


Abstract
NADPH oxidase (Nox) has a dedicated function of generating reactive oxygen species (ROS). Accumulating evidence suggests that Nox has an important role in signal transduction in cellular stress responses. We have reviewed the current evidence showing that the Nox system can be activated by a collection of chemical, physical, and biological cellular stresses. In many circumstances, Nox activation fits to the cellular stress response paradigm, in that (1) the response can be initiated by various forms of cellular stresses; (2) Nox-derived ROS may activate mitogen-activated protein kinases (extracellular signal-regulated kinase, p38) and c-Jun NH2-terminal kinase, which are the core of the cell stress-response signaling network; and (3) Nox is involved in the development of stress cross-tolerance. Activation of the cell survival pathway by Nox may promote cell adaptation to stresses, whereas Nox may also convey signals toward apoptosis in irreversibly injured cells. At later stage after injury, Nox is involved in tissue repair by modulating cell proliferation, angiogenesis, and fibrosis. We suggest that Nox may have an integral role in cell stress responses and the subsequent tissue repair process. Understanding Nox-mediated redox signaling mechanisms may be of prominent significance at the crossroads of directing cellular responses to stress, aiming at either enhancing the stress resistance (in such situations as preventing ischemia-reperfusion injuries and accelerating wound healing) or sensitizing the stress-induced cytotoxicity for proliferative diseases such as cancer. Therefore, an optimal outcome of interventions on Nox will only be achieved when this is dealt with in a timely and disease-and stage-specific manner.
I. Introduction
A. Cellular Stress Response
Cellular stress is induced by abrupt disruption of the favorite local environment for cell survival. Cells react to various stresses primarily through a number of specific and well conserved adaptive intracellular signaling pathways that attempt to alleviate damage and maintain or re-establish homeostasis, known as cellular stress response, which is present in all cell types of metazoans (Simmons et al., 2009). At the cellular level, the stress response can be initiated by external environmental factors that cause damage to biological macromolecules including lipids, proteins, and nucleic acids (Kültz, 2005). The cellular stress response entails a series of coordinated transcription and translational events leading to the accumulation of protective proteins that may counteract the stress-induced damage, and temporarily increase cellular tolerance to such damage; otherwise, cellular stress response may trigger programmed cell death (apoptosis) to remove terminally damaged cells (Welch, 1987; Kültz, 2005). On the other hand, cellular stress response can also be set off by internal deleterious changes, in forms of genomic stress and endoplasmic reticulum (ER1) stress for example (Simmons et al., 2009). Genomic stress response is induced by DNA damage caused by endogenous and exogenous reasons such as genotoxic chemicals, ionizing radiation, hypoxia, mitotic spindle damage, or ribonucleotide depletion, in which the transcription factor p53 plays a central role in orchestrating the signaling events. ER stress, also known as unfolded protein response, is caused by conditions under which the ER function is disrupted. ER is the organelle responsible for protein biosynthesis as well as protein folding and maturation for membrane incorporation and secretion (Simmons et al., 2009). The chaperone protein BiP/GRP78 mediates ER stress response, which is the sensor for accumulation of misfolded proteins in the ER. BiP/GRP78 regulates expression of an array of ER stress-responsive chaperone proteins that are capable of refolding proteins and removing damaged proteins. If ER stress persists and results in irreversible damage, the apoptosis process is then initiated (Simmons et al., 2009). More details of the signaling mechanisms in genomic and ER stresses are covered in a number of review articles (Yang et al., 2003; Coates et al., 2005; Lin et al., 2008).
The cellular stress response is an evolutionarily conserved mechanism with extraordinary physiological and pathophysiological significance (Welch, 1992; Kültz, 2005). Strikingly, the format of cellular stress response is very similar throughout prokaryotic and eukaryotic organisms. Proteomic analysis across human, yeast, and bacterium has raised that the cellular stress response can be characterized by the induction of a limited number (∼300) of highly conserved proteins (Kültz, 2005). It is noteworthy that among the 44 proteins with known functions, 40% of them are related to regulation of the intracellular redox status. It is noticed that increased reactive oxygen species (ROS) generation seems to be a common response in cells exposed to stresses; thus, it is argued that redox regulation may represent a critical second messenger system that is upstream of the cell stress signaling network (Kültz, 2005).
B. NADPH Oxidase
Oxidoreductases catalyze the transfer of electrons from the reductant (electron donor) to the oxidant (electron acceptor). In aerobic organisms, these chemical reactions are always associated with chances of formation of superoxide, the primary ROS molecule in biological systems, which is produced when molecular oxygen, instead of the specific substrate, is reduced by acquisition of an electron. Several oxidoreductases have been identified as potential sources of superoxide in mammalian cells. These include cyclooxygenase (Kukreja et al., 1986), lipoxygenase (Kukreja et al., 1986; Roy et al., 1994), cytochrome P450 enzymes (Puntarulo and Cederbaum, 1998), nitric-oxide synthase (Pou et al., 1992), xanthine oxidase (Berry and Hare, 2004), mitochondrial NADH:ubiquinone oxidoreductase (complex I) (Lambert and Brand, 2004), and NADPH oxidase (Griendling et al., 2000). Unlike other oxidoreductases, NADPH oxidase is a distinct enzymatic source of cellular ROS generation, because this enzyme is a “professional” ROS producer (Lambeth, 2004), whereas the other enzymes produce ROS only as by-products along with their specific catalytic pathways.
The NADPH oxidase paradigm was largely derived from studies in phagocytic leukocytes, such as neutrophils and macrophages (Babior, 1999). The backbone of this enzyme is the cell membrane-bound cytochrome b558, which consists of two subunits gp91phox (Nox2) and p22phox. Upon cell activation, two cytosolic regulatory subunits p47phox and p67phox, as well as a small G protein Rac, translocate to the membrane and associate with the cytochrome b558. The assembled multisubunit enzyme complex then generates superoxide by one-electron reduction of oxygen via its gp91phox subunit using reduced NADPH as the electron donor (Babior, 1999). During the last decade, people had observed that NADPH oxidase activity also existed in nonphagocytic cells, and this observation resulted in the discovery of additional isoforms of NADPH oxidase subunits. Thus far six additional Nox isoforms (Nox1, -3, -4, -5, and Duox1, -2), one p47phox isoform (Noxo1) and one p67phox isoform (Noxa1) have been identified in mammalian cells (Dupuy et al., 1999; Suh et al., 1999; De Deken et al., 2000; Geiszt et al., 2000; Bánfi et al., 2001, 2003; Cheng et al., 2001), and the expression patterns of these isoforms are distinct and seem to be tissue-specific. Unlike Nox2 in phagocytic cells, the subcellular localization of other Nox isoforms in nonphagocytic cells is not restricted to the cell membrane. Moreover, their dependence on cytosolic regulatory subunits for ROS generation also varies among different Nox members (Bedard and Krause, 2007).
It is noteworthy that the genes encoding NADPH oxidase subunits, although only exist in eukaryotes but not in prokaryotes, are evolutionarily ancient (Kawahara and Lambeth, 2007; Kawahara et al., 2007). Nox (Duox) genes are present in both plants and animals and can be traced back to lower organisms such as slime molds and fungi (Groom et al., 1996; Keller et al., 1998; Bayne et al., 2001; Bedard et al., 2007; Kawahara et al., 2007). Given the very early appearance in evolution and its wide distribution in different cell types in animals (especially in mammals), NADPH oxidase is likely to have a fundamental role in maintaining normal cell functions. In mammals, however, most of the primary biological significance of NADPH oxidase is largely unclear (Bedard and Krause, 2007). In contrast, wealthy evidence has linked this enzyme to cellular oxidative stresses that may eventually contribute to various pathophysiological conditions and diseases (Griendling et al., 2000; Lassègue and Clempus, 2003; Jiang et al., 2004; Lambeth, 2007).
Several lines of studies have suggested that one important physiological function of NADPH oxidase in mammalian cells is the modulation of multiple redox-sensitive intracellular signaling pathways by generating ROS molecules, including inhibition of protein tyrosine phosphatases, activation of certain redox-sensitive transcription factors, and modulation of the functions of some ion channels (Bedard and Krause, 2007). Compared with other signaling mechanisms, however, a prominent feature of NADPH oxidase/ROS-mediated signaling is the heterogeneity of its activating stimuli. By searching the literature, it is noted that at the cellular level, NADPH oxidase can be activated by a large collection of chemical, physical, environmental, and biological factors. Here we reviewed the current experimental evidence showing the diversity of stimuli that can activate NADPH oxidase. These stimuli may enhance NADPH oxidase function by up-regulating the gene expression of the enzyme, whereas many of them can activate the enzyme system acutely before alterations in transcription occur. We interpret these data by grouping these stimuli into a set of cellular stresses and suggest that NADPH oxidase may be an important component of the cellular stress signal transduction network. NADPH oxidase/ROS-mediated signaling might therefore represent a cellular “alarm system” that can alert the cells and prime the cells either to be adapted to the stress or to undergo apoptosis. In the following sections, we use “Nox” as an abbreviatation for NADPH oxidase, whereas Nox with a suffix refers to specific Nox subunits.
II. NADPH Oxidase Is Activated by Various Cellular Stresses
As mentioned above, current experimental results have demonstrated that Nox-dependent ROS generation can be stimulated by a large group of stimuli (summarized in Table 1). Analysis on the situations in which the Nox system is activated has revealed that many of these stimuli can be classified as a collection of cellular stresses. Moreover, the responsiveness of Nox to cellular stresses is not only restricted to mammals but can be found in a range of eukaryotic organisms. As the current evidence indicates that the intracellular signaling function of Nox is solely mediated by the generated ROS, we in some instances used the phrases such as “Nox-mediated signaling” or “Nox as signaling molecules” as a convenient description for this nature.
Activation of NADPH oxidase by various cellular stress stimuli
A. Chemical Factors
Nox can be activated by both inorganic and organic chemicals that may be potentially cytotoxic. In human endothelial and vascular smooth muscle cells, short-term exposure (within 30 min) to the heavy metal lead resulted in a significant rise in both superoxide and hydrogen peroxide production, whereas long-term lead exposure (60 h) resulted in Nox2 up-regulation (Ni et al., 2004). In renal epithelial cells, treatment with excessive zinc acutely induced translocation of p67phox from the cytosol to the membrane fraction and ROS generation in 3 h, and the latter was sensitive to the Nox inhibitor diphenyleneiodonium (DPI) (Matsunaga et al., 2005). It is noteworthy that DPI is a relatively nonspecific flavoprotein inhibitor and can inhibit a number of other oxidases than Nox. Likewise, in cultured neuronal cells, cadmium exposure for 24 h induced ROS generation and activation of the stress kinases c-Jun N-terminal kinase (JNK) and p38, all of which were inhibited by DPI (Rockwell et al., 2004). Nox has also been shown to be responsive to the nonmetallic poison arsenic, in the form of arsenite, as evidenced in fibroblasts (Chen et al., 1998), endothelial (Barchowsky et al., 1999; Smith et al., 2001), and smooth muscle cells (Lynn et al., 2000) and in preosteoclastic monocytes (Szymczyk et al., 2006). The enzymatic activity of Nox has also been shown to be stimulated by organic solvents. In perfused rat liver, a 20-min exposure to ethyl or butyl alcohol activated the Nox system in sinusoidal Kupffer cells (Hasegawa et al., 2002). In addition, some environmental cytotoxic substances, such as cigarette smoke and diesel exhaust particles, can activate Nox, which ought to be relevant to cells lining the airway. For example, people have found that these stimuli could induce Nox activity in airway epithelial cells (Lavigne and Eppihimer, 2005; Amara et al., 2007). It is noteworthy that cigarette smoke has also been shown to stimulate Nox-dependent ROS production in both vascular endothelial (Jaimes et al., 2004; Orosz et al., 2007) and smooth muscle cells (Orosz et al., 2007).
An important class of endogenously formed cytotoxic molecules is the oxidized lipoproteins. Oxidized low-density lipoprotein (oxLDL) causes endothelial cell activation and injury that have critical roles in the pathogenesis of atherosclerosis. In vascular smooth muscle cells, short-term treatment with lysophosphatidylcholine (a major active constituent found in oxLDL) for 3 min stimulated extracellular signal-regulated kinase (ERK)1/2 activation, which was dependent on Nox activity (Yamakawa et al., 2002). Likewise, in human endothelial cells, oxLDL up-regulated Nox2 expression and ROS generation (Rueckschloss et al., 2001; Dandapat et al., 2007), and inhibition of Nox activity with different pharmacological inhibitors suppressed oxLDL-induced endothelial proliferation, morphogenesis and activation of ERK (Heinloth et al., 2000; Dandapat et al., 2007). Moreover, oxidized high-density lipoproteins were also shown to be able to trigger endothelial Nox activation (Matsunaga et al., 2003).
The mechanisms by which cytotoxic chemicals activate Nox, especially short-term mechanisms, are not totally understood. In both vascular endothelial and smooth muscle cells, lysophosphatidylcholine induced protein kinase C (PKC) activation (Kugiyama et al., 1992; Motley et al., 2002), which was a critical upstream activator of Nox via phosphorylation of p47phox in both phagocytic and nonphagocytic cells (Babior, 1999; Li and Shah, 2003). Using a smooth muscle cell line, Yamakawa et al. (2002) demonstrated that p47phox translocated from the cytosol to the membrane after 2 min of stimulation with lysophosphatidylcholine, and inhibition of p47phox function with a dominant-negative mutant suppressed lysophosphatidylcholine-induced, ROS-dependent ERK activation. Taking these results together, it is suggested that PKC may have a pivotal role in transducing the cellular stress signals to Nox after exposure to toxic chemicals. In vascular endothelial cells, it has been shown that arsenite-induced Nox activation involves membrane translocation of the small GTPase Rac1, although the signaling mechanisms for this process are not clear (Smith et al., 2001). It is noteworthy that there is evidence that both elevation in intracellular Ca2+ concentration ([Ca2+]i) and activation of protein kinase C may induce translocation of cytosolic Rac to the plasma membrane (Price et al., 2003).
B. Physical Challenges
Exposure of different types of cells to ionizing radiation has been shown to induce intracellular superoxide and hydrogen peroxide generation as a result of Nox activation or up-regulation (Narayanan et al., 1997; Collins-Underwood et al., 2008; Tateishi et al., 2008). Radiation-induced ROS generation may trigger secondary cellular responses, such as expression of inflammatory molecules, activation of the redox-sensitive transcription factors (such as nuclear factor-κB), activation of mitogen-activated protein kinases (MAPKs) [including ERK1/2 and p38] and the stress kinase JNK, and cell apoptosis (Azzam et al., 2002; Collins-Underwood et al., 2008; Tateishi et al., 2008). In addition, people have found that acute exposure of keratinocytes to both UVA and UVB results in activation of Nox and generation of ROS (Wang and Kochevar, 2005; Van Laethem et al., 2006; Valencia and Kochevar, 2008). Given that keratinocytes are directly exposed to ambient UV lights, these studies suggest that a rapid activation of Nox by UV irradiation in these cells may have a distinct physiological importance (discussed below). How irradiation activates Nox is not totally understood. In keratinocytes, there is evidence that UVA-induced activation of Nox1 is accompanied by an increase in [Ca2+]i, which may stimulate Nox1 function indirectly by activating the small GTPase Rac (Valencia and Kochevar, 2008).
Physical forces may also activate the Nox system. Sustained oscillatory shear stress, which was thought to be an essential detrimental challenge to vascular endothelial cells, caused endothelial Nox activation and ROS production (Hwang et al., 2003). On the other hand, exaggerated pulsatile stretch and cyclic strain, such as those found in arterial hypertension, have been shown to stimulate Nox activity in both smooth muscle and endothelial cells (Hishikawa and Lüscher, 1997; Hishikawa et al., 1997; Matsushita et al., 2001; Grote et al., 2003; Mata-Greenwood et al., 2005). Moreover, physical stretch-induced activation of Nox has also been observed in retinal pericytes (Suzuma et al., 2007) and pulmonary epithelial cells (Chapman et al., 2005). In retinal pericytes, cyclic stretch-stimulated intracellular ROS generation and JNK phosphorylation, which were sensitive to Nox inhibition and blunted by a dominant-negative PKC-δ. In addition, physical stretch has been shown to activate PKC-δ and increase PKC association with p47phox, supporting an essential role of PKC in physical force-induced Nox activation (Suzuma et al., 2007).
The Nox system can also be activated by a variety of other physical challenges, such as changes in temperature, osmotic pressure, and pH. These observations are summarized in Table 1. In renal tubular epithelial cells, prolonged cold storage at 4°C induced ROS generation by Nox and subsequent activation of ERK1/2 (Karhumäki et al., 2007). Similar observations of cold storage-induced Nox activation were also reported in liver Kupffer cells (Shibuya et al., 1997). Exposure of cells to abnormal osmotic pressure stimulated Nox-dependent ROS production in animal cells (Cazalé et al., 1998; Rouet et al., 2006; Martins et al., 2008). In NIH3T3 mouse fibroblasts, hypotonic medium induced Nox-dependent ROS production, and this effect seemed to exhibit a dose-response relationship; i.e., the activity of Nox increased as a function of the magnitude of osmotic stress and reached a plateau thereafter (Friis et al., 2008). Moreover, alterations of the intracellular pH have also been linked to modulation of the Nox activity. In phagocytic cells, Nox is electrogenic, for a proton is generated in the cytoplasm when an electron is transported across the plasma membrane to generate superoxide; therefore, an outward H+ current that is independent of the Nox subunit is required to compensate this charge imbalance and prevents subsequent acidification of the cytoplasm (Henderson et al., 1988; DeCoursey et al., 2000). Abramov et al. (2005) reported that in brain astrocytes, inducing intracellular acidosis, which was thought to facilitate H+ efflux by increasing the cross-membrane gradient, enhanced the rate of ROS generation by Nox, whereas intracellular alkalosis had a suppressive effect. However, this phenomenon has not been observed in blood-borne phagocytic cells (Morgan et al., 2005).
C. Cellular Environments
Stresses imposed by adverse cellular environments, particularly altered nutritional conditions and oxygen tension, have been demonstrated to be able to activate the Nox system. An example of the involvement of Nox in mediating nutrient deprivation-induced stress responses was found in the slime mold (Dictyostelium discoideum), in which loss of the functions of different Nox or p22phox subunits resulted in defective starvation-induced transformation and spore production (Lardy et al., 2005). On the other hand, forced expression of exogenous NoxA in the NoxA-null strain restored the spore formation function (Lardy et al., 2005). Similar observations were also obtained in animal cells, in which serum withdrawal from the culture medium induced Nox activation in a Rac-dependent manner (Lopes et al., 2002). Collectively, these results suggest that nutritional stress-induced Nox activation is a conserved response in eukaryotes and is likely to have a critical role in promoting cellular adaptation and tolerance to the harsh conditions. Apart from nutrition restriction, Nox can also be activated by both hypoxia and hyperoxia (Parinandi et al., 2003; Schäfer et al., 2003; Chowdhury et al., 2005; Mollen et al., 2007). Brueckl et al. (2006) demonstrated that hyperoxia induced a rapid activation of Nox in lung capillary endothelial cells, which involved intracellular Ca2+ elevation and Rac1 activation.
D. Inflammatory Stimuli
Various endogenous or exogenous inflammatory signals that alert the cells to nearby “dangers” are the most important biological factors that can trigger cellular Nox activation. One example is the bacterial endotoxin lipopolysaccharide (LPS), which not only activates leukocyte Nox, as expected, but also has significant affects on nonphagocytic Nox. In human embryonic kidney 293 cells, Park et al. (2004) have shown that the C-terminal region of Nox4 interacts with the cytoplasmic tail of toll-like receptor 4, the cognate receptor for LPS; this interaction is essential in LPS-mediated ROS generation. In brain microglia and astrocytes, LPS stimulated rapid ROS production, and expression of inducible nitric-oxide synthase and cytokines, in a Nox-dependent manner (Pawate et al., 2004). It has been reported that in human airway epithelial cells, LPS stimulated Nox activity, and this process might require PKC activation (Yan et al., 2008). During inflammation, a variety of lipid products is generated, such as prostanoids and leukotrienes. It is noteworthy that many of these secondary inflammatory mediators have been shown to be capable of activating the Nox system in different cell lines (Katsuyama et al., 2002; Luchtefeld et al., 2003; Muzaffar et al., 2004b). Another group of inflammatory mediators that have been implicated in Nox activation are different cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1, interferon-γ (Lo et al., 1998; Frey et al., 2002; Muzaffar et al., 2004a; Li et al., 2005; Wu et al., 2007; Yang et al., 2007a; Kamizato et al., 2009), and the proinflammatory hormone angiotensin II (Hannken et al., 1998; Zafari et al., 1998; Sano et al., 2001; Gorin et al., 2003; Li and Shah, 2003; Zimmerman et al., 2005; Hingtgen et al., 2006; An et al., 2007; Zhang et al., 2010).
E. Stress-Related Humoral and Neural Factors
Limited evidence suggests that some stress-responsive neural transmitters and hormones (e.g., norepinephrine and endogenous opioid peptides) (Charmandari et al., 2005) may induce Nox activation in different cells. In adult rat cardiac myocytes, norepinephrine treatment induced a short-term ERK1/2 activation within 5 min that was sensitive to various Nox inhibitors and antioxidants (Xiao et al., 2002). Likewise, treatment of the same cells with the α-adrenoceptor agonist phenylephrine for 5 min induced significant ROS production that was suppressed by Nox inhibition (Tanaka et al., 2001). In HEK293 cells, treatment with various μ-opioid receptor agonists induced a phospholipase D2-dependent activation of Nox and subsequent ROS production (Koch et al., 2009). However, the effects of μ-receptor agonists in neuronal cells are unknown. In contrast, the effects of glucocorticoids, another important systemic stress response effector (Charmandari et al., 2005), on Nox activation seemed to be controversial and cell-specific (Marumo et al., 1998; Iuchi et al., 2003).
F. Evidence from Plant Cells
Evidence showing that Nox can be activated by cell exposure to heavy metals such as lead and cadmium has also been obtained in plant cells (Yeh et al., 2007; Pourrut et al., 2008). In plant cells, such heavy metals result in an increase in the [Ca2+]i, which may be involved in the chemically induced Nox activation (Yeh et al., 2007; Pourrut et al., 2008). Moreover, lead-induced Nox activity in plant root cells was shown to be suppressed by the calcium channel blocker lanthanum and the calmodulin inhibitor W7, highlighting the implication of Ca2+-dependent mechanisms in chemically induced Nox activity (Pourrut et al., 2008). In plant cells, activation of the oxidase by hypo-osmotic stress seemed to be dependent on protein phosphorylation and opening of cell membrane Ca2+ channels (Cazalé et al., 1998). Moreover, potassium deprivation in plant root cells triggered ROS generation (Shin and Schachtman, 2004). Inhibition of the plant Nox rhd2 prevented the potassium deficiency-induced gene expression program; conversely, exogenous hydrogen peroxide restored the potassium deprivation-induced gene expression in rhd2 mutant cells (Shin and Schachtman, 2004).
G. (Patho)Physiological Significance of Stress-Induced NADPH Oxidase Activation
In the scope of (patho)physiology, cellular stresses imposed by various toxic chemicals and detrimental physical factors may result in different forms of tissue injury and dysfunction of the affected organs. Activation of Nox-mediated redox signaling may have a critical role in coordinating the responses of the cell to deal with the adverse affects, either by activating stress kinases and promoting stress tolerance or by removing the seriously damaged cells by inducing apoptosis. Moreover, experimental evidence also suggests that Nox-dependent redox mechanisms may also be involved in the subsequent wound healing and tissue regeneration processes. These aspects will be elaborated below.
III. Signal Output from NADPH Oxidase
A. Apoptosis Signal-Regulating Kinase-1
A well studied signaling protein that is involved in cellular stress reactions and is modulated by redox mechanisms is apoptosis signal-regulating kinase-1 (ASK-1), a serine/threonine protein kinase (a member of the MAPK kinase kinase family) that activates both p38 and JNK pathways (Matsukawa et al., 2004). ASK1 is activated in response to various exogenous and endogenous cellular stresses and mediates cell apoptosis. In resting cells, ASK-1 binds to the repressor protein thioredoxin. This binding is dependent on the presence of a reduced form of an intramolecular disulfide bridge between two cysteine residues of thioredoxin. Upon oxidation of thioredoxin by ROS molecules such as H2O2, thioredoxin dissociates from and liberates ASK-1, which is then activated by oligomerization and threonine autophosphorylation (Matsukawa et al., 2004). A direct link between ASK-1 and Nox has been revealed by several studies. In a human astrocyte cell line, it was shown that ROS production from Nox4 induced activation of ASK-1 after bacterial LPS treatment, which was important in mediating subsequent p38 activation and altered gene expression (Chiang et al., 2006). Likewise, Van Laethem et al. (2006) reported that UVB irradiation elicited ROS generation and ASK-1 activation in human keratinocytes; inhibition of Nox prevented UVB-induced activation of ASK-1 and subsequent p38 phosphorylation and apoptosis. Moreover, ASK-1 overexpression sensitized the keratinocytes to UVB-induced apoptosis (Van Laethem et al., 2006). Similar observations were also obtained in alveolar macrophages (Liu et al., 2006). It was found that ADP-induced ROS generation via Nox resulted in dissociation of ASK-1 from thioredoxin, and the activation of ASK-1 was enhanced by an inhibitor of thioredoxin reductase (Liu et al., 2006). These results together suggest that ASK-1 is an important effector of Nox in the redox signaling involved in cellular stress responses (Fig. 1).

Signaling network of NADPH oxidase-dependent redox modulation during cellular stress responses. Depending on the cell type and specific stress conditions, these signaling pathways may result in cellular adaptation and tolerance to the stress, or cell apoptosis. Trx, thioredoxin.
B. Protein Tyrosine Phosphatases
Protein tyrosine phosphatases (PTPs) are another family of intracellular targets of Nox-derived ROS. For example, in adipose cells, overexpression of the wild-type Nox4 inhibited PTP1B catalytic activity and reversed the inhibitory effects of ectopically expressed PTP1B on insulin-stimulated glucose uptake (Mahadev et al., 2004). In pancreatic cancer cells, the activity of a low-molecular-weight PTP has been shown to be negatively regulated by Nox4 (Lee et al., 2007). Similar effects of Nox1 on low-molecular-weight PTP activity has also been reported in rat kidney fibroblasts (Shinohara et al., 2007). Another example of PTP that is inhibited by Nox-derived ROS is SHP-1 and -2. In lymphocytes, Nox5 seemed to have a major role in ROS production, and inhibition of Nox function resulted in an increase in SHP-1 activity (Kamiguti et al., 2005). Moreover, there is evidence that in cardiac fibroblasts, SHP-2 was transiently oxidized during Nox activation (Chen et al., 2006a). In addition, the activity of the dual lipid/tyrosine phosphatase PTEN (phosphatase and tensin homolog) is also influenced by Nox. Overexpression of Nox1 in fibroblasts induced a transient inactivation of PTEN as a result of cysteine oxidation and formation of a disulfide bond between the Cys124 and Cys71 in the catalytic domain (Kwon et al., 2004).
PTPs have an essential role in down-regulating receptor tyrosine kinase (RTK)-mediated signaling by dephosphorylating RTKs. Enhanced RTK tyrosine phosphorylation may lead to activation of the ERK pathway. Indeed, activation of ERK1/2 by NADPH-derived ROS has been reported in a variety of cells, including vascular smooth muscle cells (Bhunia et al., 1997) and endothelial cells (Datla et al., 2007), fibroblasts (Go et al., 2004), kidney mesangial cells (Gorin et al., 2003; Wagner et al., 2007), and pulmonary epithelial cells (Ranjan et al., 2006; Amara et al., 2007), although mechanisms other than PTP inactivation may also be involved in mediating these effects of ROS. Based on these data, we suggest that modulation of RTK signaling via PTP inactivation by Nox-derived ROS may have a significant contribution in mediating the activation of ERK MAPK pathway observed in various cellular stress responses (Fig. 1). Apart from the ERK pathway, some evidence indicates that PTPs may also be involved in modulating the activation of p38 MAPK and JNK. Saxena et al. (1999) have shown in T lymphocytes that the PTP member hematopoietic protein-tyrosine phosphatase (HePTP) negatively regulates the activities of ERK and p38, but not JNK, via direct interactions with these kinases, leading to a suppression of the antigen receptor signaling during the early phase of T-cell activation. On the other hand, Lee and Esselman (2002) reported that stimulation of Jurkat T cells with hydrogen peroxide induced phosphorylation of ERK, p38, and JNK, which was accompanied by inhibition of the activities of PTPs CD45, SHP-1, and HePTP. They also demonstrated that JNK activation might be triggered by SHP-1 inactivation, whereas HePTP might be responsible for ROS-induced p38 activation.
A significant feature of the mode of modulation of RTK signaling by Nox is signal divergence. It is clear that Nox activation may potentially affect the activities of multiple PTPs. Subsequently, a single PTP member may be involved in modulating diverse RTK-mediated pathways. For example, it has been shown that PTP1B is implicated in modulating signaling events mediated by receptors of epidermal growth factor, PDGF, and insulin (Liu and Chernoff, 1997; Haj et al., 2003; Shi et al., 2004). Several studies have provided evidence suggesting that a single member of Nox can modulate multiple RTK pathways. In vascular endothelial cells, we recently found that overexpression of Nox4 enhanced the angiogenic responses induced by vascular endothelial growth factor (Datla et al., 2007). Likewise, Nox4 has also been shown to enhance PDGF-induced proliferation in the kidney mesangial precursor cells, insulin-induced glucose uptake in the adipose cells, and insulin-like growth factor-1-induced cell survival in pancreatic cancer cells (Mahadev et al., 2004; Lee et al., 2007; Wagner et al., 2007). Although these results were obtained in different cell types, these observations nonetheless suggest that the signal-diverging effect of Nox in cellular stress responses might have a pivotal role in orchestrating the functions of multiple RTK pathways, which might be collectively necessary for maintaining sufficient strength and duration of downstream signalings such as MAPK activation. Therefore, Nox enzymes may represent an important intracellular signal amplifier for the initial cellular stress stimulus.
C. Dual Specificity Mitogen-Activated Protein kinase Phosphatases
Activation of the MAPK pathways is also tightly regulated by a family of dual-specificity MAPK phosphatases (DS-MKPs) (Camps et al., 2000; Farooq and Zhou, 2004). Maximum MAPK activation requires phosphorylation on both a threonine and a tyrosine residue located in the consensus motif—pTXpY—within the activation loop of the kinase domain. DS-MKPs are encoded by a subclass of the PTP gene superfamily. DS-MKPs can directly interact with MAPKs and dephosphorylate both of the essential phosphothreonine and phosphotyrosine residues, resulting in inactivation of MAPKs. DS-MKPs provide a negative feedback regulatory mechanism that prevents sustained activation of MAPKs, even in the continued presence of activating stimuli (Camps et al., 2000; Farooq and Zhou, 2004). Thirteen mammalian DS-MKPs have been identified that exhibit differential substrate specificity over ERK, p38, or JNK (see Farooq and Zhou, 2004). Several studies have shown that the activities of many DS-MKPs are sensitive to redox modulation. For example, by using recombinant enzymes, people have shown that the DS-MKP vaccinia H1-related was rapidly inactivated by H2O2 at low micromolar concentrations (Denu and Tanner, 1998). Similar inactivation of DS-MKP activity by H2O2 has also been observed with MKP-3 (Seth and Rudolph, 2006). Hydrogen peroxide rapidly and reversibly inactivates MKP-3 by forming a sulfenic acid intermediate on the active site cysteine (Seth and Rudolph, 2006). Experimental evidence also suggests that ROS-mediated modulation of DS-MKPs may be an important mechanism underlying the enhanced MAPK activation under oxidative conditions. Hou et al. (2008) demonstrated that low-glucose challenge in pancreatic β cells induced a sustained activation of JNK and subsequent apoptosis that was associated with MKP-1 oxidation. In addition, ROS scavengers or DS-MKP overexpression prevented JNK activation and cell death. In hepatocytes, ethanol treatment induced ROS production, sustained JNK activation, and increased cell apoptosis (Venugopal et al., 2007). It is noteworthy that these authors reported that ROS triggered a remarkable MKP-1 degradation via PKC-dependent mechanisms, although the MKP-1 activity was not directly assessed. So far, there is no information about the role of Nox in modulating DS-MKP activities, and further investigations in this direction may provide novel insights into the mechanisms by which Nox mediates cell stress responses.
D. Phosphoinositide 3-Kinase/Akt Pathway
The antiapoptotic kinase Akt is another downstream target of the Nox signaling, as evidenced by a number of studies showing that Nox-derived ROS may induce Akt activation in a variety of cells (Mahadev et al., 2001; Gorin et al., 2003; Chen et al., 2006b; Hingtgen et al., 2006; Wagner et al., 2007; Wang et al., 2007b; Mofarrahi et al., 2008). This effect of Nox-derived ROS on Akt may be the result of PTP inactivation and enhanced RTK signaling, because the phosphoinositide 3-kinase (PI3K)/Akt pathway is a proximal downstream target of RTKs (Fayard et al., 2005). Alternatively, Nox-derived ROS may enhance Akt activation by inactivating the phosphatase PTEN, which is a negative regulator of the PI3K activity. There is evidence that H2O2 accumulation in response to peptide growth factors in cells expressing Nox can transiently inactivate PTEN as a result of cysteine oxidation, leading to enhanced activation of the PI3K/Akt pathway (Kwon et al., 2004). Moreover, Seo et al. (2005) provided evidence showing that in neuroblastoma cells, Nox inhibition diminished insulin-induced PI3K activation and Akt phosphorylation, suggesting that insulin-induced Akt activation in these cells required a concomitant inhibition of PTEN by Nox-derived ROS to achieve a maximal response.
IV. Cell Survival versus Apoptosis
It is intriguing that activation of Nox is involved in distinct pathways that seem to mediate two opposite cellular responses, namely cell survival/growth versus apoptosis. That Nox can act as a double-edged sword in determining cell fate is well evidenced by studies in the kidney, where Nox promotes growth of certain types of cells but triggers apoptosis in others (Jiang, 2009). How Nox preferentially transduces one signal pathway over another is poorly understood. It is conceivable that this is likely to be determined by both extrinsic (e.g., the nature and strength of the stimulus) and intrinsic factors (e.g., the predominant Nox isoforms expressed in a particular cell type, and the differential availability of pro- and antiapoptotic kinases).
It has been shown in vascular endothelial cells that atherogenic remnant lipoprotein particles, the inflammatory cytokine TNF-α, and homocysteine all induced endothelial apoptosis in a Nox-dependent manner (Shin et al., 2004; Dong et al., 2005a; Basuroy et al., 2009); on the other hand, activation of Nox by the endogenous cytochrome P450 product hydroxyeicosatetraenoic acid or the endothelium-derived vasoactive peptide endothelin-1 protected against cell apoptosis during serum starvation (Dong et al., 2005b; Dhanasekaran et al., 2009). Likewise, UV irradiation-induced activation of Nox in skin keratinocytes was associated with increased apoptosis (Van Laethem et al., 2006); in contrast, Rygiel et al. (2008) provided evidence that deletion of the guanine nucleotide exchange factor of Tiam1 (activator of Rac GTPase) impaired Nox function, decreased ERK phosphorylation, and increased keratinocyte apoptosis after growth factor deprivation or heat shock, suggesting that normal Nox function was critical in maintaining keratinocyte survival. It is noteworthy that in these situations, the amount of ROS generated by the activated Nox may have a critical role in determining the eventual cellular outcome. It is proposed that temporally and spatially regulated ROS generation is essential for the specificity of redox signaling, whereas prolonged ROS generation in a diffused manner may act in favor of the apoptotic pathway by inducing nonspecific oxidative damages (Terada, 2006). It is suggested that the balance between the amount and location of ROS generation and the availability of antioxidant enzymes that can confine the ROS within specific subcellular compartments may be a determinant in specifying the cellular outcomes after Nox activation during cellular stresses (Terada, 2006).
Differential expression of various Nox isoforms may also contribute to the disparate effects of this enzyme on cell viability. For example, Nox2 and Nox4 are often coexpressed in a variety of nonphagocytic cells of a mesoderm origin, and both of them contribute to cellular ROS production. However, some studies have suggested that these two isoforms have distinct features in terms of the expression level, subcellular localization and the ROS species generated, although the evidence is still incomplete (Van Buul et al., 2005; Martyn et al., 2006; Petry et al., 2006). Indeed, a study by Anilkumar et al. (2008) demonstrated that Nox4 and Nox2 mediated distinct effects on the MAPK and Akt pathways in transfected HEK293 cells. Specifically, Nox4 overexpression induced basal activation of ERK1/2 and JNK, whereas Nox2-transfected cells showed a modest increase in p38 activation. Moreover, insulin stimulation increased the phosphorylation of p38 and Akt only in Nox4-expressing cells, whereas JNK activation was specifically observed in Nox2-expressing cells (Anilkumar et al., 2008). Finally, differences in susceptibilities of the pro-and antiapoptotic kinase pathways within the cell to redox modulation may also have a role in determining the cell fate after Nox activation in a stress response. In a study in mouse embryonic fibroblasts, Dolado et al. (2007) demonstrated that tumorigenic transformation of the cells by expressing a constitutively active form of Ras rendered the cells resistant to ROS-mediated p38 activation and apoptosis compared with nontransformed cells; this seemed to be caused by up-regulation of an ASK1-binding protein, glutathione transferase μ-1, which was proposed to inhibit ROS-induced ASK-1 activation by retaining the binding of ASK-1 with its inhibitor thioredoxin. Such a mechanism that uncouples p38 activation from ROS production may potentially bypass the proapoptotic effect of Nox, leaving the prosurvival arm of this enzyme intact. It remains to be determined, however, whether similar mechanisms might be involved in the bidirectional regulation of cell functions by Nox during stress responses. It is noteworthy that induction of apoptosis via Nox activation in irreversibly damaged cells may have fundamental importance at the whole organism level, in that this may substantially reduce the chance of tumorigenesis (Van Laethem et al., 2006; Dolado et al., 2007).
V. Mechanisms of NADPH Oxidase Activation
A. Up-Regulation of Expression
If Nox can act as an intracellular stress transducer to signal cell stress responses, then how Nox system is activated by diverse stress stimuli needs to be addressed. One mechanism is by up-regulation of expression of the enzyme. Increased expression of Nox subunits may occur at the transcriptional or post-transcriptional levels, as evidenced by the numerous studies using mRNA as the endpoint measurement. On the other hand, Peshavariya et al. (2009b) recently showed that Nox expression could also be modulated at the translational level, at least in vitro. Several transcription factors and their cognate binding sites have been identified in the promoter sequences of mammalian Nox subunit genes, among which those responsive to inflammatory cytokines are well documented. In both phagocytic and nonphagocytic cells, there is evidence that Nox2 transcription is regulated by the interferon-γ/Janus tyrosine kinase/signal transducer and activator of transcription pathway, and the consensus signal transducer and activator of transcription 1 binding site, γ-activated sequence, located in the Nox2 promoter, has been shown to be essential in mediating interferon-γ-induced Nox2 transcription (Kumatori et al., 2002; Manea et al., 2010). The γ-activated sequence motif has also been identified in Nox1, Nox4, p22phox, p47phox, and p67phox promoters and may be involved in interferon-γ modulation of the transcription of these subunits (Kuwano et al., 2006; Moriwaki et al., 2006; Manea et al., 2010). AP-1 is a well characterized transcription factor that is activated in response to a large range of stimuli and has a critical role in modulating gene expression under cellular stress conditions (Wisdom, 1999). Binding sites for AP-1 have been found in the promoters of Nox1, p22phox, NOXO1, and p67phox (Gauss et al., 2002; Cevik et al., 2008; Kuwano et al., 2008; Manea et al., 2008). Introduction of mutations at the AP-1 site consistently abolished prostanoid-induced transcriptional activation of the Nox1 promoter in a luciferase assay in vascular smooth muscle cells (Cevik et al., 2008). Several lines of study have shown that Nox1 gene transcription in nonphagocytic cells is modulated by members of the GATA family transcription factors, especially GATA-6 (Brewer et al., 2006; Adachi et al., 2008; Valente et al., 2008). Moreover, there is evidence that Nox4 transcription in smooth muscle cells is modulated by the transcription factor E2F, a critical protein in driving cell cycle progression and promoting cell proliferation (Zhang et al., 2008). However, the role of GATA- and E2F-regulated Nox expression during cellular stress remains unclear.
B. Protein Kinase C-Mediated Phosphorylation
As mentioned above, many stress stimuli can acutely activate Nox enzyme activity before alterations in gene expression. Our understanding of the mechanisms by which cellular stresses activate Nox remains incomplete, although it is clear that different stresses can ultimately result in the hierarchical activation of stress kinases such as p38 and JNK, which are highly conserved stress signal transducers in all eukaryotic cells (Kyriakis and Avruch, 2001). In phagocytic cells, PKC-mediated p47phox phosphorylation is essential for Nox activation (for review, see Babior, 1999). In contrast to phagocytic cells, the mechanism(s) of Nox activation in nonphagocytic cells is still poorly understood. It was reported that angiotensin II stimulation in endothelial cells promoted p47phox phosphorylation and further enhanced the oxidase activity (Li and Shah, 2003). Quagliaro et al. (2003) demonstrated that intermittent high-glucose challenge in endothelial cells resulted in Nox activation that was sensitive to PKC inhibitors. In cultured rat astrocytes, hypo-osmotic cell swelling induced rapid Nox activation accompanied by serine phosphorylation of p47phox; this response was blocked by a PKC inhibitor (Reinehr et al., 2007). It was also demonstrated that exposure of mouse cortical brain slices to hypo-osmosis stimulated ROS production, which was absent in brain slices from p47phox knockout animals, suggesting that PKC-induced p47phox phosphorylation had a pivotal role in osmotic stress-induced Nox activation in astrocytes (Reinehr et al., 2007). It is noteworthy that several lines of study have demonstrated that PKC activation can be observed after exposing cells to a variety of stress stimuli (for example, cytotoxic chemicals, physical stretch, osmotic shock, hypoxia, and inflammatory cytokines) (Kugiyama et al., 1992; Rzymkiewicz et al., 1996; Goldberg et al., 1997; Liu and Huang, 1997; Schanne et al., 1997; Wyatt et al., 1997; Deng and Poretz, 2002; Motley et al., 2002; Suzuma et al., 2002; Yeon et al., 2002; Liu et al., 2003), and PKC may be viewed as a stress sensor in some circumstances (Barnett et al., 2007). If PKC is indeed involved in Nox activation in acute stress responses, the specific roles of different PKC isoforms need to be defined.
C. Intracellular Ca2+
Elevation of the intracellular Ca2+ concentration may also act as an upstream signal to activate Nox during cellular stresses. As with PKC, increased intracellular Ca2+ has been observed in cells exposed to cytotoxic chemicals (Stoll and Spector, 1993; Liu and Huang, 1997; Yeh et al., 2007; Pourrut et al., 2008), osmotic challenges (Cazalé et al., 1998; Yellowley et al., 2002), physical stretches (Davis et al., 1992; Sokabe et al., 1997), hypoxia (Berna et al., 2001), and inflammatory mediators (Chen et al., 1997; García et al., 1999). It is noteworthy that an increase in intracellular Ca2+ can be a secondary response after PKC activation and vice versa (Schanne et al., 1997; Barnett et al., 2007). The Nox5 isoform contains multiple EF hand Ca2+ binding domains in the N-terminal region, allowing its activation through calcium sensing (Bánfi et al., 2001). It was shown that calcium binding caused a marked conformational change in the N terminus of Nox5, resulting in a direct interaction between the regulatory N terminus and the catalytic C terminus; this intramolecular interaction was likely to be responsible for Ca2+-induced Nox5 activation (Bánfi et al., 2004).
Plant cells exclusively express Nox5-like isoforms (Bedard et al., 2007). Using in vivo Ca2+ imaging, several lines of study have shown that activation of Nox by cellular stresses in plant cells is accompanied by Ca2+ entry into the cells (Kadota et al., 2004; Garnier et al., 2006; Yeh et al., 2007). At the functional level, blockade of Ca2+ entry prevented Nox activation induced by various stresses (Jiang and Zhang, 2003; Olmos et al., 2003; Balestrasse et al., 2008; Pourrut et al., 2008). Moreover, it was shown that direct treatment with Ca2+ in plant cells could stimulate Nox activity and superoxide production (Jiang and Zhang, 2003). The above evidence collectively indicates that activation of the plant Nox isoforms is likely to be regulated by Ca2+ binding, although further studies are required to directly confirm this mechanism. It was shown that the plant Nox isoform RbohB could be phosphorylated by the calcium-dependent protein kinases CDPK4 and CDPK5 at serines 82 and 97 in a calcium-dependent manner; moreover, the lost function of RbohB after gene knockdown was reconstituted by the wild-type RbohB, but not by a S82A/S97A mutant, suggesting that RbohB activation was dependent on Ca2+-induced phosphorylation but independent of Ca2+ binding to the EF hand domains (Kobayashi et al., 2007). More recently, Ogasawara et al. (2008) demonstrated that ROS production from RbohD in plant cells induced by Ca2+ influx required a conformational change in the EF hand region as a result of Ca2+ binding. These authors also demonstrated that RbohD was phosphorylated in response to Ca2+, and inhibition of protein phosphatases significantly enhanced the Ca2+-induced ROS production, suggesting that Ca2+ binding and phosphorylation had synergistic effects in activating the plant Nox. It is noteworthy that some studies have indicated that there might be a feed-forward mechanism in regulation of the Nox5 function by calcium in plant cells. Evidence has shown that, apart from the Ca2+-dependent Nox activation, Nox-derived ROS can in turn promote Ca2+ entry into the cells (Foreman et al., 2003; Mori and Schroeder, 2004; Yang et al., 2007b). Animal Nox5 is found in fetal tissues, T and B lymphocytes, and sperm precursors in testis (Bedard and Krause, 2007). In contrast to the plant Nox isoforms, the role of Nox5 in cellular stress responses in animal cells is poorly understood. However, the feed-forward mechanism in Ca2+-mediated regulation of Nox5 function has also been observed in human cells, where hydrogen peroxide promotes the influx of calcium, and this calcium influx subsequently stimulates the activity of Nox5 and generation of ROS (El Jamali et al., 2008).
Activation of conventional PKC isoforms α, β, and γ is Ca2+-dependent; thus, Ca2+ influx and PKC activation may be inter-related in the process of Nox activation. On the other hand, calcium and PKC may work together to activate the small GPTase Rac. Rac is activated in response to various stimuli and has been implicated in Nox activation and ROS generation in response to cellular stresses in both plant and animal cells (Park et al., 2000; Kim et al., 2007). Increase in intracellular Ca2+ activates Rac in a PKC-dependent manner (Price et al., 2003). However, no direct evidence shows that the Ca2+-PKC-Rac pathway is involved in Nox activation in either plant or animal cells. It is noteworthy that Wong et al. (2007) reported that in plant cells, there was a physical interaction between Rac and the N-terminal region of Rboh containing the EF-hand motifs. In addition, this direct Rac-Rboh interaction seemed to be regulated by the intracellular Ca2+ concentration and be important for Nox activation (Wong et al., 2007). Taken together, it is suggested that PKC and calcium may be the master regulators of Nox function during stress responses (Fig. 2).

Ca2+ influx and PKC activation may have pivotal roles in stimulating NADPH oxidase activity during cellular stress responses. Stress stimuli may trigger Ca2+ entry via membrane Ca2+ channels and activate PKC by mechanisms that are not entirely understood. Ca2+ entry via membrane ion channels and PKC activation may reciprocally modulate each other. PKC-mediated phosphorylation of p47phox has an essential role in activating the electron transport functions of Nox members, such as Nox2, that rely on cytosolic regulatory subunits. Ca2+ and PKC may also work together to activate NADPH oxidase by stimulating the small GPTase Rac. Certain Nox members such as Nox5 and Duox have Ca2+ binding domains and can be directly activated by Ca2+. NADPH oxidase-derived ROS may further elevate intracellular Ca2+ by enhancing the membrane ion channel activity and/or promoting Ca2+ release from intracellular stores. The activity of Nox members that do not require cytosolic subunits, for example Nox4, may be regulated at the transcriptional level. Moreover, NADPH oxidase may also be activated via direct protein-protein interactions with the adaptor proteins involved in Toll-like receptor and tumor necrosis factor receptor (TNFR) signaling.
D. NADPH Oxidase Activation by Direct Protein-Protein Interactions
Several lines of experiments have suggested that the enzymatic activity of Nox members can be stimulated by direct protein-protein interactions. In HEK293 cells ectopically expressing the Toll-like receptor (TLR) 4 and accessory proteins, Park et al. (2004) provided evidence that stimulation with LPS induced direct association of TLR4 and Nox4, which further triggered ROS production and nuclear factor-κB activation. Further, it was shown that TLR4-Nox4 interaction was mediated via the TIR domain of TLR4 and the C-terminal region of Nox4. However, it was noted that the TIR domain in TLR4 bound to other signaling molecules such as MyD88 and IRAK (Park et al., 2004); thus, it was not clear whether the TLR4-Nox4 interaction was direct or indirect. More recently, another group demonstrated a similar physical interaction between Nox2 and TLR2 in phagocytic cells and showed this interaction essential for efficient innate immune responses of macrophages to mycobacteria infection (Yang et al., 2009). TNF-receptor-associated factor (TRAF) family proteins are essentially involved in transducing signals for Toll-like, TNF, and interleukin-1 receptors; and there is evidence that Nox subunits may bind to TRAFs. In human microvascular endothelial cells, TNF-α induced association of phosphorylated p47phox with TRAF4, which was required for ROS-dependent ERK1/2 activation (Li et al., 2005). In B cells, it was shown that stimulation of CD40 receptors triggered association of TRAF3 with p40phox, a regulatory cytosolic subunit found in phagocytic cells (Ha and Lee, 2004). Moreover, overexpression of a dominant-negative form of TRAF3 blunted ROS production in response to CD40 stimulation. However, there is no evidence of interactions between TRAF proteins and the Nox subunits.
The mechanisms by which Nox is activated after the interactions with membrane receptors or associated signaling proteins are largely unknown. Yazdanpanah et al. (2009) presented evidence showing that riboflavin kinase might have an integral role in coupling the recruitment of Nox and p22phox subunits to the TNF receptor complex and subsequent activation of the enzymatic function of Nox in HeLa cells. Riboflavin kinase converts riboflavin to FMN and FAD, of which the latter is the cofactor of Nox required for the electron transfer function of Nox subunits. The data of Yazdanpanah et al. (2009) suggested that TNF-α-induced physical interaction between TNF receptor and riboflavin kinase was mediated by the adaptor protein TRADD, whereas the riboflavin kinase bound to Nox via p22phox. It is proposed that TNF-induced Nox activation relies on riboflavin kinase-dependent FAD generation near the vicinity of the receptor-Nox complex. These results present a novel mechanism of acute Nox activation by physical interactions with membrane receptors, although this needs to be further confirmed in other experimental systems.
E. NADPH Oxidase Activation by Reactive Oxygen Species Molecules
It is noted that there is evidence showing that Nox, although a ROS producer itself, can be activated in turn by ROS molecules. In vascular smooth muscle cells and fibroblasts, Li et al. (2001) have demonstrated that exogenous hydrogen peroxide acutely activated cellular superoxide production via Nox. Similar effects of hydrogen peroxide were also observed in isolated mouse pulmonary arteries (Rathore et al., 2008). It is noteworthy that the latter study further revealed that redox-dependent regulation of the Nox activity was functionally important in transducing stress signals. In the pulmonary artery preparation, acute hypoxia for 5 min increased Nox activity, which was attenuated after inhibition of the mitochondrial ROS generation with rotenone or myxothiazol, suggesting that, like exogenously applied hydrogen peroxide, ROS from endogenous sources could also activate the Nox enzymes. Moreover, pharmacological and genetic inhibition of Nox blocked hypoxia-induced increase in intracellular Ca2+ in pulmonary artery smooth muscle cells (Rathore et al., 2008). In addition to these observations, ROS-mediated modulation of Nox function was also demonstrated in plant cells, showing that in plant root hair cells, Nox-derived ROS induced Ca2+ influx into the cytoplasm, and this Ca2+ influx, in turn, further enhanced the activity of Nox (Takeda et al., 2008).
VI. NADPH Oxidase and the Stress Response Signaling Paradigm
A. The Cellular Stress Response Paradigm
As mentioned above, the signaling mechanisms underlying the cellular response to various exogenous stresses are evolutionarily conserved (Kültz, 2005). Some features of the activators and effectors of the Nox system satisfy the general characteristics of cellular stress response paradigm. First, cellular stress response lacks stress specificity (i.e., a conserved set of responses can be induced by diverse stress stimuli, exemplified by the induction of heat shock proteins in different stress conditions). This feature is perfectly resembled by the scenario of Nox activation, which can be triggered by various forms of cellular stresses, as discussed in section II, although in other situations not necessarily related to cellular stress, specific Nox proteins seem to be activated in a highly controlled and specific manner, different for each Nox isoform. Second, in many cells, stimulation of the Nox activity results in activation of MAPK/JNK pathways, the core of the cell stress signaling network (Kyriakis and Avruch, 2001). MAPKs are widely involved in modulating fundamental biological functions, including gene expression, cell cycle control, cell survival and apoptosis, cell motility, and differentiation. Using a functional genomic approach, Nahm et al. (2002) reported that exposure of the kidney medullary epithelial cells to hypertonicity induced significant up-regulation of 12 genes, many of which were stress response genes. It is noteworthy that this transcriptional program in response to stress was blunted by inhibitors of p38 and/or ERK MAPKs, suggesting that MAPKs had a pivotal role in mediating stress-induced cellular reactions. Likewise, the stress kinase JNK is also tightly involved in modulating cell proliferation, gene expression, and apoptosis during stress responses (Leppä and Bohmann, 1999). Although the MAPKs pathways have been recognized as an essential cellular stress signaling module, the mechanisms that couple extrinsic stress stimuli to the activation of MAPKs are poorly understood (Kyriakis and Avruch, 2001). We propose that Nox may represent a signal transducer between the two ends.
B. NADPH Oxidase-Mediated Stress Tolerance
Another important feature of the general cell stress response is the preconditioning phenomenon induced by mild stress stimuli; i.e., prior exposure to a nonlethal stress challenge can result in an increased tolerance to stresses of the same type (so called stress-hardening) or others (cross-tolerance) (Kültz, 2005). Stress hardening may be best exemplified by the well studied phenomenon of ischemic preconditioning in the heart, and evidence has indeed suggested that ROS-mediated redox mechanisms may have a central role in mediating the preconditioning effects (Penna et al., 2009). It is noteworthy that a number of studies have revealed that Nox enzymes may have a significant role in stress-induced preconditioning. For instance, in Langendorff perfused hearts, ischemic preconditioning significantly attenuated the infarct size in wild-type but not Nox-deficient tissues. Furthermore, inhibition of PKC completely abrogated the Nox activation and the preconditioning effect induced by ischemia, suggesting a pivotal role of PKC-dependent Nox activation in mediating the stress tolerance (Bell et al., 2005). In myocardium, administration of angiotensin II exhibited significant preconditioning effects; inhibition of Nox activity blunted angiotensin II-induced preconditioning against myocardial ischemia-reperfusion injury in both in vitro and in vivo experiments (Kimura et al., 2005; Das et al., 2006a). Moreover, this Nox-mediated protective effect seemed to involve activation of the MAPK pathways (Kimura et al., 2005; Das et al., 2006b). In addition, inhibition of Nox activity in vivo also blocked the preconditioning effects induced by physical exercise or tachycardia against myocardial infarction (Sánchez et al., 2008).
The involvement of Nox in stress-induced tolerance is not limited to cardiomyocytes. Kim et al. (2009) reported that exposure to a low level of ionic irradiation protected the kidney from later ischemia-reperfusion injury. The irradiation-induced preconditioning was accompanied by Nox activation and was blocked by superoxide scavengers. In perfused liver, it was shown that pretreatment with H2O2 reduced ischemia-reperfusion–triggered hepatocyte injuries, an effect that was abolished by Nox inhibition (Tejima et al., 2007). Moreover, it has been demonstrated that preconditioning with a systemic administration of alcohol or LPS induced stress tolerance in the brain against neuroexcitotoxicity– or ischemia-reperfusion–mediated injuries; importantly, these preconditioning effects were lost in Nox2-deficient animals or after Nox inhibition (Kawano et al., 2007; Kunz et al., 2007; Wang et al., 2007a). It is noteworthy that similar findings on the importance of Nox in the development of cross-tolerance to cellular stresses have also been obtained in plant cells (Pastori and Foyer, 2002). Taken together, it is suggested that the Nox enzymes may act as a signal transducer mediating stress-hardening and cross-tolerance during cell stress responses.
C. Role of NADPH Oxidase in Endoplasmic Reticulum and Genomic Stresses
Under normal conditions, BiP/Grp78 is located in the ER lumen and is bound to the ER-residing proteins PRKR-like endoplasmic reticulum kinase, inositol requiring enzyme 1α (IRE1α), and the transcription factor ATF6, which are the core mediators of ER stress response. Upon ER stress, BiP/Grp78 is displaced by unfolded proteins, and the subsequent release of PRKR-like endoplasmic reticulum kinase, IRE1α, and ATF6 induces global deceleration of protein biosynthesis and expression of several ER stress-related chaperones. Moreover, activation of ATF6 induces up-regulation of the chaperone protein CHOP, which is implicated in ER stress-associated apoptosis (Lin et al., 2008; Simmons et al., 2009). There is evidence that ER stress is accompanied by ROS production; and this redox process has an integral role in modulating both of the proadaptive and proapoptotic effects of ER stress (Santos et al., 2009). It is noteworthy that several studies have shown that Nox is activated by ER stress and is involved in modulating ER stress signaling. In human aortic smooth muscle cells, the cytotoxic lipid derivative 7-ketocholesterol up-regulated Nox4 expression and induced ER stress, as evidenced by the expression of Bip/GRP78 and CHOP via IRE1 activation, whereas silencing of IRE1 or inhibition of JNK normalized Nox4 expression, suggesting that 7-ketocholesterol promoted Nox4 expression through the IRE1/JNK/AP-1 signaling pathway (Pedruzzi et al., 2004). Moreover, the authors demonstrated that silencing of Nox4 expression with siRNA significantly reduced the 7-ketocholesterol-induced ROS production and abolished the apoptotic response. It has been shown that endothelial cells exhibited ER-specific production of H2O2 after treatment with the ER stress inducers tunicamycin and the viral protein Tat (Wu et al., 2010). Knocking down of Nox4 or expression of ER-targeted catalase blocked the ER-specific H2O2 production and the ER stress response (expression of Bip/GRP78 and CHOP). ER stressors also triggered Nox4-dependent activation of Ras, leading to sustained ERK pathway activation. It is noteworthy that the Nox4/Ras/ERK pathway provoked cell autophagy, which prevented ER stress-induced cell death, suggesting that Nox4-dependent redox process represented a homeostatic protective mechanism rather than oxidative stress during endothelial ER stress (Wu et al., 2010). In addition to the above studies in cell culture, the involvement of Nox in ER stress response has also been demonstrated in vivo. In a diabetic model, it was found that deficiency of the NADPH oxidase component Rac1 or systemic administration of the Nox inhibitor apocynin suppressed the expression of ER stress markers in the myocardium (Li et al., 2010).
Genotoxic agents and other factors such as irradiation cause genomic stress (also called genotoxic stress) by inducing alterations of the genetic material, mainly the genomic DNA, in the affected cells. Several serine/threonine protein kinases (including DNA-dependent protein kinase, ataxia telangiectasia mutated, and ataxia telangiectasia and Rad3 related) have been identified as the intracellular sensor for genomic stress; activation of these kinases initiates complex signaling events leading to cell cycle arrest or apoptosis, allowing the damaged DNA to be repaired and preventing propagation of the genomic defects (Yang et al., 2003; Coates et al., 2005). During genomic stress, phosphorylation and activation of the transcription factor p53 have a pivotal role in orchestrating the cellular response. Once activated, p53 regulates the expression of a variety of target genes involved in DNA repair, cell cycle regulation, and apoptosis (Simmons et al., 2009). Genomic stress is associated with increased ROS generation, and the majority of evidence points to a role of Nox and ROS in triggering DNA damage (Lynn et al., 2000; Chiera et al., 2008; Herbert et al., 2008; Wang et al., 2010). In contrast, studies into the role of Nox/ROS in transducing genomic stress signals are sparse. It is noteworthy that ERK, p38 and JNK are all activated by various genotoxic stresses and can phosphorylate p53 (Yang et al., 2003; Coates et al., 2005). Therefore, it is probable that Nox-derived ROS may be involved in the activation of these stress kinases, whereas it is likely that in this case, Nox is not an effector of the genomic stress sensors but is activated by the initial DNA damage inducers such as UV irradiation. In a pharmacological study on an anticancer agent 6-((2-(dimethylamino)ethyl)amino)-3-hydroxy-7H-indeno(2,1-c)quinolin-7-one dihydrochloride (TAS-103), a topoisomerase inhibitor, Mizutani et al. (2002) reported that TAS-103-induced DNA damage in tumor cell lines was ROS-independent, whereas TAS-103-induced apoptosis required ROS, suggesting that redox signaling mechanisms were involved in the genotoxic stress-triggered cell death. Moreover, Nox inhibition was shown to prevent TAS-103-induced apoptosis. It is noteworthy that inhibitors of poly(ADP-ribose) polymerase, an enzyme involved in DNA repair, also prevented both TAS-103-induced ROS generation and apoptosis. The authors proposed that TAS-103-induced DNA damage resulted in poly(ADP-ribose) polymerase hyperactivation, and the subsequent NAD+ depletion might contribute to the activation of Nox; the enhanced ROS generation was then involved in mediating apoptosis (Mizutani et al., 2002). However, the significance of this mechanism in the general genomic stress signaling network is uncertain.
D. Cross-Talk between Mitochondria and NADPH Oxidase in Stress Response
Mitochondria are essential for cellular energy homeostasis, intracellular Ca2+ regulation, thermogenesis, and apoptosis. Like ER, this organelle is also involved in dealing with cellular stresses (Manoli et al., 2007). Moreover, mitochondria represent another major source of ROS in the cells. At the cellular level, mitochondria-associated ROS production, thermogenesis, and apoptosis are thought to serve as protective mechanisms against infection or other cell damages during stress (Manoli et al., 2007). Limited evidence has suggested that mitochondria may have a cross-talk with the Nox system (for review, see Daiber, 2010). In a study in vascular smooth muscle cells, it was observed that induction of mild mitochondrial dysfunction abolished the responsiveness of Nox to angiotensin II, although the baseline hydrogen peroxide production was enhanced (Wosniak et al., 2009). The biological mechanisms of this phenomenon are not clear. It is noteworthy that there is evidence that mitochondrial dysfunction and subsequent Ca2+ release to the cytoplasm may activate multiple signaling pathways, including protein kinase C, MAPKs, and JNK (Manoli et al., 2007). However, the role of Nox in this process remains elusive. In another study, the authors provided more direct evidence showing that hypoxia-induced mitochondrial ROS formation might act as a signal to further stimulate Nox activation and ROS production, which was blocked after PKC inhibition, suggesting that stress-induced mitochondrial dysfunction could activate Nox via PKC (Rathore et al., 2008). A cross-talk between Nox and mitochondria during cellular stress response is further supported by a study in 293T cells (Lee et al., 2006). In this study, it was observed that serum deprivation promoted an acute phase (0–4 h) of ROS production from mitochondria and an additional delayed and sustained ROS production from Nox1. Mitochondrial ROS production occurred within a few minutes in response to serum withdrawal, and this initial ROS release stimulated Nox1 by activating PI3K and Rac1. Moreover, it was found that serum deprivation-induced cell apoptosis was suppressed by either blockers of the mitochondrial electron transfer chain or genetic manipulation of the PI3K/Rac1/Nox1 pathway, suggesting that the maintenance of cellular stress-induced redox signaling required the Nox system, whereas the mitochondria might act as a stress sensor (Lee et al., 2006). Overall, the above data indicate that the Nox system works downstream of mitochondria in stress signaling. Conversely, it is also noted that Nox-derived ROS may contribute to inducing mitochondrial signaling during cellular stress. For example, Nox activation and subsequent ROS release may promote opening of mitochondrial ATP-sensitive potassium channels, leading to depolarization of mitochondrial membrane potential followed by mitochondrial ROS formation and respiratory dysfunction (Daiber, 2010). It is noteworthy that there is evidence to suggest that Nox-mediated mitochondrial ATP-sensitive potassium channel opening may be involved in mediating the stress tolerance in cardiac muscle cells induced by angiotensin II (Kimura et al., 2005).
VII. NADPH Oxidase and Tissue Regeneration
In multicellular organisms, cellular stresses may result in various forms of cell loss and tissue injury. We have reviewed the current evidence suggesting that the Nox-mediated redox signaling may have a pivotal role in coordinating the biological responses at the single cell level in dealing with endogenous and exogenous stresses. At the physiological level, the ability of organisms to repair the damaged tissue and restore the relevant biological functions (i.e., wound healing and tissue regeneration) is also a fundamentally important aspect of biology. In the following sections, we review our current knowledge on the role of Nox-dependent redox mechanisms in wound healing and tissue regeneration.
A. NADPH Oxidase and Wound Healing
Activation of the cell survival pathway by Nox may promote cell adaptation to stresses, whereas Nox may also convey signals toward apoptosis in irreversibly injured cells. Moreover, recent results have suggested that after tissue injuries, NADPH oxidase may participate in the process of wound healing and tissue regeneration (for review, see Chan et al., 2009). After skin wounding, factors involved in hemostasis, coagulation, and inflammation may create a local (stress) environment that favors Nox activation. Nox-derived ROS have been implicated in promoting keratinocyte proliferation (Nakai et al., 2008). Many studies have shown that Nox activation has a positive role in regulating proliferation of collagen-producing cells, including fibroblasts, kidney mesangial cells, and stellate cells of the liver and pancreas (Chan et al., 2009). Deposition of extracellular matrices is a critical component of tissue repair by forming scars. However, this process is often detrimental in parenchymal organs such as liver and kidney, causing fibrosis. Consistent with this, there is evidence that blocking Nox activation or down-regulating Nox expression may prevent the development of fibrosis in the kidney, liver, and heart under stress conditions (Chan et al., 2009).
In human airway epithelial cell monolayer cultures, scraping-generated artificial wounds were closed faster with cells treated with nontoxic concentrations of LPS (Koff et al., 2006). This effect of LPS was inhibited by ROS scavengers, Nox inhibitors, and siRNA against Duox1, suggesting that Nox-dependent redox signaling was involved in this repairing process. In our previous studies, a positive effect of Nox in in vitro cellular wound healing response was also observed in vascular endothelial cells, in which we demonstrated that the Nox4 isoform, but not Nox2, was markedly up-regulated upon scratch wounding of the endothelial monolayer (H. M. Peshavariya, F. Jiang, and G. J. Dusting, unpublished data), whereas Nox4 gene silencing markedly suppressed the wound closure in the cultured cells (Datla et al., 2007; Peshavariya et al., 2009a). Likewise, it was demonstrated that inhibition of Nox with DPI suppressed arsenite-induced wound healing response in monolayers of an immortal mouse endothelial cell line; overexpression of a dominant-negative Rac1 was sufficient to abolish arsenic-induced superoxide-production, indicating a specific involvement of Nox (Qian et al., 2005).
In vivo, hydrogen peroxide is actively generated at the wound site, which may act as a redox signaling molecule to promote wound healing (Sen et al., 2002). In a recent study performed in zebrafish larvae, which expressed a transgene encoding a H2O2-sensitive fluorescent protein as ROS indicator, it was demonstrated that upon local injury of the tail fin, there was a rapid and sustained increase in H2O2 production at the wound margin, which peaked at 20 min after wounding (Niethammer et al., 2009). Moreover, H2O2 production at the wound margin occurred before the recruitment of leukocytes, suggesting that the source of H2O2 was the tail-fin epithelial cells, not leukocytes. This finding contrasts with the prevailing view that the ROS molecules found at the wound site are mainly produced by the inflammatory leukocyte oxidative bursts (Niethammer et al., 2009). Furthermore, these authors demonstrated that Duox1 was the Nox isoform responsible for the early ROS generation after tissue wounding. It was observed that at the maximum of ROS production, an H2O2 gradient was formed around the wound margin, reaching a distance of approximately 100 to 200 μm. It is noteworthy that knockdown of Duox significantly reduced wound-induced H2O2 production and suppressed the consequent recruitment of leukocytes to the wound (Niethammer et al., 2009). These results raise the possibility that ROS molecules may exert signaling actions beyond the single cellular context and may play an important paracrine signaling role during the wound repair process (Niethammer et al., 2009; Wong and Shimamoto, 2009). This notion is supported by another recent study in plants that has shown that upon local tissue wounding or cellular stresses (such as heat, cold, high-intensity light, and salinity stress), ROS accumulated in the extracellular spaces in a Nox-dependent manner (Miller et al., 2009). Moreover, these authors reported that such a focal cellular redox event could travel at a rate of 8.4 cm/min toward remote areas, indicating a continuous long-distance, cell-to-cell propagation of the redox signal in plants (Miller et al., 2009).
An involvement of Nox homologs in the wound-healing process in plants has also been well documented. For example, in pea seedlings, H2O2 could be systemically induced by wounding, and the Nox inhibitor DPI significantly inhibited this wounding-induced H2O2 burst (Liu et al., 2005). In tomato, the plant Nox homolog Rboh has been shown to be required for the wound-induced expression of proteinase inhibitor II (Sagi et al., 2004). Moreover, Kumar et al. (2007) found that the Nox homolog RbohA might be important for wound-induced oxidative burst and wound healing in potato tubers. Taken together, these data suggest that activation of the Nox system during the wound healing process is likely to be an evolutionarily conserved phenomenon.
B. NADPH Oxidase and New Blood Vessel Development
Another biological response that is critical for tissue repair is angiogenesis, a process that restores functional microcirculation. Mounting evidence suggests that Nox-mediated signaling has a critical role in modulating the angiogenic functions of vascular endothelial cells (Ushio-Fukai, 2006). Nox2 and Nox4 isoforms are functionally important in modulating vascular endothelial cell biology. We and others have found that both Nox2 and Nox4 are responsible for ROS generation in endothelial cells, and knocking down either Nox2 or Nox4 expression with siRNA significantly suppresses endothelial proliferation (Petry et al., 2006; Peshavariya et al., 2009a). Despite these similarities between Nox2 and Nox4 in endothelial cells, we demonstrated that Nox4 protein expression was highly up-regulated in proliferating (subconfluent) cells but diminished in quiescent (confluent) cells, whereas the level of Nox2 remained unchanged (Peshavariya et al., 2009a). Moreover, we found that knocking down of Nox2, but not Nox4, in untreated endothelial cells induced apoptosis, although Nox4 overexpression conferred an antiapoptotic effect (Datla et al., 2007; Peshavariya et al., 2009a). In vitro, we have shown that Nox4 is a positive regulator of angiogenic activities of human microvascular endothelial cells, and this effect is associated with enhanced activation of the ERK pathway (Datla et al., 2007). Similar actions of Nox4 have also been reported in human pulmonary artery and lung microvascular endothelial cells (Pendyala et al., 2009). These observations together suggest that Nox4 might be a more suitable target for manipulating postnatal angiogenesis than Nox2, given the regulated expression of Nox4 in proliferating endothelial cells and that the basal Nox2 expression seems to be important in maintaining normal endothelial cell survival. In vivo angiogenesis has been found to be compromised in Nox2-deficient mouse (Tojo et al., 2005). The evidence for a role of Nox4 in angiogenesis in vivo is currently not available.
In addition to endothelial cells, Nox has been implicated in modulating the functions of pericytes. Pericyte recruitment after the initial capillary sprouting has an essential role in angiogenesis by facilitating vessel remodeling, maturation, and stabilization (Gerhardt and Betsholtz, 2003). On the other hand, it is thought that pericytes may suppress excessive endothelial cell proliferation in the late phase of angiogenesis (Hellström et al., 2001; Kondo et al., 2005). Expression of Nox1, together with p22phox and all cytosolic regulatory subunits, has been detected in microvascular pericytes (Manea et al., 2005). Activation of Nox in pericytes by stress stimuli has been shown to promote apoptosis (Cacicedo et al., 2005; Suzuma et al., 2007). These data indicate that Nox might have a dual role in angiogenesis (i.e., promoting the initial capillary sprouting while suppressing vessel maturation and stabilization at later stages).
Current data indicate that Nox might also be involved in embryonic new blood vessel development (vasculogenesis). As mentioned, Nox-dependent redox modulation results in inactivation of certain PTPs and thereby enhances receptor tyrosine kinase signaling. This leads to the activation of ERK and Akt, which are directly downstream of RTKs. Nox may also stimulate Akt activation by inactivating the phosphatase PTEN, a direct negative regulator of the PI3K/Akt pathway. In mouse embryos deficient in the gene encoding MEK1 (the immediate upstream kinase of ERK1/2), disrupted vascularization of the labyrinthine region of placenta was observed that was associated with a marked decrease in the number of vascular endothelial cells (Giroux et al., 1999). In the chicken chorioallantoic membrane model of vasculogenesis, it was found that fibroblast growth factor-1-induced vasculogenesis was sensitive to Akt inhibition (Forough et al., 2005). More direct evidence has been reported in cultured embryoid bodies, in which PDGF-induced vascular sprouting and branching of capillary-like structures, and the accompanying ROS generation and activation of ERK1/2, were blunted by Nox inhibitors (Lange et al., 2009).
C. NADPH Oxidase and Stem-Cell Function
Probably a more constructive aspect of Nox activation in tissue repair lies in that Nox-mediated redox signaling may modulate stem-cell expansion and differentiation. Sauer et al. (2000) have demonstrated that Nox function is dynamically regulated during embryonic stem-cell differentiation. Nox-derived ROS have been implicated in cardiomyocyte differentiation from mouse embryonic stem cells, and this effect of Nox seems to be mediated by modulating MAPK functions (Schmelter et al., 2006; Wo et al., 2008). More recent studies have provided evidence that Nox activation may also be involved in inducing embryonic stem-cell differentiation to the vascular smooth muscle lineage (Xiao et al., 2009). In addition, Nox seemed to be involved in modulating the proliferation of adult stem (precursor) cells, because reduction of p22phox expression or inhibition of Nox4 activity suppressed the proliferation rate of skeletal muscle precursor cells, which was accompanied by decreased ERK and Akt phosphorylation and reduced cyclin D1 expression (Mofarrahi et al., 2008). It is noteworthy that it has been observed that Nox4 gene silencing reduced ROS production in adipocyte precursors and suppressed insulin-induced differentiation of precursors into mature adipocytes but at the same time promoted precursor cell proliferation. This raises the possibility that Nox-mediated redox signaling in stem cells might act as a crucial switch in regulating proliferation or differentiation. Readers are referred to a recent review article for further discussions in this area (Ushio-Fukai and Urao, 2009).
VIII. Role of NADPH Oxidase-Mediated Stress Response in Vivo
Nox-mediated cellular stress response, stress tolerance, and subsequent repairing processes seem to be conserved, fundamental biological functions that aid cells and organisms to cope with adverse conditions and injuries. These mechanisms are activated in a range of pathophysiological conditions and may have complex, even contradictory roles in different diseases or different stages of the same disease. In some situations, NADPH oxidase-mediated stress tolerance and activation of cell survival pathways are beneficial in preventing unnecessary cell loss and accelerating injury repair, whereas these effects are most detrimental for proliferative diseases such as cancer. The double-facet role of Nox suggests that indiscriminately treating this enzyme as a source of oxidative stress is not an appropriate option.
A. Cardiovascular System
In the heart, Nox2 mediates the cardiomyocyte protective effects induced by preceding ischemic insults (Bell et al., 2005). On the other hand, the same group reported that, after cardiac infarction, Nox2 contributed to the cardiomyocyte apoptosis and hypertrophy (Looi et al., 2008). Moreover, there is evidence that Nox2 is involved in inducing myocardial fibrosis in a chronic setting (Hayashi et al., 2008; Looi et al., 2008). All these data suggest that the same Nox member may have differential affects on the development of pathologic conditions at different stages of ischemic heart disease. However, at present, there is no study that examines the effects of ectopically expressed Nox isoforms on the cell fate in normal and ischemic cardiomyocytes. Healing of injured blood vessels, such as endothelial denudation occurring during catheter-based interventional treatment, involves activation of both endothelial and smooth muscle cells. Re-endothelialization of the luminal surface is the prerequisite for restoration of normal blood vessel functions. As mentioned above, Nox-dependent signaling has a positive role in promoting endothelial cell proliferation, migration, and wound closure, thereby accelerating the recovery of the endothelial layer. It is noteworthy that recent studies have shown that Nox may also has a stimulating effect on mobilization of endothelial progenitor cells from the bone marrow (Schröder et al., 2009), which contribute to re-endothelialization and inhibition of neointimal growth after endothelial injury (Hristov et al., 2007). In Nox2 knockout mice, hypoxia and erythropoietin failed to induce the mobilization of endothelial progenitors, in contrast to wild-type and Nox1 knockouts (Schröder et al., 2009). In cultured endothelial progenitor cells, erythropoietin-induced ROS production, proliferation, and migration were all blunted by Nox2 deficiency, and inhibition of the phosphatase SHP-2 restored these responses, suggesting an involvement of ROS-dependent PTP inactivation. In addition, it was shown that re-endothelialization of the injured mouse carotid artery was enhanced by hypoxia as well as by erythropoietin, and this effect was not observed in Nox2-deficient animals (Schröder et al., 2009). In contrast to the beneficial effects of Nox in endothelial activation, Nox isoforms expressed in smooth muscle cells have been implicated in the development of neointimal hyperplasia, an outcome of aberrant healing process in muscular blood vessels. Wire injury-induced neointimal formation in the femoral artery was reduced in Nox1-deficient mice (Lee et al., 2009). Nox1-deficient smooth muscle cells exhibited reduced proliferation and migration responses, whereas Nox1 overexpression had opposite effects (Lee et al., 2009). Taking these results together, it is suggested that the precise role of Nox-mediated stress response in cardiovascular diseases needs to be examined in a gene-, cell-, and time-specific manner.
Nox-mediated angiogenic response is also a double-edged sword in different diseases. Inhibiting angiogenesis is a promising strategy for treatment of diseases such as cancer and proliferative diabetic retinopathy. On the other hand, therapeutic angiogenesis aims at promoting new blood vessel growth to treat ischemic disorders such as occlusive coronary and peripheral arterial diseases. Nox2 deficiency has been shown to delay the blood flow recovery in ischemic hind limbs (Tojo et al., 2005). It is noteworthy that using the same animal model, Urao et al. (2008) reported that Nox2 deficiency-induced impairment of ischemic angiogenesis was associated with reduced endothelial progenitor mobilization from the bone marrow in response to ischemic insults. It is noteworthy that it was shown that the impaired blood flow recovery in Nox2 knockout mice was rescued by transplantation of wild-type bone marrow cells into Nox2-deficient animals, indicating a therapeutic potential of targeting Nox in endothelial progenitors in treating ischemic cardiovascular diseases (Urao et al., 2008). Activation of Nox and enhanced angiogenesis may also have implications in tissue engineering, in which creation of functional vasculature is crucial for maintaining the viability of three-dimensional tissue blocks. We observed previously that inhibition of Nox function suppressed the neovascular invasion in a tissue engineering chamber and resulted in lumen malformation of the newly formed vessels (Hachisuka et al., 2008; Jiang et al., 2008).
In contrast to these beneficial roles of Nox-stimulated angiogenesis, there is evidence that Nox also contributes to pathological angiogenesis in other disease conditions. In a mouse model of oxygen-induced retinopathy, it was observed that treatment with the Nox inhibitor apocynin suppressed oxygen-induced retinal neovascularization and VEGF induction (Al-Shabrawey et al., 2005). Moreover, targeted suppression of p22phox expression in the retinal pigment epithelial cells reduced laser-induced vascular proliferative lesions (Li et al., 2008).
Nox may contribute to tumor growth by promoting angiogenesis. Arbiser et al. (2002) reported that prostate cancer cells overexpressing Nox1 exhibited enhanced tumor growth after in vivo inoculation, and the tumor became highly vascularized, accompanied by an up-regulation of VEGF and its receptors. Conversely, targeting of Nox may be an effective way to suppress tumor angiogenesis. For example, specific targeting of Nox4 with siRNA transfection and treatment with DPI significantly decreased angiogenesis by 60% in ovarian cancer cells implanted to the chicken chorioallantoic membrane (Xia et al., 2007). More recently, it has been shown that inhibition of Nox was effective in suppressing the growth of hemangioma, an endothelial cell-derived, benign tumor exclusively found in infants (Bhandarkar et al., 2009).
B. Inflammation
Nox was initially discovered in phagocytic cells and implicated in innate immunity and inflammation. Nox in nonphagocytic cells is responsive to various inflammatory stress stimuli such as LPS and proinflammatory cytokines and is well recognized as a critical mediator of inflammatory responses. However, several very recent studies by independent groups cast questions on this generalization. Using Nox2 and p47phox mice, it was found that LPS-induced systemic inflammatory responses (expression of inflammatory molecules in the lung, heart, and liver) were significantly enhanced by genetic deficiency of Nox (Zhang et al., 2009). Using the same approach, Segal et al. (2010) demonstrated that, when challenged with either intratracheal zymosan or LPS, Nox2- and p47phox-deficient mice developed exaggerated and progressive lung inflammations compared with wild-type mice. Moreover, it was shown that the putative antiinflammatory effect was mainly mediated by Nox in blood cells (Segal et al., 2010). Likewise, exposure of Nox2- and p47phox-deficient mice to cigarette smoke induced increased inflammatory response in the lung, despite decreased ROS production (Yao et al., 2008). The mechanisms underlying these actions of Nox are currently unclear. Experiments in isolated macrophages demonstrated that in the absence of functional Nox, zymosan failed to activate Nrf2 (Segal et al., 2010), a key stress-responsive transcription factor that mediated expression of an array of genes involved in stress defense. It is reasonable to speculate that the enhanced inflammatory responses in the absence of Nox might be related to the loss of basal cellular stress response and tolerance capacity mediated by ROS.
C. Cancer Cell Survival and Proliferation
A number of studies have shown that Nox-mediated redox modulation has a stimulating role in cancer cell proliferation and survival (for review, see Chan et al., 2009). Activation of ROS-dependent cell stress responses may confer tolerance of cancer cells to exogenous challenges such as chemotherapies and irradiation. Thus it is argued that a pro-oxidant intracellular milieu may be an acquired cancer trait to facilitate survival and growth by fueling proliferative capacity at the expense of death signalings (Tang et al., 2007). Thus, manipulating the intracellular redox state might potentially serve as a strategy for increasing their sensitivity to chemotherapeutic drugs or irradiation. In prostate cancer cells, it has been shown that androgen can induce elevated basal levels of ROS, thereby resulting in adaptation to radiation-induced cytotoxic stress (Lu et al., 2010). It is noteworthy that androgen-induced ROS generation in prostate cancer cells was dependent on Nox2 and Nox4; preradiation treatment with Nox inhibitors sensitized the cells to radiation-induced cytotoxicity (Lu et al., 2010). In another study, stable expression of the constitutively active mutant V12-H-Ras in virus transformed human lung cells dramatically increased intracellular production of superoxide via activation of Nox and induced resistance to TNF-α-mediated apoptosis (Liu et al., 2001). Consistently, treatment with ROS scavengers in V12-H-Ras cells reduced the TNF-α resistance and sensitized the cells for apoptosis (Liu et al., 2001). Moreover, several lines of evidence show that inhibition of Nox functions alone may trigger spontaneous apoptosis in pancreatic cancer cells, which exhibit properties of aggressive growth and unresponsiveness to drug treatments, indicating an essential role of Nox in maintaining a basal survival signal in these cells (Vaquero et al., 2004; Mochizuki et al., 2006). In addition, limited evidence suggests that redox signaling may also be involved in modulating the balance between self-renewal and differentiation of cancer stem or progenitor cells. Smith et al. (2000) demonstrated that factors that promoted self-renewal produced a relatively reduced status in cancer progenitors, whereas stimuli that promoted differentiation caused progenitors to become more oxidized. Little is known about the expression and functions of Nox in cancer stem cells. Although cancer stem cells are thought to be more quiescent and resistant to chemotherapy (Visvader and Lindeman, 2008), along with the notion that stem cells and the differentiated progenies might have different intracellular redox conditions (Ogasawara and Zhang, 2009), it will be interesting to see whether disruption of the redox balance in cancer stem cells by manipulating Nox enzymes may enhance their sensitivity to therapy-induced cytotoxicity in these cells. Then again, the effect of Nox activation on cancer cells is not unambiguous either. Ample evidence shows that activation of Nox in cancer cells may facilitate cell apoptosis. For instance, Carmona-Cuenca et al. (2008) provided evidence that up-regulation of Nox4 was required for TGF-β-induced caspase activation and cell death in hepatoma cells.
IX. Concluding Remarks—NADPH Oxidase at the Crossroads
Current experimental evidence suggests that Nox-mediated redox signaling may represent a novel stress signaling module, in which Nox enzymes respond to various cellular stresses and amplify the initial stress signal by facilitating multiple RTK-mediated pathways, resulting in a robust activation of MAPKs and/or JNK, the master regulators of the cellular stress response. Stress response is an evolutionarily conserved mechanism by which cells respond to and defend against abrupt adverse changes of the cellular environment (Welch, 1992). Nox activation during cellular stress can confer a survival signal to counterbalance the general cytostatic effects of various stresses via activation of the ERK1/2 and/or Akt pathways. Therefore, modulating Nox functions may represent a new approach to preventing stress-induced cell loss, such as that occurring during ischemia-reperfusion injury. The rationale of this argument is further supported by the reported observations that Nox-mediated cytoprotective “preconditioning” actions can be induced by various forms of cellular stresses in a panel of animal cells (Bell et al., 2005; Kimura et al., 2005; Das et al., 2006a,b; Tejima et al., 2007; Sánchez et al., 2008; Kim et al., 2009). However, this task is complicated because massive ROS production from Nox may also induce oxidative damage, which may be lethal to the cells. Hence, analyses on the spatial and temporal patterns of Nox activation during stresses, as well as the specific Nox isoform(s) involved in different cells and stress conditions, are essential to advance in this area.
At the other end, Nox-mediated stress tolerance can be detrimental in the killing of cancer cells. Many studies have shown that Nox activation may facilitate cancer cell proliferation and survival. Therefore, learning how to effectively shut down this signaling route should be a priority; alternatively, switching the Nox signaling from being a prosurvival pathway to a proapoptotic one might be a solution to stress tolerance of cancer cells. However, the challenge is that currently we know little about how these potential opposite effects of Nox in cell stress responses are orchestrated at the cellular level. It would seem that this would be influenced by the ROS species generated, the intensity of the stress signal, sites of ROS production, differentiation status of the cell, and interactions with other signaling pathways (Gechev et al., 2006; Terada, 2006). Obviously, these issues should be addressed in a cell-specific way because of the highly distinct expression patterns of Nox isoforms in different cell lineages. Moreover, another important question remaining is how the outcome of the Nox signaling initiated by a particular stress is affected by other concurrent stresses.
In summary, we propose that Nox-dependent ROS generation in response to cellular stresses may act as an alert system that primes the cells into a relatively stress-resistant status by integrating and amplifying the stress signals, providing preemptive protection against further cellular challenges (Pastori and Foyer, 2002) (see Fig. 3). The double-edged sword property of Nox adds another level of complexity for therapeutic targeting of this enzyme. The mechanisms of Nox-mediated redox signaling in different stress conditions may be paramount at the crossroads of directing the cellular responses to stress, aiming at either enhancing stress resistance in situations such as preventing ischemia-reperfusion injuries and accelerating wound healing, or sensitizing the stress-induced cytotoxicity for proliferative diseases such as cancer. Therefore, an optimal outcome of interventions on Nox will be achieved only when this is handled in a timely and disease- and stage-specific manner.

An integral role of NADPH oxidase in the cellular stress response signaling circuit. NADPH oxidase may be acutely activated and/or up-regulated by certain cellular stress sensing mechanisms. NADPH oxidase-dependent redox processes modulate the signaling cascades of the conserved stress kinase network and amplify its signal output, thereby priming the cells to a relatively stress-resistant status, providing preemptive protection against further cellular challenges. In the long run, activation of the NADPH oxidase system may facilitate the tissue repair process by promoting cell proliferation, differentiation, migration, and fibrosis. On the other hand, NADPH oxidase may also promote apoptosis and elimination of severely damaged cells. The final cellular outcome of the interactions between NADPH oxidase and stress kinases is supposed to be determined by multiple factors (e.g., the nature and strength of the stress, the predominant NADPH oxidase isoforms expressed in a particular cell type, the ROS species generated, the differentiation status of the stressed cells, the differential availability of pro- and antiapoptotic kinases) in a spatial and temporal context-dependent manner.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Jiang, Zhang, and Dusting.
Acknowledgments
This work was partially supported by the National 973 Basic Research Program of China [Grants 2010CB732605, 2009CB521900] (to F.J. and Y.Z., respectively); the National High-Tech Research and Development Program of China [Grant 2006AA02A406] (to Y.Z.), the National Health and Medical Research Council (NHMRC) of Australia [Grant 566911] (to G.J.D.); the National Heart Foundation of Australia [Grant-in-Aid G06M2662] (to G.J.D.); a Shandong University Qilu Scholar Professorship (to F.J.); and a NHMRC of Australia Principal Research Fellowship (to G.J.D.).
Footnotes
This article is available online at http://pharmrev.aspetjournals.org.
doi:10.1124/pr.110.002980.
↵1 Abbreviations:
- AP-1
- activator protein-1
- ASK-1
- apoptosis signal-regulating kinase-1
- ATF6
- activating transcription factor 6
- [Ca2+]i
- intracellular Ca2+ concentration
- DPI
- diphenyleneiodonium
- DS-MKP
- dual specificity MAPK phosphatases
- ER
- endoplasmic reticulum
- ERK
- extracellular signal-regulated kinase
- HEK
- human embryonic kidney
- HePTP
- hematopoietic protein-tyrosine phosphatase
- IRE1α
- inositol requiring enzyme 1α
- JNK
- c-Jun N-terminal kinase
- LPS
- lipopolysaccharide
- MAPK
- mitogen-activated protein kinase
- Nox
- NADPH oxidase
- oxLDL
- oxidized low density lipoprotein
- PDGF
- platelet-derived growth factor
- PI3K
- phosphoinositide 3-kinase
- PKC
- protein kinase C
- PTEN
- phosphatase and tensin homolog
- PTP
- protein tyrosine phosphatases
- ROS
- reactive oxygen species
- RTK
- receptor tyrosine kinase
- siRNA
- short interfering RNA
- TAS-103
- 6-((2-(dimethylamino)ethyl)amino)-3-hydroxy-7H-indeno(2,1-c)quinolin-7-one dihydrochloride
- TLR
- Toll-like receptor
- TNF
- tumor necrosis factor
- TRAF
- TNF-receptor-associated factor.
- © 2011 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





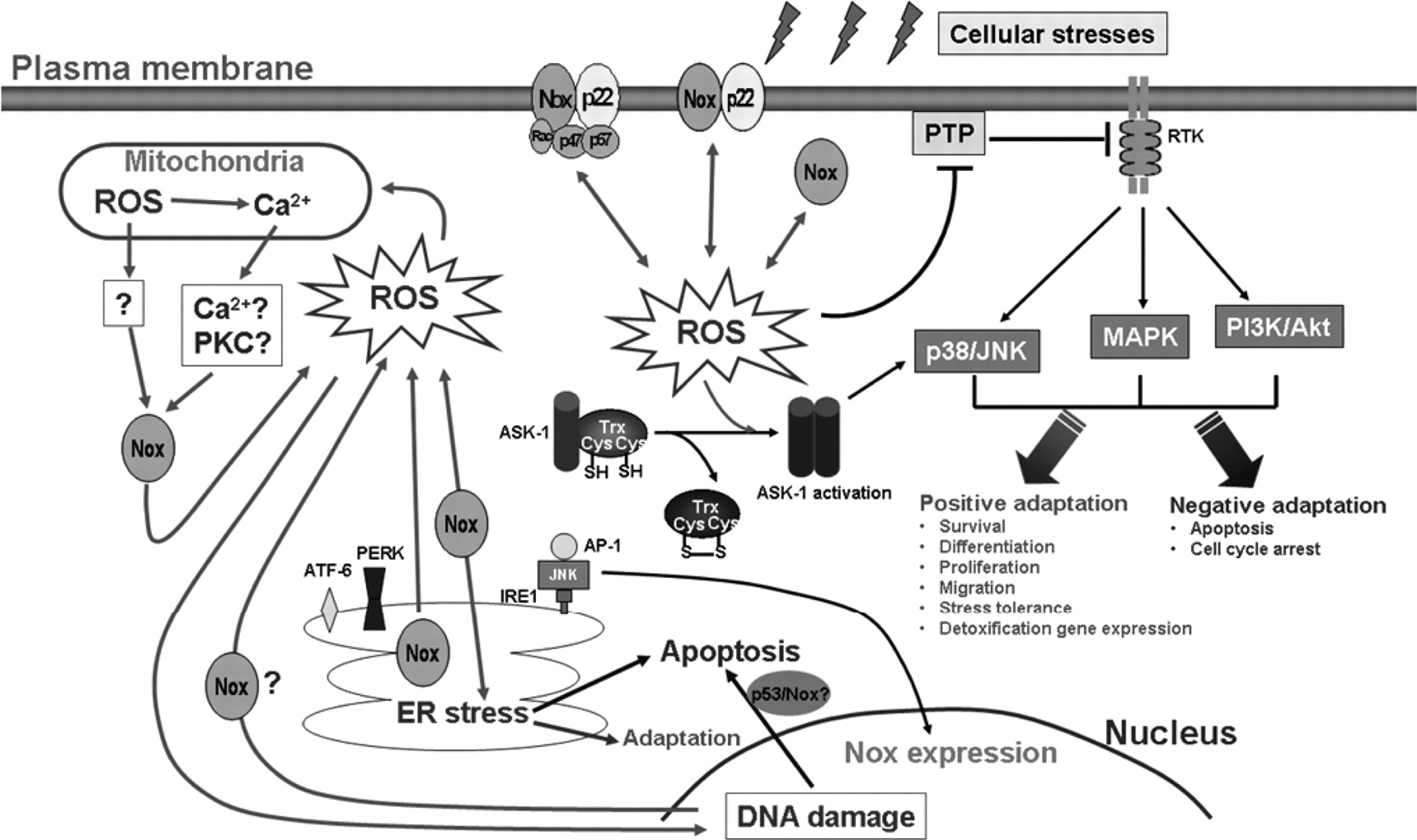{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- I. Introduction
- II. NADPH Oxidase Is Activated by Various Cellular Stresses
- III. Signal Output from NADPH Oxidase
- IV. Cell Survival versus Apoptosis
- V. Mechanisms of NADPH Oxidase Activation
- VI. NADPH Oxidase and the Stress Response Signaling Paradigm
- VII. NADPH Oxidase and Tissue Regeneration
- VIII. Role of NADPH Oxidase-Mediated Stress Response in Vivo
- IX. Concluding Remarks—NADPH Oxidase at the Crossroads
- Authorship Contributions
- Acknowledgments
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters