Fe(II), being more efficient than the positive control EDTA; 3) in contrast to DFO and 311, in the presence of Fe(II), Triapine resulted in marked plasmid DNA degradation; and 4) incubation with Triapine resulted in depletion of the cellular reductant, glutathione. These results suggested that apart from the ability of Triapine to bind cellular iron and induce iron depletion, the production of free radicals by its iron complex may also impart antitumor activity (Chaston et al., 2003).
ii. Animal studies with Triapine.
Triapine has recently been examined in animal studies and clinical trials and has shown some promising results (Finch et al., 2000; Giles et al., 2003; Murren et al., 2003; Wadler et al., 2004; Yen et al., 2004). For instance, this ligand shows selective antitumor activity and is fully inhibitory to cancer cells that show resistance to hydroxyurea (Cory et al., 1994; Finch et al., 2000; Yuan et al., 2004). The activity of Triapine (5 mg/kg) in vivo against the L1210 leukemia growing in CD2F1 mice was far greater than that found for hydroxyurea (70 mg/kg) (Finch et al., 2000). The effect of Triapine in inhibiting DNA synthesis in tumor cells was more marked than that found in normal cells of the intestinal mucosa and bone marrow. In addition, Triapine was active in nude mice bearing mouse M109 mouse lung carcinoma and A2780 ovarian carcinoma xenografts (Finch et al., 2000). Interestingly, Triapine could also penetrate the blood-brain barrier and effectively killed >95% of L1210 leukemia cells.
iii. Clinical Trials with Triapine.
A phase I clinical trial demonstrated that intravenous administration of Triapine caused prolonged stabilization of disease or decreased serum tumor markers associated with stable disease in four patients. However, there was no objective responses with treatment with the drug (Wadler et al., 2004). High doses of the drug (160 mg/m2/day) resulted in dose-limiting toxicities, including neutropenia, hyperbilirubinemia, nausea, or vomiting. Lower doses of Triapine administered as a 96-h iv infusion at 120 mg/m2/day every 2 weeks was found to be well tolerated (Wadler et al., 2004).
Other studies have examined the combination of Triapine with gemcitabine in patients with advanced cancer (Yen et al., 2004). These studies were based on the fact that combination therapy in vitro with gemcitabine and Triapine was shown to cause additive or synergistic cytotoxicity (Chen et al., 2002). Gemcitabine (2′,2′-difluoro-2′-deoxycytidine) is a nucleoside analog that, when phosphorylated to its triphosphate form, acts to inhibit DNA synthesis and topoisomerase I, and its diphosphorylated form is a potent inhibitor of RR (Heinemann et al., 1990; Ross and Cuddy, 1994; Pourquier et al., 2002). Of the 22 patients examined after treatment with gemcitabine and Triapine, 3 were observed to have an objective response, and 1 patient had evidence of tumor reduction (Yen et al., 2004). It is of interest to note that in this trial, Triapine was suggested to cause oxidation of hemoglobin to methemoglobin (Yen et al., 2004). This property may have led to or contributed to the hypoxia, acute hypotension, and EKG changes in patients receiving Triapine and to the asymptomatic myocardial infarction observed in one individual (Yen et al., 2004). Clearly, the ability of Triapine to redox cycle provides additional cytotoxicity to its iron-chelating ability. On the other hand, deleterious effects caused by the potential oxidation of heme-containing proteins must be considered when designing future compounds of this class.
b. Development of hybrid chelators derived from thiosemicarbazones and aroylhydrazones: the 2-hydroxy-1-naphthylaldehyde-3-thiosemicarbazone series.
Considering the high activity of the HCTs, a new series of chelators were synthesized by Schiff base condensation using the 2-pyridylcarboxaldehyde moiety from the HCT class of chelators and a series of acid hydrazides (Becker and Richardson, 1999; Richardson et al., 2001). These chelators were designated as the analogs of PCIH and demonstrated significant potential for the treatment of iron overload disorders as they showed high iron chelation efficacy but low antiproliferative activity (Becker and Richardson, 1999; Richardson et al., 2001). The failure of the 2-pyridylcarboxaldehyde moiety to confer antitumor activity suggested that the thiosemicarbazide moiety of HCTs may be significant in mediating their antiproliferative effects. Further studies were implemented to incorporate these observations into the design of new chelators (Lovejoy and Richardson, 2002). As described above, the 2-hydroxy-1-naphthylaldehyde moiety is associated with high antiproliferative activity of the PIH analogs (see section VI.B.4.c.). It was reasoned that chelators featuring both the thiosemicarbazide and 2-hydroxy-1-naphthylaldehyde moieties could show high antiproliferative activity. Hence, thiosemicarbazide and 2-hydroxy-1-naphthylaldehyde were condensed to yield a “hybrid” chelator, namely 2-hydroxy-1-naphthylaldehyde-3-thiosemicarbazone (NT), the parent ligand of the NT series of compounds (Fig. 16A) (Lovejoy and Richardson, 2002).
To gain further insight into structure-activity relationships, other NT series chelators were synthesized with increasingly lipophilic thiosemicarbazides to assess the effect of lipophilicity (Fig. 16A) (Lovejoy and Richardson, 2002). These studies identified NT, 2-hydroxy-1-naphthaldehyde-4-methyl-3-thiosemicarbazone (N4mT), and 2-hydroxy-1-naphthaldehyde-4,4-dimethyl-3-thiosemicarbazone (N44mT) as chelators having very high antiproliferative activity (IC50 = 0.5-1.5 μM; Lovejoy and Richardson, 2002). These ligands were significantly (p < 0.0001) more active than DFO (IC50 = 22 μM) and have efficacy comparable to that of 311 (IC50 = 0.3 μM; Lovejoy and Richardson, 2002). The most active chelators (NT, N4mT, and N44mT) demonstrated greater ability to inhibit the growth of neoplastic cells compared with a range of normal cells (Lovejoy and Richardson, 2002). These ligands had much less effect on the proliferation of the mortal MRC5 fibroblast cell line (IC50 > 25 μM), in contrast to immortal SK-N-MC cells, in which these chelators showed potent antiproliferative effects (IC50 = 0.5-1.5 μM; Lovejoy and Richardson, 2002).
A significant structure-activity relationship was identified in the NT series of ligands, which lost antiproliferative activity as the substituents on the terminal nitrogen atom of the thiosemicarbazide became increasingly lipophilic. For instance, replacing methyl groups with phenyl substituents resulted in a marked decrease in antiproliferative activity (Lovejoy and Richardson, 2002).
To determine the role of iron chelation in the antiproliferative effects of the NT series, the ability of the ligands to mobilize 59Fe from SK-N-MC cells and prevent 59Fe uptake from 59Fe-Tf was determined (Lovejoy and Richardson, 2002). Broadly, the ligands can be grouped into those with high or relatively low iron chelation efficacy. The latter group had activity similar to that of DFO and included the chelators NT and 2-hydroxy-1-naphthaldehyde-2-methyl-3-thiosemicarbazone (N2mT) (Lovejoy and Richardson, 2002). The high iron chelation efficacy group comprised the remaining NT analogs with N44mT being the most efficient hybrid chelator, having iron chelation efficacy comparable to that of 311 (Lovejoy and Richardson, 2002). Interestingly, the parent analog NT demonstrated little ability to inhibit iron uptake and increase iron mobilization despite its high antiproliferative activity (Lovejoy and Richardson, 2002). This suggested that NT had additional modes of antiproliferative activity or, like DFO, formed an iron complex that had impaired membrane permeability (Lovejoy and Richardson, 2002).

A, chemical structures of the NT class, including NT, N2mT, N4mT, N44mT, 2-hydroxy-1-naphthaldehyde-4-ethyl-3-thiosemicarbazone (N4eT), 2-hydroxy-1-naphthaldehyde-4-allyl-3-thiosemicarbazone (N4aT), and 2-hydroxy-1-naphthaldehyde-4-phenyl-3-thiosemicarbazone (N4pT). B, chemical structures of chelators of the DpT series, including DpT, Dp2mT, di-2-pyridylketone-4-methyl-3-thiosemicarbazone (Dp4mT), Dp44mT, di-2-pyridylketone-4-ethyl-3-thiosemicarbazone (Dp4eT), di-2-pyridylketone-4-allyl-3-thiosemicarbazone (Dp4aT), and di-2-pyridylketone-4-phenyl-3-thiosemicarbazone (Dp4pT).
As described above for aroylhydrazone ligands, the mechanism of these chelators in inhibiting cell growth has been examined by assessing their effects on cell cycle control molecules such as GADD45, cdk2, and the cyclins (Gao and Richardson, 2001). Examining the effects of the most cytotoxic members of the NT series, namely NT and N4mT, these chelators increased the expression of the growth arrest and DNA-damage gene, GADD45, to a significant but lesser extent than 311 (Lovejoy and Richardson, 2002). Furthermore, NT and N4mT were effective in decreasing the expression of cdk2 and cyclin D1, D2, and D3, which are required for G1/S progression. Hence, the effect of the chelators in inhibiting proliferation may be mediated, at least in part, through these mechanisms in addition to their ability to inhibit RR. Thus, as discussed above in section VI.B.4. on aroylhydrazones, iron chelators affect multiple molecular targets to induce cytotoxicity.
In summary, several of the NT analogs exhibited considerable antiproliferative activity that showed selectivity against tumor cells in vitro (Lovejoy and Richardson, 2002). These compounds are now currently being evaluated in a number of tumor models in animals.
c. Development of hybrid chelators derived from thiosemicarbazones and aroylhydrazones: the di-2-pyridylketone thiosemicarbazone analogs.
More recently, even more effective chelators known as the DpT series have been designed (Fig. 16B) (Yuan et al., 2004). The design of these ligands was based on the high activity of Triapine and the previously discussed PKIH group of ligands (Becker et al., 2003; Chaston et al., 2003). One of the most active compounds of the DpT series is the chelator, di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT), although many other analogs also showed high efficacy as antiproliferative agents (Fig. 16B) (Yuan et al., 2004). As a relative control compound, we also synthesized the analog, di-2-pyridylketone 2-methyl-3-thiosemicarbazone (Dp2mT; Fig. 16B), in which methylation of the hydrazone bridge at the 2-position prevented iron binding (Yuan et al., 2004). Hence, Dp2mT provides a relevant negative control for Dp44mT. Indeed, in cell culture, Dp2mT possesses little activity in terms of iron chelation efficacy and antiproliferative activity (Yuan et al., 2004).
The chelator, Dp44mT, was far more effective than DFO in both entering cells and inducing iron release and preventing iron uptake from Tf (Yuan et al., 2004). These chelators are selective and markedly inhibit the growth of a variety of tumor cells compared with normal MRC-5 fibroblasts (Yuan et al., 2004). Strikingly, studies in vivo showed that Dp44mT significantly decreased tumor weight in mice bearing the chemotherapy-resistant M109 lung carcinoma to 47% of the control after only 5 days of treatment (Yuan et al., 2004). Hence, over this treatment period, the chelator acted relatively selectively to inhibit tumor growth (Yuan et al., 2004).
More recent studies have assessed the effects of Dp44mT on the expression of the iron-regulated growth and metastasis suppressor gene, Ndrg1 (Le and Richardson, 2004). This ligand was highly effective at up-regulating Ndrg1 being far more effective than chelators with much lower antiproliferative activity, such as DFO, L1, and dipyridyl (Le and Richardson, 2004). As an appropriate negative control for the highly active ligand, Dp44mT, we incubated cells with its structural analog, Dp2mT, which cannot effectively bind iron. Hence, the marked antiproliferative activity of Dp44mT may be at least partly related to its ability to up-regulate Ndrg1 expression. The up-regulation of Ndrg1 was shown to be both dependent and independent upon hypoxia-inducible factor-1α (Le and Richardson, 2004). However, the hypoxia-inducible factor-1α-independent mechanism was not identified and remains the subject for future work. It is probable that Ndrg1 is not the only target responsible for the growth arrest observed after iron chelation; the relative contributions of Ndrg1 and inhibition of RR, among other mechanisms, are yet to be elucidated.
VII. Conclusions
Considering the essential role of iron in cellular proliferation and its potential to mediate deleterious oxidative damage when in excess, iron-chelating agents provide a promising form of treatment for both iron overload disease and cancer therapy. Due to the limitations in the activity of the clinically used siderophore, DFO, a vast array of iron chelators have been developed from both natural and synthetic sources in an attempt to replace it. Among the wide variety of analogs that have evolved from predecessors, a novel range of orally effective iron chelators have been developed of which many show great promise as prospective clinical drugs. Through the analysis of their structure-activity relationships, these novel ligands provide an insight into the role various chemical moieties contribute to their modes of action. For instance, through the hydroxylation of DFT analogs, 4′-(OH)-DADFT has been isolated as an effective and nontoxic ligand that is ready for clinical evaluation. In addition, various synthetic analogs, including ICL670A and Triapine, are currently being appraised in humans for iron overload disease and cancer, respectively, with promising results. Although many of these analogs show great potential, further in vitro and in vivo studies must be undertaken to elucidate their extra- and intracellular effects. This would aid in the determination of their molecular mechanisms and modes of action to develop more potent iron chelators.
Acknowledgments
We gratefully acknowledge Dr. Neil Davies, Dr. Jonathan Howard, and Louise Dunn of the Iron Metabolism and Chelation Program for critical evaluation of the manuscript prior to submission. Children's Cancer Institute Australia for Medical Research is affiliated with the University of New South Wales and Sydney Children's Hospital.
Footnotes
-
↵1 Abbreviations: DFO, desferrioxamine; NB, neuroblastoma; ROS, reactive oxygen species; FA, Friedreich's ataxia; DMT1, divalent metal transporter 1; Tf, transferrin; TfR1, transferrin receptor 1; LIP, labile iron pool; IRP, iron-regulatory protein; IRE, iron responsive element; RR, ribonucleotide reductase; PKIH, di-2-pyridylketone isonicotinoyl hydrazone; DFT, desferrithiocin; SAR, structure-activity relationship; DMDFT, desmethyldesferrithiocin; DADFT, desazadesferrithiocin; DADMDFT, desazadesmethyl-desferrithiocin; 4-(OH)-DADMDFT, 4′-hydroxydesazadesferrithiocin; BDU, (S,S)-1,11-bis[5-(4-carboxy-4,5-dihydrothiazol-2-yl)-2,4-dihydroxyphenyl]-4,8-dioxaundecane; DpT, di-2-pyridylketone thiosemicarbazone; D-Exo, desferri-exochelin(s); ICL670A, 4-[3,5-bis-(hydroxyphenyl)-1,2,4-triazol-1-yl]-benzoic acid; CP94, 1,2-diethyl-3-hydroxypyridin-4-one; CP117, 1-(2′-trimethylacetoxyethyl)-2-ethyl-3-hydroxypyridin-4-one; PIH, pyridoxal isonicotinoyl hydrazone; SIH, salicylaldehyde isonicotinoyl hydrazone; cdk, cyclin-dependent kinase; Ndrg1, N-myc downstream regulated gene 1; PCIH, 2-pyridylcarboxaldehyde isonicotinoyl hydrazone; PCTH, 2-pyridylcarboxaldehyde thiophenecarboxyl hydrazone; PCBH, 2-pyridylcarboxaldehyde benzoyl hydrazone; PCBBH, 2-pyridylcarboxaldehyde m-bromobenzoyl hydrazone; PCAH, 2-pyridylcarboxaldehyde p-aminobenzoyl hydrazone; PCHH, 2-pyridylcarboxaldehyde p-hydroxybenzoyl hydrazone; PK3BBH, di-2-pyridylketone 3-bromobenzoyl hydrazone; PKAH, di-2-pyridylketone p-aminobenzoyl hydrazone; PKHH, di-2-pyridylketone p-hydroxybenzoyl hydrazone; HCT, α-(N)-heterocyclic carboxaldehyde thiosemicarbazone; NT, 2-hydroxy-1-naphthylaldehyde-3-thiosemicarbazone; N4mT, 2-hydroxy-1-naphthaldehyde-4-methyl-3-thiosemicarbazone; N44mT, 2-hydroxy-1-naphthaldehyde-4,4-di-methyl-3-thiosemicarbazone; N2mT, 2-hydroxy-1-naphthaldehyde-2-methyl-3-thiosemicarbazone; Dp44mT, di-2-pyridylketone-4,4,-di-methyl-3-thiosemicarbazone; Dp2mT, di-2-pyridylketone 2-methyl-3-thiosemicarbazone; CP365, 1-ethyl-2-(1′hydroxyethyl)-3-hydroxypyridin-4-one; CP117, 1-[2′-(pivaloyloxy)ethyl]-2-ethyl-3-hydroxy-4-pyridinone; CP165, 1-[3′-(pivaloyloxy)propyl-2-methyl-3-hydroxy-4-pyridinone; CP102, 1-(2′-hydroxyethyl)-2-ethyl-3-hydroxypyridin-4-one; CP41, 1-(2′-hydroxypropyl)-2-methyl-3-hydroxypyridin-4-one; CP502, 1,4-dihydro-3-hydroxy-N-1,6-trimethyl-4-oxo-2-pyridinecarboxamide; CP38, 1-(2′-carboxyethyl)-2-methyl-3-hydroxy-4-pyridinone.
-
Article, publication date, and citation information can be found at http://pharmrev.aspetjournals.org.
-
doi:10.1124/pr.57.4.2.
-
D.R.R. was supported by a fellowship and grants from the National Health and Medical Research Council of Australia, the Australian Research Council, and the Muscular Dystrophy Association USA. D.S.K. was supported by an Australian postgraduate scholarship.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
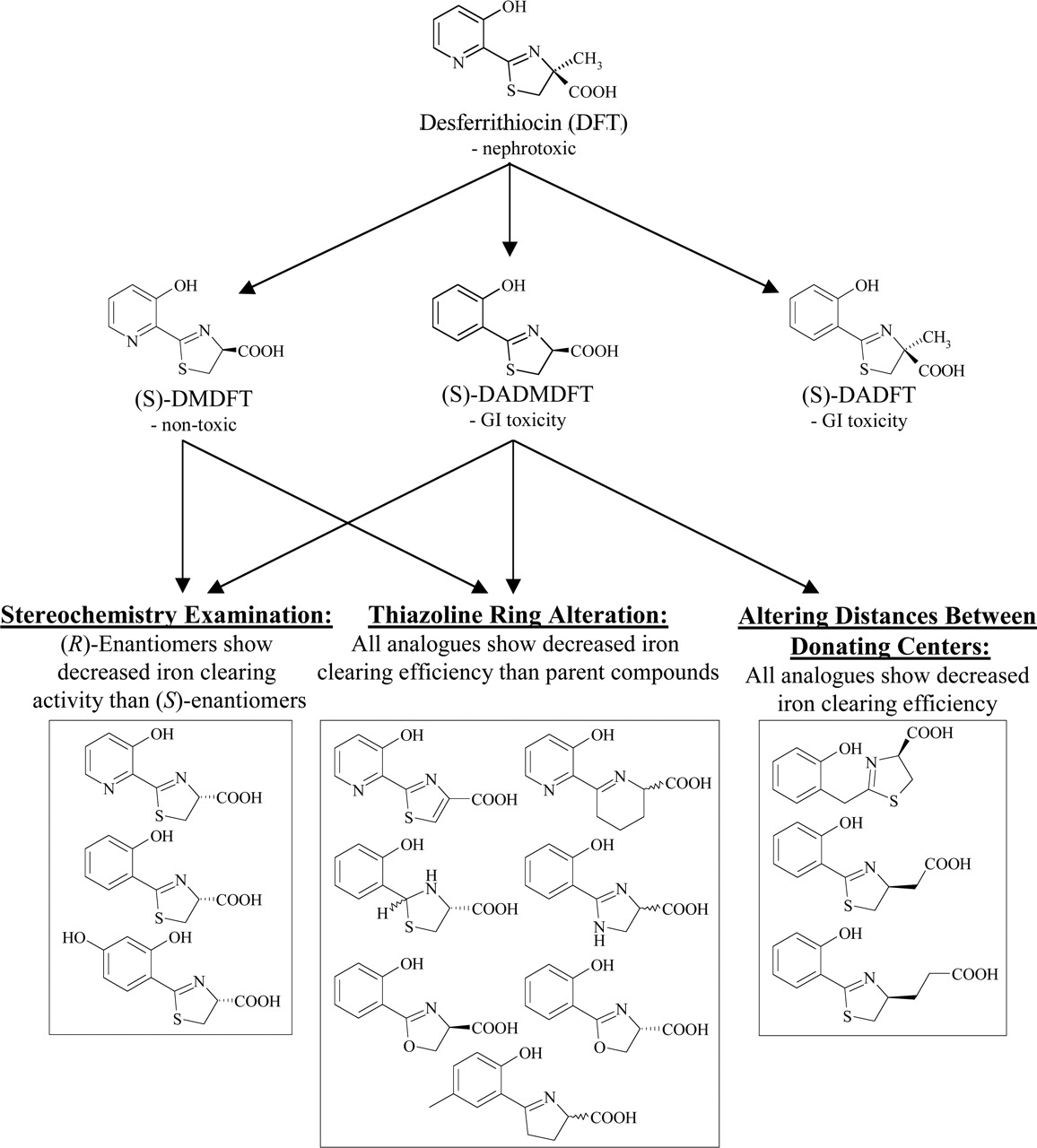{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
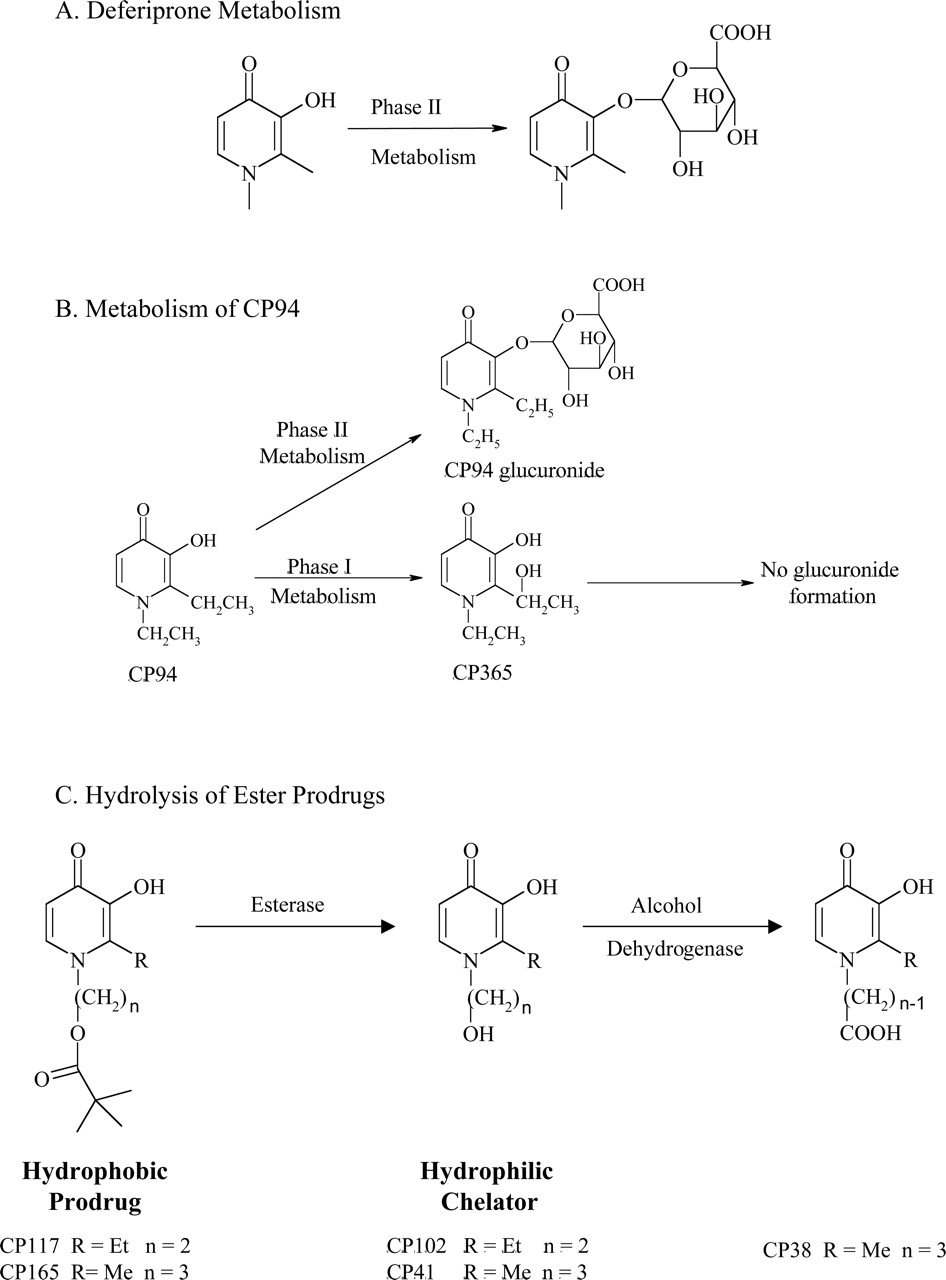{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}